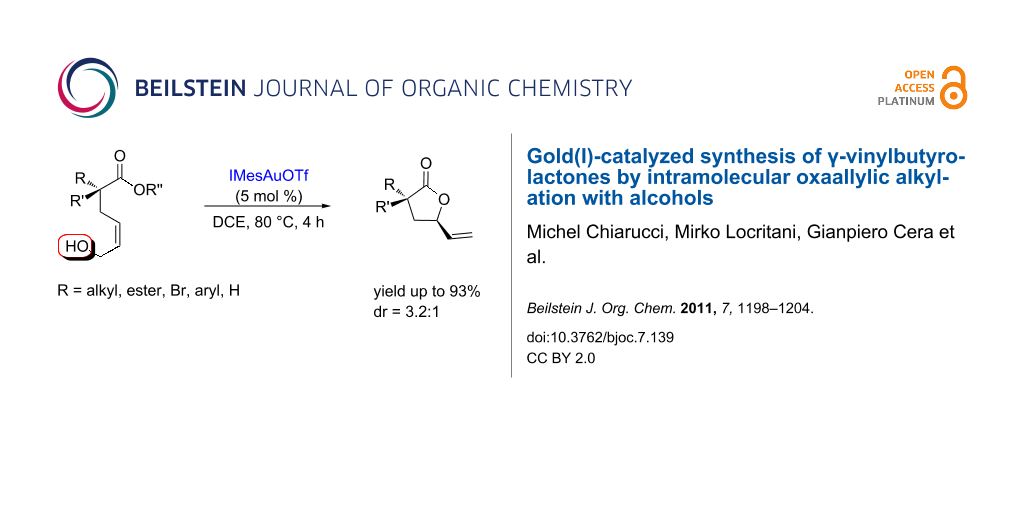
Abstract
Gold(I)-N-heterocyclic carbene (NHC) complexes proved to be a reliable catalytic system for the direct synthesis of functionalized γ-vinylbutyrolactones by intramolecular oxaallylic alkylation with primary alcohols. Good isolated chemical yields were obtained for a range of malonyl and acetate derivatives. The good performance in reagent-grade solvents and the functional group/moisture tolerance make this catalytic process a promising route for the synthesis of architecturally complex polycyclic structures.

Graphical Abstract
Introduction
Allylic alcohols are highly desirable, readily available, cheap, and environmental sustainable reaction partners for allylic alkylation reactions in the presence of C- as well as X-based (X: heteroatom) nucleophiles [1,2]. Despite their undoubted synthetic/economic advantages (i.e., water is the only stoichiometric byproduct produced), the intrinsic lower reactivity of allylic alcohols compared to allyl halides/acetates/carbonates generally necessitates harsher reaction conditions and/or the need for activating agents (i.e., Brønsted or Lewis acids) [3,4].
Recently, late-transition metal (LTM) catalysis (i.e., Hg, Pd, Pt, Au, and Ru) has received growing attention in organic synthesis and enables unprecedented manipulations of unfunctionalized hydrocarbons under mild reaction conditions [5-9]. In this context, electrophilic LTM activation of carbon–carbon unsaturations, adjacent to alcoholic moieties (i.e., allylic, benzylic, and propargylic alcohols, usually referred to as π-activated alcohols), deserves a particular mention [10-13].
As a part of our ongoing interest in the gold-catalyzed allylic functionalization of C- and heteroatom-based nucleophiles with alcohols [14-17], we previously observed the formation of synthetically useful vinylbutyrolactones [18-22] as minor products in the Friedel–Crafts-type allylic alkylation of arenes [23]. The wide impact of functionalized γ-lactones on the synthesis of naturally occurring compounds [24-26] prompted us to optimize a direct synthesis of vinylbutyrolactones by direct gold activation of allylic alcohols [27-31] with esters [32-37].
In this direction, we targeted malonyl alcohols 1 as a readily available class of model acyclic precursors to create chemical diversity through an oxaallylic ring-closing reaction (Figure 1).
![[1860-5397-7-139-1]](/bjoc/content/figures/1860-5397-7-139-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Working hypothesis for the present gold-catalyzed oxaallylic alkylation reaction.
Figure 1: Working hypothesis for the present gold-catalyzed oxaallylic alkylation reaction.
It should be noted that the synthesis of such a class of heterocyclic compounds has been the subject of several investigations. Among them, a multi-step synthetic pathway with final TBAF-promoted cyclization was proposed by Lepore [38] and, almost simultaneously, Poli and Prestat described a Pd-catalyzed Tsuji–Trost-type allylic alkylation procedure to obtain valuable precursors (i.e., lactams and lactones) of podophyllotoxin analogs [39,40]. However, to the best of our knowledge, no examples of metal-catalyzed lactonization through direct activation of allylic alcohols have been described so far.
Results and Discussion
At the outset of our investigation, we focused our attention on the non-enolizable allyl alcohol (Z)-1a, as a model candidate for the intramolecular oxaallyl alkylation. Our choice was dictated by the well-known reluctance of disubstituted malonyl derivatives to provide vinylbutyrolactones. This aspect was convincingly highlighted in a recent report by Chen and coworkers that described an analogous catalytic approach based on allyl acetate derivatives [41].
At this stage, an extended survey of reaction parameters (metal source, solvent, and temperature) was conducted in order to ascertain the optimal catalytic conditions (Table 1).
Table 1: Optimization of the reaction conditions for the lactonization of 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-139-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cat (%) | Solvent | Yield (%)b | (trans:cis)c |
|---|---|---|---|---|
| 1 | [P(t-Bu)2o-biphenyl](AuCH3CN)SbF6 (5) | DCE | 42 | nd |
| 2 | [P(Cy)2o-biphenyl-2,4,6(iPr)3]AuNTf2 (5) | DCE | 82 | 1.5:1 |
| 3 | PPh3AuNTf2 (5) | DCE | 52 | 1.1:1 |
| 4 | [(PPh3Au)3O]BF4 (2) | DCE | Trace | nd |
| 5 | AuCl3 (5) | DCE | <20 | nd |
| 6 | [biphepAu2Cl2/AgOTf] (2.5) | DCE | 56 | 1.3:1 |
| 7 | [dppf(AuNTf2)2] (2.5) | DCE | 98 | 1.2:1 |
| 8d | [dppf(AuNTf2)2] (0.5) | DCE | 96 | 1.4:1 |
| 9d,e | IMesAuOTf (5) | DCE | 94 | 2.1:1 |
| 10f | IMesAuOTf (5) | DCE | Trace | nd |
| 11 | IMesAuOTf (5) | Toluene | 31 | 1.9:1 |
| 12 | IMesAuOTf (5) | CH3CN | Trace | nd |
| 13 | IMesAuOTf (5) | THF | 79 | 1.9:1 |
| 14 | AgOTf (5) | DCE | 35 | nd |
| 15 | TsOH (10) | DCE | 64 | 1.3:1 |
| 16d,e,g | IMesAuOTf (5) | DCE | Trace | nd |
aAll the reactions were carried out under nitrogen atmosphere at 80 °C for 16 h, unless otherwise stated. bIsolated yield after flash chromatography. cDetermined by GC on the reaction crude. The relative configuration was determined by NOE experiments on the single diastereoisomers separated by flash chromatography. dUnder no moisture restriction, with reagent-grade solvent. eReaction time: 4 h. fAt room temperature. IMes: 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene. gIn the presence of K2CO3 (1 equiv). nd: not determined.
Initial attempts to perform the lactonization reaction of 1a were carried out by means of a silver-free cationic complex [P(t-Bu)2o-biphenyl](AuCH3CN)SbF6 (5 mol %). The desired butyrolactone 2a was obtained selectively under reflux in DCE for 16 h (entry 1), although only in low yield. With the less bulky triphenylphosphine ligand, the corresponding cationic gold(I) complex (i.e., PPh3AuNTf2) led to an increase in the isolated yield up to 52%, although the diastereoselection remained elusive (≈ 1:1, entry 3). After demonstrating that the Au(III) catalysis promoted the cyclization in lower extent compared to the Au(I) counterpart (entry 5 versus entries 1–3), we also observed that dinuclear [dppf(AuNTf2)2] provided 2a with almost complete conversion (entry 7). The possibility to reduce the loading of the catalyst (0.5 mol %) further, without the need for moisture restriction, was successfully verified by the isolation of 2a in 96% isolated yield (entry 8). Interestingly, the diastereoselection of the protocol was slightly improved (up to 2.1:1) and the reaction time shortened to 4 h, by employing the carbene-based gold complex IMesAuCl/AgOTf (5 mol %, entry 9) [42,43]. Therefore, by addressing NHCAuOTf as the optimal catalytic system, the impact of the reaction media on the chemical output of the process was investigated (entries 10–13). Here, although 2a was also isolated in good yield in reagent-grade THF (yield = 79%, dr = 1.9:1, entry 13), DCE was employed as the solvent of choice.
In order to confirm that the catalysis was indeed due to the presence of gold, a control experiment with AgOTf (5 mol %) was performed on compound 1a. Under comparable reaction conditions (80 °C, 16 h), lactone 2a was isolated in poor yield (35%). Finally, the hypothetical cocatalysis by Brønsted acids (BA) was verified by means of experimental controls with TsOH (entry 15), and also in the presence of an acid scavenger (entry 16). Here, the desired cyclic compound 2a was obtained in lower yield (64%) with concomitant substantial decomposition of the starting allylic alcohol. Such evidence confirms the allylic SN1 mechanism for the present methodology [44].
The high chemoselectivity guaranteed by the gold catalysts is worthy of note, as it channels the reaction toward the allylic alkylation mechanism without any contamination deriving from transesterification reactions. This evidence is reasonably rationalized in terms of the high π-acidity and poor oxophilicity of the Au(I) species [37].
With the optimal catalytic systems in hand (IMesAuOTf or [dppf(AuNTf2)2], DCE, 80 °C), we verified the generality of the method by subjecting a range of malonyl alcohols 1b–j to the gold-catalyzed lactonization (Table 2).
Table 2: Proving the scope of the gold-catalyzed intramolecular allylation of 1.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-139-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 1 | Catalytic system | Product | Yield (%)b | trans:cisc |
|---|---|---|---|---|---|
| 1 | (E)-1a | A |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-139-i5.svg?max-width=637&scale=1.0)
2a |
72 | 1.3:1 |
| 2 | (Z)-1b | A |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-139-i6.svg?max-width=637&scale=1.0)
2b |
95 | 1:1 |
| 3 | (Z)-1c | A |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-139-i7.svg?max-width=637&scale=1.0)
2c |
66 | 1.1:1 |
| 4 | (Z)-1d | A |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-139-i8.svg?max-width=637&scale=1.0)
2d |
67 | 1.4:1 |
| 5 | (Z)-1e | A |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-139-i9.svg?max-width=637&scale=1.0)
2e |
85 | 1:1 |
| 6 | (Z)-1f | A, B |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-139-i10.svg?max-width=637&scale=1.0)
2f |
94, 35 | 1.5:1, 1.5:1 |
| 7 | (Z)-1g | Ad, Be |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-139-i11.svg?max-width=637&scale=1.0)
2g |
63, 12 | 1:1.4, 1:1.4 |
| 8 | (Z)-1h | A |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-139-i12.svg?max-width=637&scale=1.0)
2h |
54 | 3.2:1 |
| 9 | (Z)-1i | A |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-139-i13.svg?max-width=637&scale=1.0)
2i |
45 | 1.4:1 |
| 10 | (Z)-1j | Af |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-139-i14.svg?max-width=637&scale=1.0)
2j |
93 | 1.1:1 |
aAll the reactions were carried out in reagent-grade solvents under air (80 °C, 0.3 M). Catalytic systems: A = IMesAuCl/AgOTf (5 mol %), DCE, 80 °C, 7–9 h. B = [dppf(AuNTf2)2] (2.5 mol %), THF, 80 °C, 16 h. bIsolated yield after flash chromatography. cDetermined by GC on the reaction crude. dDihydronaphthalene derived from undesired Friedel–Crafts alkylation (yield = 14%). eA considerable amount of Friedel–Crafts dihydronaphthalene (yield = 67%) was isolated [23]. f10 mol % of catalyst was used.
The impact of the carbon–carbon double bond configuration on both chemical and stereochemical outputs of the process was initially investigated. Here, by subjecting (E)-1a to the reaction conditions A (i.e., IMesAuCl/AgOTf, DCE, 80 °C) the corresponding lactone 2a was isolated in comparable yield (72%, entry 1) and similar diastereomeric ratio. Here, although the impact of the C─C double bond configuration on the stereochemical outcome of SN2′-type gold-catalyzed intramolecular O- [45] and N-alkylations [37,46] with allylic alcohols was demonstrated, we consider it likely that an allylic SN1 mechanism is involved in the present methodology, due to the similar optical outcomes obtained in the presence of BA metal-free catalysts (entry 15, Table 1).
Then, enolizable substrates carrying different malonyl residues (1b–e) were taken into account. In all cases the cyclization occurred smoothly leading to the disappearance of the acyclic precursors within 7–9 h reaction time (entries 2–5). Interestingly, in this case no appreciable differences in reaction rate were observed between substrates carrying labile and nonlabile ester alkyl groups.
In some specific cases, both catalytic systems were tested and a direct comparison of performances can be made. Clear evidence was gained for the higher activity of the catalytic system A in the expected oxaallylic alkylation process. As an example, when multiple reactive channels were available (i.e., lactonization and Friedel–Crafts-type alkylation, 1g) dppf-based species (catalytic system B) led to a complex mixture of crude reaction products (entry 7), while carbene–gold complex provided mainly the butyrolactone 2g. Moreover, the methodology proved to be tolerant toward several functional groups/atoms at the methylene carbon atom of the malonyl derivative. In particular, 3,5-trans-3-bromo-γ-vinylbutyrolactone 2h was isolated with 54% yield and in 76:24 diastereoisomeric ratio (entry 8). A protected carbonyl moiety was also tested leading to the corresponding lactone 2i in moderate yield (45%, entry 9). Finally, the methodology proved to be adaptable allowing a double lactonization event with 1j, hence providing spiro-lactone 2j in 93% yield.
Apart from the generality on malonyl substrates, we decided to explore the applicability of the present methodology to less reactive monoester analogs [47]. In this context, readily available alcohols 3a,b were subjected to cyclization in the presence of the gold catalytic system A. In both cases lactones 4a,b were isolated in good to excellent yields (93 and 75%, respectively, Scheme 1).
![[1860-5397-7-139-i1]](/bjoc/content/inline/1860-5397-7-139-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Gold-catalyzed synthesis of γ-lactones 4 from the corresponding monoesters 3.
Scheme 1: Gold-catalyzed synthesis of γ-lactones 4 from the corresponding monoesters 3.
Finally, the 1,3-ketoester 3c was also subjected to the optimized conditions, but a complex reaction mixture was observed with concomitant decomposition of the starting material.
The mechanistic proposal for the formation of γ-vinylbutyrolactones 2 is depicted in Scheme 2. As previously mentioned, the formation of an allylic cationic species (II) is assumed, upon coordination of the gold catalyst to the allylic alcohol (I). In Scheme 2, the possible coordination modes for [Au+] to the allylic alcohol are reported. As a matter of fact, although we have previously demonstrated the C=C···Au interaction in the presence of allylic alcohols [16], a concomitant [Au]···OH contact cannot be ruled out [48,49]. Subsequently, the direct nucleophilic attack by the carboxylate unit would lead to an oxonium intermediate III [50,51] that, after dealkylation, resulted in the final lactone 2. Control experiments have been performed to indentify the presence of a Brønsted acid cocatalysis in the ring-closing procedure (see [52] and entry 15 in Table 1). Regeneration of the active cationic gold species or assistance in the formation of the reactive allylic carbocation intermediate II are key steps in which the Brønsted cocatalysis could be exerted [52]. Finally, the mandatory role of enol tautomer (or gold–enolate intermediates) [53-55] in the nucleophilic attack was excluded; non-enolizable compounds being suitable candidates for the cyclization reaction.
![[1860-5397-7-139-i2]](/bjoc/content/inline/1860-5397-7-139-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanistic sketch of the gold-promoted oxaallylic alkylation reaction.
Scheme 2: Mechanistic sketch of the gold-promoted oxaallylic alkylation reaction.
Conclusions
In conclusion, we have documented an unprecedented example of gold-catalyzed lactonization with primary allylic alcohols. Cationic NHCAu carbene gold complexes allowed the preparation of a range of functionalized malonyl esters by direct activation of the allylic alcohol by gold. The methodology appears highly chemoselective toward the allylic lactonization, with the possibility to extend the protocol also to acetate derivatives.
Supporting Information
| Supporting Information File 1: Experimental details and characterization of the synthesized compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
References
-
Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501. doi:10.1039/b823217b
Return to citation in text: [1] -
Bandini, M. Angew. Chem., Int. Ed. 2011, 50, 994. doi:10.1002/anie.201006522
And references therein.
Return to citation in text: [1] -
Kimura, M.; Tamaru, Y. Mini-Rev. Org. Chem. 2009, 6, 392. doi:10.2174/157019309789371631
Return to citation in text: [1] -
Hartwig, J. F. Allylic Substitution. In Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010; pp 967–1014.
Return to citation in text: [1] -
Nevado, C.; Echavarren, A. M. Synthesis 2005, 167. doi:10.1055/s-2005-861781
Return to citation in text: [1] -
Widenhoefer, R. A.; Han, X. Eur. J. Org. Chem. 2006, 4555. doi:10.1002/ejoc.200600399
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Chianese, A. R.; Lee, S. J.; Gagné, M. R. Angew. Chem., Int. Ed. 2007, 46, 4042. doi:10.1002/anie.200603954
Return to citation in text: [1] -
Abu Sohel, S. M.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m
Return to citation in text: [1] -
Defieber, C.; Ariger, M. A.; Moriel, P.; Carreira, E. M. Angew. Chem., Int. Ed. 2007, 46, 3139. doi:10.1002/anie.200700159
Return to citation in text: [1] -
Tanaka, S.; Seki, T.; Kitamura, M. Angew. Chem., Int. Ed. 2009, 48, 8948. doi:10.1002/anie.200904671
Return to citation in text: [1] -
Yamamoto, H.; Ho, E.; Namba, K.; Imagawa, H.; Nishizawa, M. Chem.–Eur. J. 2010, 16, 11271. doi:10.1002/chem.201001656
Return to citation in text: [1] -
Miyata, K.; Kutsana, H.; Kawakami, S.; Kitamura, M. Angew. Chem., Int. Ed. 2011, 50, 4649. doi:10.1002/anie.201100772
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9533. doi:10.1002/anie.200904388
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A.; Gualandi, A.; Quinto, T.; Savoia, D. ChemCatChem 2010, 2, 661. doi:10.1002/cctc.201000077
Return to citation in text: [1] -
Bandini, M.; Monari, M.; Romaniello, A.; Tragni, M. Chem.–Eur. J. 2010, 16, 14272. doi:10.1002/chem.201002606
Return to citation in text: [1] [2] -
Bandini, M.; Gualandi, A.; Monari, M.; Romaniello, A.; Savoia, D.; Tragni, M. J. Organomet. Chem. 2011, 696, 338. doi:10.1016/j.jorganchem.2010.09.065
Return to citation in text: [1] -
Hon, Y.-S.; Chen, H.-F.; Kao, C.-Y.; Luo, C.-Z. Tetrahedron 2010, 66, 8468. doi:10.1016/j.tet.2010.08.035
Return to citation in text: [1] -
Park, B. R.; Kim, S. H.; Kim, Y. M.; Kim, J. N. Tetrahedron Lett. 2011, 52, 1700. doi:10.1016/j.tetlet.2011.01.153
Return to citation in text: [1] -
He, H.; Dai, L.-X.; You, S.-L. Org. Biomol. Chem. 2010, 8, 3207. doi:10.1039/b924770j
Return to citation in text: [1] -
Csuk, R.; Barthel, A.; Schwarz, S.; Kommera, H.; Paschke, R. Bioorg. Med. Chem. 2010, 18, 2549. doi:10.1016/j.bmc.2010.02.042
Return to citation in text: [1] -
Elford, T. G.; Hall, D. G. Synthesis 2010, 893. doi:10.1055/s-0029-1218664
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A.; Kotrusz, P.; Tragni, M.; Troisi, S.; Umani-Ronchi, A. Adv. Synth. Catal. 2009, 351, 319. doi:10.1002/adsc.200800628
Return to citation in text: [1] [2] -
Ward, R. S. Recent Advances in the Chemistry of Lignans. In Studies in Natural Products Chemistry, Volume 24, Bioactive Natural Products, Part E; Rahman, A.-u., Ed.; Elsevier: Amsterdam, 2000; pp 739–798.
Return to citation in text: [1] -
Ward, R. S. Nat. Prod. Rep. 1999, 16, 75. doi:10.1039/a705992b
Return to citation in text: [1] -
Saleem, M.; Kim, H. J.; Ali, M. S.; Lee, Y. S. Nat. Prod. Rep. 2005, 22, 696. doi:10.1039/b514045p
Return to citation in text: [1] -
Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395. doi:10.1038/nature05592
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g
Return to citation in text: [1] -
Aponick, A.; Biannic, B. Synthesis 2008, 3356. doi:10.1055/s-0028-1083160
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Palmes, J. A. Org. Lett. 2009, 11, 121. doi:10.1021/ol802491m
Return to citation in text: [1] -
Rao, W.; Chan, P. W. H. Org. Biomol. Chem. 2008, 6, 2426. doi:10.1039/b805067h
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Biannic, B. Org. Lett. 2008, 10, 669. doi:10.1021/ol703002p
Return to citation in text: [1] -
Aponick, A.; Biannic, B.; Jong, M. R. Chem. Commun. 2010, 46, 6849. doi:10.1039/c0cc01961e
Return to citation in text: [1] -
Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2011, 13, 1334. doi:10.1021/ol103175w
Return to citation in text: [1] [2] [3] -
Silvestri, M. A.; He, C.; Khoram, A.; Lepore, S. D. Tetrahedron Lett. 2006, 47, 1625. doi:10.1016/j.tetlet.2005.12.114
Return to citation in text: [1] -
Vitale, M.; Prestat, G.; Lopes, D.; Madec, D.; Poli, G. Synlett 2006, 2231. doi:10.1055/s-2006-949651
Return to citation in text: [1] -
Vitale, M.; Prestat, G.; Lopes, D.; Madec, D.; Kammerer, C.; Poli, G.; Girnita, L. J. Org. Chem. 2008, 73, 5795. doi:10.1021/jo800707q
And references therein.
Return to citation in text: [1] -
Wang, Y.-H.; Zhu, L.-L.; Zhang, Y.-X.; Chen, Z. Chem. Commun. 2010, 46, 577. doi:10.1039/b913348h
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776. doi:10.1039/b711132k
Return to citation in text: [1] -
Nolan, S. P. Acc. Chem. Res. 2011, 44, 91. doi:10.1021/ar1000764
Return to citation in text: [1] -
Different Lewis acids were tested (10 mol %), furnishing 2a in lower yields in comparison to IMesAuOTf and [dppf(AuNTf2)2]. Zn(OTf)2: traces, [codPt(OTf)2]: 65% (dr = 1.7:1), In(OTf)3: 87% (dr = 1.1:1), Bi(OTf)3: 80% (dr = 1.9:1).
Return to citation in text: [1] -
Aponick, A.; Biannic, B. Org. Lett. 2011, 13, 1330. doi:10.1021/ol200203k
Return to citation in text: [1] -
Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2010, 12, 1184. doi:10.1021/ol902923e
Return to citation in text: [1] -
Gierasch, T. M.; Shi, Z.; Verdine, G. L. Org. Lett. 2003, 5, 621. doi:10.1021/ol027116f
The usefulness of compounds of 4-type in obtaining libraries of diversity oriented-synthesis has been documented.
Return to citation in text: [1] -
Georgy, M.; Boucard, V.; Campagne, J.-M. J. Am. Chem. Soc. 2005, 127, 14180. doi:10.1021/ja0534147
Return to citation in text: [1] -
Georgy, M.; Boucard, V.; Debleds, O.; Dal Zotto, C.; Campagne, J.-M. Tetrahedron 2009, 65, 1758. doi:10.1016/j.tet.2008.12.051
Return to citation in text: [1] -
Liu, L.-P.; Xu, B.; Mashuta, M. S.; Hammond, G. B. J. Am. Chem. Soc. 2008, 130, 17642. doi:10.1021/ja806685j
Return to citation in text: [1] -
Liu, L.-P.; Hammond, G. B. Chem.–Asian J. 2009, 4, 1230. doi:10.1002/asia.200900091
Return to citation in text: [1] -
In this context, when K2CO3 (1 equiv) was used as a scavenger, the desired product 2a was obtained only in trace amount. Interestingly, the use of stoichiometric amounts of TFA led to the formation of the O-allyl trifluoroacetate derivative exclusively (yield = 63%).
Return to citation in text: [1] [2] -
Yao, X.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 6884. doi:10.1021/ja0482637
Return to citation in text: [1] -
Hashmi, A. S. K.; Schäfer, S.; Wölfle, M.; Gil, C. D.; Fischer, P.; Laguna, A.; Blanco, M. C.; Gimeno, M. C. Angew. Chem., Int. Ed. 2007, 46, 6184. doi:10.1002/anie.200701521
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078
Return to citation in text: [1]
| 1. | Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501. doi:10.1039/b823217b |
| 2. |
Bandini, M. Angew. Chem., Int. Ed. 2011, 50, 994. doi:10.1002/anie.201006522
And references therein. |
| 14. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9533. doi:10.1002/anie.200904388 |
| 15. | Bandini, M.; Eichholzer, A.; Gualandi, A.; Quinto, T.; Savoia, D. ChemCatChem 2010, 2, 661. doi:10.1002/cctc.201000077 |
| 16. | Bandini, M.; Monari, M.; Romaniello, A.; Tragni, M. Chem.–Eur. J. 2010, 16, 14272. doi:10.1002/chem.201002606 |
| 17. | Bandini, M.; Gualandi, A.; Monari, M.; Romaniello, A.; Savoia, D.; Tragni, M. J. Organomet. Chem. 2011, 696, 338. doi:10.1016/j.jorganchem.2010.09.065 |
| 44. | Different Lewis acids were tested (10 mol %), furnishing 2a in lower yields in comparison to IMesAuOTf and [dppf(AuNTf2)2]. Zn(OTf)2: traces, [codPt(OTf)2]: 65% (dr = 1.7:1), In(OTf)3: 87% (dr = 1.1:1), Bi(OTf)3: 80% (dr = 1.9:1). |
| 10. | Defieber, C.; Ariger, M. A.; Moriel, P.; Carreira, E. M. Angew. Chem., Int. Ed. 2007, 46, 3139. doi:10.1002/anie.200700159 |
| 11. | Tanaka, S.; Seki, T.; Kitamura, M. Angew. Chem., Int. Ed. 2009, 48, 8948. doi:10.1002/anie.200904671 |
| 12. | Yamamoto, H.; Ho, E.; Namba, K.; Imagawa, H.; Nishizawa, M. Chem.–Eur. J. 2010, 16, 11271. doi:10.1002/chem.201001656 |
| 13. | Miyata, K.; Kutsana, H.; Kawakami, S.; Kitamura, M. Angew. Chem., Int. Ed. 2011, 50, 4649. doi:10.1002/anie.201100772 |
| 37. | Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2011, 13, 1334. doi:10.1021/ol103175w |
| 5. | Nevado, C.; Echavarren, A. M. Synthesis 2005, 167. doi:10.1055/s-2005-861781 |
| 6. | Widenhoefer, R. A.; Han, X. Eur. J. Org. Chem. 2006, 4555. doi:10.1002/ejoc.200600399 |
| 7. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335 |
| 8. | Chianese, A. R.; Lee, S. J.; Gagné, M. R. Angew. Chem., Int. Ed. 2007, 46, 4042. doi:10.1002/anie.200603954 |
| 9. | Abu Sohel, S. M.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m |
| 41. | Wang, Y.-H.; Zhu, L.-L.; Zhang, Y.-X.; Chen, Z. Chem. Commun. 2010, 46, 577. doi:10.1039/b913348h |
| 3. | Kimura, M.; Tamaru, Y. Mini-Rev. Org. Chem. 2009, 6, 392. doi:10.2174/157019309789371631 |
| 4. | Hartwig, J. F. Allylic Substitution. In Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010; pp 967–1014. |
| 42. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776. doi:10.1039/b711132k |
| 43. | Nolan, S. P. Acc. Chem. Res. 2011, 44, 91. doi:10.1021/ar1000764 |
| 27. | Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395. doi:10.1038/nature05592 |
| 28. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x |
| 29. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l |
| 30. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319 |
| 31. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g |
| 38. | Silvestri, M. A.; He, C.; Khoram, A.; Lepore, S. D. Tetrahedron Lett. 2006, 47, 1625. doi:10.1016/j.tetlet.2005.12.114 |
| 24. | Ward, R. S. Recent Advances in the Chemistry of Lignans. In Studies in Natural Products Chemistry, Volume 24, Bioactive Natural Products, Part E; Rahman, A.-u., Ed.; Elsevier: Amsterdam, 2000; pp 739–798. |
| 25. | Ward, R. S. Nat. Prod. Rep. 1999, 16, 75. doi:10.1039/a705992b |
| 26. | Saleem, M.; Kim, H. J.; Ali, M. S.; Lee, Y. S. Nat. Prod. Rep. 2005, 22, 696. doi:10.1039/b514045p |
| 39. | Vitale, M.; Prestat, G.; Lopes, D.; Madec, D.; Poli, G. Synlett 2006, 2231. doi:10.1055/s-2006-949651 |
| 40. |
Vitale, M.; Prestat, G.; Lopes, D.; Madec, D.; Kammerer, C.; Poli, G.; Girnita, L. J. Org. Chem. 2008, 73, 5795. doi:10.1021/jo800707q
And references therein. |
| 23. | Bandini, M.; Eichholzer, A.; Kotrusz, P.; Tragni, M.; Troisi, S.; Umani-Ronchi, A. Adv. Synth. Catal. 2009, 351, 319. doi:10.1002/adsc.200800628 |
| 18. | Hon, Y.-S.; Chen, H.-F.; Kao, C.-Y.; Luo, C.-Z. Tetrahedron 2010, 66, 8468. doi:10.1016/j.tet.2010.08.035 |
| 19. | Park, B. R.; Kim, S. H.; Kim, Y. M.; Kim, J. N. Tetrahedron Lett. 2011, 52, 1700. doi:10.1016/j.tetlet.2011.01.153 |
| 20. | He, H.; Dai, L.-X.; You, S.-L. Org. Biomol. Chem. 2010, 8, 3207. doi:10.1039/b924770j |
| 21. | Csuk, R.; Barthel, A.; Schwarz, S.; Kommera, H.; Paschke, R. Bioorg. Med. Chem. 2010, 18, 2549. doi:10.1016/j.bmc.2010.02.042 |
| 22. | Elford, T. G.; Hall, D. G. Synthesis 2010, 893. doi:10.1055/s-0029-1218664 |
| 32. | Aponick, A.; Biannic, B. Synthesis 2008, 3356. doi:10.1055/s-0028-1083160 |
| 33. | Aponick, A.; Li, C.-Y.; Palmes, J. A. Org. Lett. 2009, 11, 121. doi:10.1021/ol802491m |
| 34. | Rao, W.; Chan, P. W. H. Org. Biomol. Chem. 2008, 6, 2426. doi:10.1039/b805067h |
| 35. | Aponick, A.; Li, C.-Y.; Biannic, B. Org. Lett. 2008, 10, 669. doi:10.1021/ol703002p |
| 36. | Aponick, A.; Biannic, B.; Jong, M. R. Chem. Commun. 2010, 46, 6849. doi:10.1039/c0cc01961e |
| 37. | Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2011, 13, 1334. doi:10.1021/ol103175w |
| 37. | Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2011, 13, 1334. doi:10.1021/ol103175w |
| 46. | Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2010, 12, 1184. doi:10.1021/ol902923e |
| 23. | Bandini, M.; Eichholzer, A.; Kotrusz, P.; Tragni, M.; Troisi, S.; Umani-Ronchi, A. Adv. Synth. Catal. 2009, 351, 319. doi:10.1002/adsc.200800628 |
| 53. | Yao, X.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 6884. doi:10.1021/ja0482637 |
| 54. | Hashmi, A. S. K.; Schäfer, S.; Wölfle, M.; Gil, C. D.; Fischer, P.; Laguna, A.; Blanco, M. C.; Gimeno, M. C. Angew. Chem., Int. Ed. 2007, 46, 6184. doi:10.1002/anie.200701521 |
| 55. | Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078 |
| 52. | In this context, when K2CO3 (1 equiv) was used as a scavenger, the desired product 2a was obtained only in trace amount. Interestingly, the use of stoichiometric amounts of TFA led to the formation of the O-allyl trifluoroacetate derivative exclusively (yield = 63%). |
| 52. | In this context, when K2CO3 (1 equiv) was used as a scavenger, the desired product 2a was obtained only in trace amount. Interestingly, the use of stoichiometric amounts of TFA led to the formation of the O-allyl trifluoroacetate derivative exclusively (yield = 63%). |
| 48. | Georgy, M.; Boucard, V.; Campagne, J.-M. J. Am. Chem. Soc. 2005, 127, 14180. doi:10.1021/ja0534147 |
| 49. | Georgy, M.; Boucard, V.; Debleds, O.; Dal Zotto, C.; Campagne, J.-M. Tetrahedron 2009, 65, 1758. doi:10.1016/j.tet.2008.12.051 |
| 50. | Liu, L.-P.; Xu, B.; Mashuta, M. S.; Hammond, G. B. J. Am. Chem. Soc. 2008, 130, 17642. doi:10.1021/ja806685j |
| 51. | Liu, L.-P.; Hammond, G. B. Chem.–Asian J. 2009, 4, 1230. doi:10.1002/asia.200900091 |
| 47. |
Gierasch, T. M.; Shi, Z.; Verdine, G. L. Org. Lett. 2003, 5, 621. doi:10.1021/ol027116f
The usefulness of compounds of 4-type in obtaining libraries of diversity oriented-synthesis has been documented. |
| 16. | Bandini, M.; Monari, M.; Romaniello, A.; Tragni, M. Chem.–Eur. J. 2010, 16, 14272. doi:10.1002/chem.201002606 |
© 2011 Chiarucci et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)