
Abstract
The conformational properties of 1,3-diindolylureas and thioureas were studied by a combination of heteronuclear NMR spectroscopy and quantum mechanics calculations. NOE experiments showed that the anti–anti conformer along the C7–N7α bonds was predominant in DMSO-d6 solution in the absence of anions. Anion-induced changes in the 1H and 15N chemical shifts confirm the weak binding of chloride anions with negligible conformational changes. Strong deshielding of ureido protons and moderate deshielding of indole NH was observed upon the addition of acetate, benzoate, bicarbonate and dihydrogen phosphate, which indicated that the predominant hydrogen bond interactions occurred at the urea donor groups. Binding of oxo-anions caused conformational changes along the C7–N7α bonds and the syn–syn conformer was preferred for anion–receptor complexes. The conformational changes upon anion binding are in good agreement with energetic preferences established by ab initio calculations.
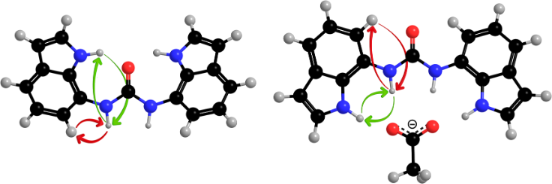
Graphical Abstract
Introduction
In the last two decades, remarkable efforts have been made in the field of the development of synthetic anion receptors, motivated by prospective applications in recognition, separation, guest inclusion and catalysis [1-13]. The fundamental role of anions in biological and chemical processes drives much of this research. Biomolecules such as the sulfate binding protein [14] and phosphate binding protein [15] employ hydrogen bonds as the key driving force to bind or transport anions through cell membranes. Hydrogen bonding interactions are extensively employed in synthetic anion receptors comprising groups such as amides, pyrroles, indoles, ureas and triazoles, as well as in ammonium, guanidinium and imidazolium moieties used as hydrogen bond donors [16-23]. Amongst neutral anion receptor systems, indole and related heterocycles, such as carbazole, 2,2'-biindole and indolo[1,2-b]carbazoles, have recently attracted significant attention [24-31]. Indole contains a single hydrogen bond donor group, which is employed in biological systems to bind anions such as chloride [32] and sulfate [14].
We have recently analyzed the conformational preferences of several 2,7-disubstituted indoles with amide substituents at C2 and urea substituents at C7, which showed preference for distinct conformations in the presence and in the absence of anions [33-35]. In addition, indole and urea groups were strongly involved in hydrogen-bonding interactions with the bound anionic guest, whilst the amide group interacted only weakly with the bound anion. These observations led to the design of diindolylureas and diindolylthioureas [36-38]. These compounds have remarkably high affinities for oxo-anions such as phosphate and sulfate for neutral receptors in DMSO-d6/0.5% water and have been shown to perturb the pKa of bound guests (Table 1) [38,39]. X-ray crystal structures of a variety of complexes with anions revealed the adoption of the syn–syn conformation in the solid state upon anion complexation. With the urea analogues, such as 1, this is accompanied by higher order complex formation with oxo-anions in the solid state. For example, with dihydrogen phosphate, three equivalents of receptor complex to a single anion, which has doubly deprotonated, resulting in the formation of a complex in which twelve hydrogen bonds stabilize the PO43− anion. In solution, the thiourea analogues such as compound 2 show significantly lower affinities for oxo-anions than do the urea analogues. We had previously proposed that this may be due to the larger size of the sulfur atom resulting in a lower propensity of these systems to adopt a planar conformation. Whilst the conformational properties of these compounds have been explored by single crystal X-ray diffraction in the solid-state, a detailed analysis of the conformational properties of the these compounds in solution, in the absence and presence of oxo-anions, has yet to be performed and may help shed light on the high affinity of these systems for oxo-anions. Therefore, in the current work the conformational preorganization of bis-indole receptors 1–4 (Figure 1), as well as the conformational changes of these systems upon binding of chloride and several oxo-anions, were studied by NMR spectroscopy and supported by energetic preferences established from ab initio calculations.
![[1860-5397-7-140-1]](/bjoc/content/figures/1860-5397-7-140-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Anion receptors 1–4 together with their atomic numbering scheme.
Figure 1: Anion receptors 1–4 together with their atomic numbering scheme.
Table 1: Stability constants of compound 1 measured in DMSO-d6/0.5% water, DMSO-d6/10% water and DMSO-d6/25% water and compound 2 in DMSO-d6/0.5% water at 298 K by 1H NMR titration techniques [37].
| Aniona | Compound 1 in DMSO-d6/0.5% water | Compound 1 in DMSO-d6/10% water | Compound 1 in DMSO-d6/25% water | Compound 2 in DMSO-d6/0.5% water |
|---|---|---|---|---|
| Cl− | 128 | 17 | – | 74 |
| CH3CO2− | >104 | 774 | 20 | 1620 |
| C6H5CO2− | >104 | 521 | precipitate | 477 |
| H2PO4− | >104 | 5170 | 160 | 1630 |
aAnions added as tetrabutylammonium salts.
Results and Discussion
Synthesis
Compounds 1–3 were synthesized following a previously reported methodology [36-39]. Compound 4 was prepared by reaction of 7-amino-N-phenyl-1H-indole-2-carboxamide (0.27 g, 1.07 mM) with 7-isothiocyanato-N-phenyl-1H-indole-2-carboxamide (0.31 g, 1.07 mM) in pyridine in 27% yield (see Supporting Information File 1 for details).
Structural features and NMR chemical shifts
The conformational properties of diindolylureas and diindolylthioureas 1–4, shown in Figure 1, were evaluated by means of NMR spectroscopy. Proton and 13C NMR resonances were assigned through 1D and 2D spectra, while 15N chemical shifts were determined by 1H–15N correlations in HSQC and HMBC spectra. Notable 1H and 15N NMR chemical shifts of 1–4 are shown in Table 2. It should be noted that only one set of signals was observed for both indole rings in all four receptors, due to the symmetry of the compounds. The full NMR data set together with 13C NMR chemical shifts is reported in Supporting Information File 1.
Table 2: Selected 1H and 15N NMR chemical shifts for 1–4 (in ppm).a
| H1 | H2β | H7α | H2 | H3 | H6 | N1 | N2β | N7α | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 10.78 | – | 8.64 | 7.35 | 6.44 | 7.08 | 136.5 | – | 102.7 |
| 2 | 11.03 | – | 9.48 | 7.36 | 6.46 | 7.03 | 136.3 | – | 124.9 |
| 3 | 11.62 | 10.29 | 8.97 | – | 7.49 | 7.59 | 134.5 | 128.6 | 104.6 |
| 4 | 11.68 | 10.26 | 9.72 | – | 7.48 | 7.39 | 134.9 | 129.0 | 126.6 |
aIn DMSO-d6 at 298 K.
Indole NH proton resonances were found between 10.8 and 11.7 ppm. Thioureido containing compounds 2 and 4 exhibited slight downfield shifts of H1 and H7α with respect to ureido receptors 1 and 3 (Table 2). N1 chemical shifts showed only minor variations as a result of structural differences in 1–4. The most significant differences in chemical shifts between the ureido and thioureido functionalized receptors were observed for H7α and N7α atoms (ΔδH = 0.8 and ΔδN = 22 ppm, Table 2). Compounds 3 and 4 contain phenylamide substituents at C2 and hence two more NH groups (Figure 1). Considerable deshielding of the H3 and H6 resonances was observed in 3 and 4 with respect to the nonsubstituted indole moieties in 1 and 2. The downfield shift of δH3 was attributed directly to the presence of the phenylamide group at C2. Deshielding of H6 (Δδ 0.4–0.5 ppm) in 3 and 4 with respect to 1 and 2, respectively, was much larger than the deshielding of H4 (Δδ 0.1 ppm), possibly due to a more efficient conjugation.
1H and 15N NMR chemical shift changes in 1 upon addition of anions
Figure 2 shows 1H chemical shift changes of 1 in the presence of one equivalent of chloride, acetate, benzoate, bicarbonate and dihydrogen phosphate anions. The protons that are most affected by anion–receptor interaction were found to be H1, H6 and H7α. Only minor ΔδH7α and negligible differences of δH1 and δH6 were observed in the presence of chloride anions (Figure 2a and Figure 2b). The very weak interactions between chloride and 1 could be due to competing interactions of the polar DMSO molecules for the hydrogen bond donor groups of the receptor, and the weak basicity of the chloride. This is supported by the stability constant determinations previously reported, and presented in Table 1. Considerable downfield shifts of δH7α were observed upon addition of acetate, benzoate and bicarbonate anions (Δδ ≈ 2 ppm, Figures 2c–2e), which suggested strong interaction of ureido NH protons with these anions. In addition, the strong deshielding of indolyl H1 protons corroborates its participation in the hydrogen bond formation with carboxylate and bicarbonate moieties (Δδ ≈ 1 ppm). Planar oxo-anions interact both with H1 and H7α due to their Y-shaped geometry. The tetrahedral geometry of the dihydrogen phosphate anion allows strong interaction with all four hydrogen bond donor groups, which is reflected in the substantial deshielding of the H1 and H7α protons (Figure 2f). Interestingly, one set of signals was observed for each type of anion on the NMR timescale, which suggested that the symmetry of the receptor 1 is preserved upon interactions with anions. The stability constant determinations presented in Table 1 also support the finding that this compound interacts selectively with oxo-anions.
![[1860-5397-7-140-2]](/bjoc/content/figures/1860-5397-7-140-2.png?scale=3.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectra of 1 in the absence of anions (a) and upon addition of one equivalent of the following anions: Chloride (b), acetate (c), benzoate (d), bicarbonate (e) and dihydrogen phosphate (f). All spectra were recorded in DMSO-d6 at 298 K. (g) 1H and 15N NMR chemical shift changes, Δδ = δ (in the presence of anion) – δ (in the absence of anions), induced by addition of one equivalent of different anions to receptor 1.
Figure 2: 1H NMR spectra of 1 in the absence of anions (a) and upon addition of one equivalent of the followi...
Anion–receptor interactions assessed by 1H chemical shift changes were corroborated by 15N NMR data. Weak shielding of N1 in 1 was observed upon addition of acetate, benzoate and bicarbonate anions, whereas addition of dihydrogen phosphate anions led to deshielding of N1 (Figure 2g). In contrast, N7α was deshielded upon addition of anions (Figure 2g). The strongest deshielding of 11.2 ppm was observed for the 1·BzO− complex.
Conformational properties of 1 and its complexes with anions
The rotational flexibility of the ureido moiety allows numerous conformations of receptor 1. Among them three major, energetically preferred, conformers are likely to be observed (Figure 3). The syn–syn conformer, where NH protons form a convergent hydrogen-bonding array, is expected to be adopted in the presence of bound anionic guests, based on the previous solid-state studies. On the other hand, this conformer is unlikely to be abundant in the absence of anions, due to repulsion between the hydrogen bond donor groups. The other two rotamers, namely anti–anti and syn–anti, can be stabilized by intramolecular NH–CO hydrogen bonds, which represent competition to anion–receptor interactions and therefore make conformational studies even more appealing.
![[1860-5397-7-140-3]](/bjoc/content/figures/1860-5397-7-140-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Three representative conformational families of rotamers of 1. Notations refer to the orientations along [C6–C7–N7α–C7β] fragments.
Figure 3: Three representative conformational families of rotamers of 1. Notations refer to the orientations ...
The conformational characteristics of diindolylurea 1 were assessed by the use of 1D difference NOE experiments in the absence and in the presence of anions. The orientation along the C7–N7α bonds was established by comparative evaluation of NOE enhancements of H7α with H1 and H6 protons. The saturation of H7α in 1 gave strong NOE at H6 (10.4%) and moderate NOE at H1 (4.3%), which suggested that the anti orientation prevails along the linkage between ureido moiety and indole ring (Figure 4a). As the observed NOE enhancements are primarily a function of the H6–H7α and H1–H7α distances, we compared their values in the optimized anti–anti (d(H6–H7α) = 2.28 Å) and syn–syn (d(H1–H7α) = 2.32 Å) structures and established a minor difference of 0.04 Å which would be reflected in a 1% change in the NOE enhancements. The observed difference between NOE enhancements in the uncomplexed form of receptor 1 was over 6%, which led us to conclude that the anti–anti conformer is predominant in the DMSO-d6 solution. In addition, the anti–anti conformer of 1 with its plane of symmetry along the carbonyl bond is in agreement with the single set of resonances in the NMR spectra. On the other hand, the syn–anti rotamer shows a twofold rotational symmetry and is expected to exhibit distinct shielding of nuclei, imposed by the orientations of H6 and H1 protons in the two indole rings with respect to the carbonyl group. However, the populations of the two distinct conformational families are averaged on the NMR time scale.
![[1860-5397-7-140-4]](/bjoc/content/figures/1860-5397-7-140-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: NOE enhancements of 1 in the absence of anions (a) and upon addition of one equivalent of acetate anions (b).
Figure 4: NOE enhancements of 1 in the absence of anions (a) and upon addition of one equivalent of acetate a...
Only minor changes in the NOE enhancements were observed upon addition of chloride anions to 1. The strongest NOE was observed between H7α and H6 (9.9%), which was of comparable magnitude to the NOE between the same protons in the absence of anions (Table 3). On the other hand, stronger NOE enhancement between H7α and H1 (7.0%) suggested predominance of the syn–anti rotamer of 1 in the presence of chloride anions in DMSO-d6 solution. Interestingly, the syn–syn rotamer was observed in the crystal structure, where conformational preferences are dictated by other forces, such as crystal packing. These observations are in agreement with minute 1H chemical shift changes and the weak stability constant of 1 for the binding of chloride anions.
Table 3: Selected NOE enhancements for 1 in the absence and in the presence of different anions (in %).
| Saturated: | H1 | H6 | H7α | |
|---|---|---|---|---|
| Enhanced: | H7α | H7α | H1 | H6 |
| no anion | 3.2 | 3.8 | 4.3 | 10.4 |
| Cl– | 4.2 | 2.2 | 7.0 | 9.9 |
| AcO– | 7.2 | 1.0 | 12.0 | 4.8 |
| BzO– | 7.2 | 0.9 | 10.4 | 4.8 |
| HCO3– | –a | 0.0 | –a | 4.7 |
| H2PO4– | 4.2 | 0.0 | 5.3 | 2.8 |
aBroad NH signals in the baseline.
Considerable changes in the NOE enhancements were observed upon addition of acetate anions to 1. The saturation of H7α resulted in a much stronger NOE at H1 (12.0%) with respect to H6 (4.8%), which suggested that addition of acetate anions led to conformational changes along the C7–N7α bond (Table 3). The syn–syn conformer is preferred for the 1·AcO− complex in solution (Figure 4b). In a similar manner, significant changes in the NOEs were observed upon addition of benzoate anions. The saturation of H7α gave much stronger NOE at H1 (10.4%) with respect to H6 (4.8%, Table 3). Broad NH proton signals prevented the study of the conformation of the 1·HCO3− complex through NOE experiments. NOE enhancements between H1 and H7α (4.2–5.3%) were considerably stronger with respect to NOE between H7α and H6 (0–2.8%) upon addition of dihydrogen phosphate to 1. The observed NOE enhancements for 1·H2PO4− complex suggest a preference for the syn–syn conformer in DMSO-d6.
Conformational analysis of 1 and its anion complexes by quantum mechanics calculations
The observations on the conformational equilibria in the absence and in the presence of anions were corroborated by quantum mechanical calculations at the B3LYP/6-311+G(d,p) level of theory by means of the Gaussian 03 [40] and Gaussian 09 [41] programs. Indole rings represent the rigid part of the anion receptors, while the substituents on the ureido moiety in 1 exhibit conformational freedom. [C6–C7–N7α–C7β] torsion angles were defined to follow energetic changes induced by reorientation of the indolyl moieties along the C7–N7α bonds. The energy surface for the [C6–C7–N7α–C7β] torsion angles, with 30° resolution, shows that the conformer with the lowest energy is in the anti–anti region (Figure 5).
![[1860-5397-7-140-5]](/bjoc/content/figures/1860-5397-7-140-5.svg?scale=2.4&max-width=1024&background=FFFFFF)
Figure 5: Surface plot of the relative potential energy of 1 as a function of the two constitutive [C6–C7–N7α–C7β] torsion angles. Individual geometries were optimized at the B3LYP/6-311+G(d,p) level of theory at 30° resolution.
Figure 5: Surface plot of the relative potential energy of 1 as a function of the two constitutive [C6–C7–N7α...
Energy minimization of the anti–anti conformer of 1 was performed without any constraints, whereas syn orientations in the syn–anti as well as the syn–syn cases were restrained along the [C6–C7–N7α–C7β] torsion angle, while other degrees of freedom were freely optimized. The relative energies for the three representative conformers are reported in Table 4. The anti–anti conformer of 1 was found to be the lowest in energy, while the syn–syn conformer showed considerably higher energy (11.6 kcal·mol−1). The angle between the two indolyl rings in the freely optimized anti–anti conformer was 53.9° (Figure 6a). The relative energies of the three representative conformers were also computed for complexes of 1 with chloride, acetate and bicarbonate anions. The syn–syn conformer exhibited the lowest relative energy for all three anion–receptor complexes (Table 4). The anti–anti conformers of anion–receptor complexes exhibited considerably higher energies between 6.2 and 7.3 kcal·mol−1. The angle between the two indolyl rings in the freely optimized syn–syn conformer of the 1·AcO− complex was found to be 21.6° (Figure 6b). The optimized structure, shown in Figure 6b, shows a single acetate anion bound to the four NH groups in the receptor 1 with N···O distances in the range from 2.75 to 2.95 Å and N–H···O angles in the range 170–176°.
Table 4: Relative energiesa (in kcal·mol−1) of receptor 1 in vacuo and in DMSO, in the absence and in the presence of anions.
| anion | conformer | in vacuo | in DMSO |
|---|---|---|---|
| no anion | anti–anti | 0.00 | 0.00 |
| syn–anti | 5.09 | 2.74 | |
| syn–syn | 11.61 | 6.60 | |
| Cl− | anti–anti | 6.50 | 1.20 |
| syn–anti | 1.84 | 0.12 | |
| syn–syn | 0.00 | 0.00 | |
| AcO− | anti–anti | 7.26 | 3.75 |
| syn–anti | 2.82 | 1.74 | |
| syn–syn | 0.00 | 0.00 | |
| HCO3− | anti–anti | 6.21 | 2.97 |
| syn–anti | 2.02 | 1.31 | |
| syn–syn | 0.00 | 0.00 | |
aRelative energies are reported with respect to the lowest energy (arbitrarily set to 0.00 kcal/mol) in the absence and in the presence of anions. Geometry optimizations were carried out at B3LYP/6-311+G(d,p).
![[1860-5397-7-140-6]](/bjoc/content/figures/1860-5397-7-140-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Freely optimized structure at the B3LYP/6-311+G(d,p) level of theory and side view showing deviation from coplanarity defined by the angle between the indolyl rings of receptor 1 in the absence of anions (a) and for the 1·AcO− complex (b).
Figure 6: Freely optimized structure at the B3LYP/6-311+G(d,p) level of theory and side view showing deviatio...
In order to evaluate the role of DMSO on the energetic preferences of rotamers, relative energies were calculated with the use of Tomasi's polarized continuum model [42,43]. Preferences amongst the three rotamers were retained (Table 4). Only small differences below 1.2 kcal·mol−1 were found between the three distinct conformers in the case of the 1·Cl− complex. In particular, the negligible energy differences between syn–syn and syn–anti rotamers are in agreement with the NOE data that suggested predominance of the syn–anti conformer upon addition of chloride. The energetic preference of the syn–syn over the anti–anti conformer of 3.8 and 3.0 kcal·mol−1 was observed for the 1·AcO− and the 1·HCO3− complexes, respectively (Table 4). The energetic preferences of the 1·AcO–complex are in excellent agreement with the NOE experiments, which showed conformational reorganizations of 1 upon addition of acetate anions.
Conformational features of receptors 2–4
The choice of thio (2 and 4) versus oxo (1 and 3) ureido functionalities, as well as the C2 functionalization of the indole scaffolds with pendant phenyl amides in 3 and 4, allows tuning of the binding affinities of the receptors. Negligible Δδ values were observed for 2 upon interaction with chloride anions (Figure 7a), which suggests a very weak interaction between chloride and 2, similar to the weak interactions observed between chloride and 1. Chemical shift changes showed that the main interaction between receptor 2 and trigonal planar anions (acetate, benzoate and bicarbonate) occurred at the H7α protons (Figure 7a). Addition of dihydrogen phosphate anions caused considerable deshielding of the H1 and H7α protons. Comparison of the Δδ values for 1 and 2 upon interaction with the anions showed that the urea derivative 1 exhibited a higher preference for anion binding relative to thiourea 2 (the data were supported by the stability constant determinations performed previously and shown in Table 1). The larger sulfur atom can prevent the receptor 2 from adopting a planar conformation, which may reduce the affinity of this receptor for anionic guests. Conformational studies of 2 with the use of NOE enhancements showed that the anti–anti conformer is the preferred conformation in the absence of anions. No apparent conformational changes were observed upon addition of chloride anions to 2. The overlap of the proton signals as well as the broad line-width of the H1 and H7α NMR resonances prevented conformational analysis of 2 upon addition of other anions used in the study. The conformational preferences of 2 were evaluated by quantum mechanical calculations at the B3LYP/6-311+G(d,p) level of theory. The freely optimized anti–anti conformer of 2 exhibited the lowest energy and the syn–syn conformer was 8.0 kcal·mol−1 higher in energy, in vacuo. Interestingly, the two indolyl rings were not coplanar, with the angle between the two indolyl planes found to be 98.9° (Figure 8a). In the case of 2·AcO− complex the syn–syn conformer exhibited the lowest energy, while the anti–anti conformer was 7.4 kcal·mol−1 higher in energy, in vacuo. The optimized structure of 2·AcO− complex is shown in Figure 8b, where the two acetate oxygen atoms are hydrogen bonded to the four NH groups, with N···O distances in the range from 2.76 to 2.94 Å and N–H···O angles in the range 168–177°. The angle between the indolyl rings in the freely optimized syn–syn conformer of the 2·AcO− complex was 68.0°.
![[1860-5397-7-140-7]](/bjoc/content/figures/1860-5397-7-140-7.png?scale=2.8&max-width=1024&background=FFFFFF)
Figure 7: 1H NMR chemical shift changes, Δδ = δ (in the presence of anions) – δ (in the absence of anions), induced by addition of one equivalent of different anions to receptors 2 (a), 3 (b) and 4 (c). Note, there is no H2β proton in 2.
Figure 7: 1H NMR chemical shift changes, Δδ = δ (in the presence of anions) – δ (in the absence of anions), i...
![[1860-5397-7-140-8]](/bjoc/content/figures/1860-5397-7-140-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Freely optimized structures at the B3LYP/6-311+G(d,p) level of theory and side view showing deviation from coplanarity defined by the angle between the indolyl rings of 2 (a) and 2·AcO− complex (b).
Figure 8: Freely optimized structures at the B3LYP/6-311+G(d,p) level of theory and side view showing deviati...
Bis-amide functionalized diindolylurea 3 exhibits two extra NH groups, which introduces additional possibilities for interactions with anions. The addition of chloride anions to 3 induced negligible chemical shifts, suggesting only weak interactions with this anion (Figure 7b). The strong deshielding of H7α and moderate deshielding of H1 protons in the 3·AcO− and 3·BzO− complexes suggests a significant interaction between the anions and the ureido functionality. Interestingly, negligible deshielding of H2β in 3 was observed and this suggests that the amide protons do not participate in the interaction with acetate and benzoate (Figure 7b). The observed Δδ values support the idea that carboxylates were strongly bound to the urea H7α protons which prevented interaction between the anions and the amide H2β protons. Analogously, large chemical shift changes of up to 1 ppm were observed for the H7α and H1 protons upon the addition of bicarbonate anions to 3. Strong deshielding of H1, H2β and H7α protons in the 3·H2PO4− complex suggests that all the NH donor groups are involved in interactions with the dihydrogen phosphate anions (Figure 7b).
The conformational properties of 3 and of its complexes with different anions were studied by NOE measurements. The saturation of the H1 protons resulted in moderately negative NOEs at the H7α and H2β protons. The cross peaks in the 2D NOESY spectra between the NH protons and bulk water suggest chemical exchange that complicated the conformational study along the C2–C2α and C7–N7α bonds. Nevertheless, strong NOE enhancements between the H2β and H3 protons suggest an orientation along the C2–C2α bond where the H2β and H3 protons are spatially close and the C2α carbonyl group is oriented towards the indole H1 proton. NOE enhancements between H2β and H3 protons were observed also in the 3·AcO− and 3·BzO− complexes, which suggests that the orientation of the carboxamide group along the C2–C2α bond is retained in 3 upon addition of carboxylate anions. This observation was supported by negligible Δδ values for the H2β protons in the 3·AcO− and 3·BzO− complexes with respect to 3. The conformational preferences and the proposed binding mode in the 3·AcO− complex are shown in Figure 9. A conformational study of 3 in the presence of bicarbonate and dihydrogen phosphate anions was hindered by the broadened and overlapped 1H signals. In the solid state compound 3 crystallized with tetrabutylammonium dihydrogen phosphate as the monohydrogen phosphate complex [38].
![[1860-5397-7-140-9]](/bjoc/content/figures/1860-5397-7-140-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Conformational preferences and proposed binding mode for the 3·AcO− 1:1 complex.
Figure 9: Conformational preferences and proposed binding mode for the 3·AcO− 1:1 complex.
Only negligible chemical shifts were observed for 4 upon addition of chloride anions (Figure 7c). Considerable deshielding of H7α protons in 4 by up to 2.3 ppm in the 4·AcO− and 4·BzO− complexes suggested that the major interactions between carboxylates and receptor 4 occurred at the ureido functionality (Figure 7c). The conformational properties of the 4·AcO– and 4·BzO– complexes could not be determined due to the broad and overlapped proton signals. Unfortunately, excessively broad and overlapped 1H signals for the 4·HCO3− and 4·H2PO4− complexes prevented unambiguous assignment of the NMR resonances and hence the conformational studies of these complexes. In the solid state compound 4 crystallized with tetrabutylammonium dihydrogen phosphate as the monohydrogen phosphate complex (see Supporting Information File 2 for more details).
Conclusion
The bis-indole receptors 1–4 were characterized by heteronuclear NMR spectroscopy. NOE based conformational analysis was supported by quantum mechanics calculations and revealed that diindolylurea 1 exhibited conformational preorganization in DMSO-d6 solution. The anti–anti conformer, which could be stabilized by intramolecular hydrogen bonds between the C7β carbonyl group and indole NH proton, was predominant for 1 in the absence of anions. The energetically minimized structure of anti–anti conformer showed a plane of symmetry along the ureido carbonyl group and deviation from coplanarity amongst the indolyl rings. Anion-induced chemical shift changes suggested weak binding of chloride anions and negligible conformational changes for 1. Addition of acetate, benzoate, bicarbonate and dihydrogen phosphate resulted in strong deshielding of the ureido protons and moderate deshielding of the indole H1, which indicated that the main hydrogen bond interaction occurred at the urea donor groups of 1. Furthermore, binding of anions caused conformational changes along the C7–N7α bonds, and the syn–syn conformer was predominant in the anion–receptor complexes according to both NOE enhancements and ab initio calculations in solution. The freely optimized syn–syn conformer of the 1·AcO− complex retained a plane of symmetry along the carbonyl bond and showed a smaller deviation from indole ring coplanarity than did the anti–anti conformer of 1. The conformational preferences for 2 were analogous to those observed for receptor 1. Unfortunately, excessively broad and overlapped 1H signals prevented a detailed conformational analysis of the anion–receptor complexes for 3 and 4.
Experimental
NMR experiments
1H, 13C and 15N NMR spectra were acquired on a Varian Unity Inova 300 MHz NMR spectrometer. All data were recorded in DMSO-d6 at 298 K. Chemical shifts were referenced to the residual solvent signal of DMSO-d6 at δ 2.50 ppm for 1H (297.801 MHz) and δ 39.50 ppm for 13C (76.190 MHz), while 15N (30.188 MHz) chemical shifts were referenced relative to external benzamide (δ 103.55 ppm). Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances, H–H coupling constants as well as by means of a series of 2D NMR experiments (COSY, gHSQC and gHMBC). The saturation delay in the 1D difference NOE experiment was 5.0 s. All anions were added as tetrabutylammonium salts except bicarbonate which was added as a tetraethylammonium salt. NOESY spectra were acquired with mixing time of 100 and 300 ms.
Ab initio calculations
Initial structures were generated by Chem3D Pro 10.0 software and energy minimization at the B3LYP/6-311+G(d,p) level was performed for 1 and 2 without any constraints for the anti–anti conformers, by means of Gaussian 03 [40] and Gaussian 09 [41]. Syn orientations in the syn–anti as well as the syn–syn conformers of 1 and 2 were restrained along the [C6–C7–N7α–C7β] torsion angle while other degrees of freedom were freely optimized. Ab initio calculations of anion–receptor complexes were carried out without any constraints for the syn–syn conformers, where anions were placed initially at the expected equilibrium distance to the H1 and H7α protons. The positions of the anions were freely optimized. Energy minimizations of the syn–anti and anti–anti conformers of the anion–receptor complexes were restrained along the [C6–C7–N7α–C7β] torsion angle while other degrees of freedom were freely optimized. The tetrabutylammonium countercation was omitted in the geometry optimization of the anion–receptor complexes. Frequency calculations verified that the optimized geometries were stable points on the potential energy surface. Relative energies in solution were calculated by means of Tomasi's polarized continuum model, where the dielectric constant of DMSO was used (ε = 46.7).
Supporting Information
| Supporting Information File 1: Experimental for the synthesis of compound 4 and details of the crystal structure of the HPO42− complex of 4, 1H and 13C NMR data for 1–4, 1D difference NOE spectra for 1 in the absence and upon addition of one equivalent of acetate anions. | ||
| Format: PDF | Size: 422.3 KB | Download |
| Supporting Information File 2: Crystallographic data of the complex of compound 4 with tetrabutylammonium dihydrogen phosphate (4·TBA2·HPO4). | ||
| Format: CIF | Size: 33.9 KB | Download |
Acknowledgements
We gratefully acknowledge the financial support of the Slovenian Research Agency (ARRS, program no. P1-0242), COST D-31, EAST-NMR (grant no. 228461) and Bio-NMR (grant no. 261863) FP7 projects. PAG thanks the EPSRC for support and the access to the crystallographic facilities at the University of Southampton.
References
-
Bianchi, A.; Bowman-James, K.; García-España, E., Eds. Supramolecular Chemistry of Anions; Wiley-VCH: New York, 1997.
Return to citation in text: [1] -
Schmidtchen, F. P.; Berger, M. Chem. Rev. 1997, 97, 1609. doi:10.1021/cr9603845
Return to citation in text: [1] -
Beer, P. D. Acc. Chem. Res. 1998, 31, 71. doi:10.1021/ar9601555
Return to citation in text: [1] -
Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675. doi:10.1021/jo982382l
Return to citation in text: [1] -
Beer, P. D.; Gale, P. A. Angew. Chem., Int. Ed. 2001, 40, 486. doi:10.1002/1521-3773(20010202)40:3<486::AID-ANIE486>3.0.CO;2-P
Return to citation in text: [1] -
Wiskur, S. L.; Ait-Haddou, H.; Lavigne, J. J.; Anslyn, E. V. Acc. Chem. Res. 2001, 34, 963. doi:10.1021/ar9600796
Return to citation in text: [1] -
Vilar, R. Angew. Chem., Int. Ed. 2003, 42, 1460. doi:10.1002/anie.200200551
Return to citation in text: [1] -
Bowman-James, K. Acc. Chem. Res. 2005, 38, 671. doi:10.1021/ar040071t
Return to citation in text: [1] -
Gale, P. A. Acc. Chem. Res. 2006, 39, 465. doi:10.1021/ar040237q
Return to citation in text: [1] -
Sessler, J. L.; Gale, P. A.; Cho, W. S. Anion Receptor Chemistry; Royal Society of Chemistry: Cambridge, 2006.
Return to citation in text: [1] -
Albrecht, M. Naturwissenschaften 2007, 94, 951. doi:10.1007/s00114-007-0282-7
Return to citation in text: [1] -
Lankshear, M. D.; Beer, P. D. Acc. Chem. Res. 2007, 40, 657. doi:10.1021/ar7000217
Return to citation in text: [1] -
Vilar, R. Eur. J. Inorg. Chem. 2008, 357. doi:10.1002/ejic.200701017
Return to citation in text: [1] -
Pflugrath, J. W.; Quiocho, F. A. Nature 1985, 314, 257. doi:10.1038/314257a0
Return to citation in text: [1] [2] -
Luecke, H.; Quiocho, F. A. Nature 1990, 347, 402. doi:10.1038/347402a0
Return to citation in text: [1] -
Bondy, C. R.; Loeb, S. J. Coord. Chem. Rev. 2003, 240, 77. doi:10.1016/S0010-8545(02)00304-1
Return to citation in text: [1] -
Gómez, D. E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. Org. Biomol. Chem. 2005, 3, 1495. doi:10.1039/b500123d
Return to citation in text: [1] -
Kang, S. O.; Begum, R. A.; Bowman-James, K. Angew. Chem., Int. Ed. 2006, 45, 7882. doi:10.1002/anie.200602006
Return to citation in text: [1] -
Gale, P. A.; García-Garrido, S. E.; Garric, J. Chem. Soc. Rev. 2008, 37, 151. doi:10.1039/b715825d
Return to citation in text: [1] -
Caltagirone, C.; Gale, P. A. Chem. Soc. Rev. 2009, 38, 520. doi:10.1039/b806422a
Return to citation in text: [1] -
Gale, P. A. Chem. Commun. 2005, 3761. doi:10.1039/b504596g
Return to citation in text: [1] -
Gale, P. A. Chem. Soc. Rev. 2010, 39, 3746. doi:10.1039/c001871f
Return to citation in text: [1] -
Gale, P. A. Chem. Commun. 2011, 47, 82. doi:10.1039/c0cc00656d
Return to citation in text: [1] -
Chmielewski, M. J.; Charon, M.; Jurczak, J. Org. Lett. 2004, 6, 3501. doi:10.1021/ol048661e
Return to citation in text: [1] -
Piatek, P.; Lynch, V. M.; Sessler, J. L. J. Am. Chem. Soc. 2004, 126, 16073. doi:10.1021/ja045218q
Return to citation in text: [1] -
Chang, K.-J.; Moon, D.; Lah, M. S.; Jeong, K.-S. Angew. Chem., Int. Ed. 2005, 44, 7926. doi:10.1002/anie.200503121
Return to citation in text: [1] -
Curiel, D.; Cowley, A.; Beer, P. D. Chem. Commun. 2005, 236. doi:10.1039/b412363h
Return to citation in text: [1] -
Pfeffer, F. M.; Lim, K. F.; Sedgwick, K. J. Org. Biomol. Chem. 2007, 5, 1795. doi:10.1039/b702804k
Return to citation in text: [1] -
Chmielewski, M. J.; Zhao, L. Y.; Brown, A.; Curiel, D.; Sambrook, M. R.; Thompson, A. L.; Santos, S. M.; Felix, V.; Davis, J. J.; Beer, P. D. Chem. Commun. 2008, 3154. doi:10.1039/b804941f
Return to citation in text: [1] -
Suk, J.-m.; Chae, M. K.; Kim, N.-K.; Kim, U.-I.; Jeong, K.-S. Pure Appl. Chem. 2008, 80, 599. doi:10.1351/pac200880030599
Return to citation in text: [1] -
Zhao, L.; Mullen, K. M.; Chmielewski, M. J.; Brown, A.; Bampos, N.; Beer, P. D.; Davis, J. J. New J. Chem. 2009, 33, 760. doi:10.1039/b818854h
Return to citation in text: [1] -
Verschueren, K. H. G.; Seljée, F.; Rozeboom, H. J.; Kalk, K. H.; Dijkstra, B. W. Nature 1993, 363, 693. doi:10.1038/363693a0
Return to citation in text: [1] -
Makuc, D.; Lenarčič, M.; Bates, G. W.; Gale, P. A.; Plavec, J. Org. Biomol. Chem. 2009, 7, 3505. doi:10.1039/b908947k
Return to citation in text: [1] -
Makuc, D.; Triyanti; Albrecht, M.; Plavec, J.; Rissanen, K.; Valkonen, A.; Schalley, C. A. Eur. J. Org. Chem. 2009, 2009, 4854. doi:10.1002/ejoc.200900721
Return to citation in text: [1] -
Makuc, D.; Albrecht, M.; Plavec, J. Supramol. Chem. 2010, 22, 603. doi:10.1080/10610278.2010.506544
Return to citation in text: [1] -
Caltagirone, C.; Gale, P. A.; Hiscock, J. R.; Brooks, S. J.; Hursthouse, M. B.; Light, M. E. Chem. Commun. 2008, 3007. doi:10.1039/b806238b
Return to citation in text: [1] [2] -
Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236. doi:10.1002/chem.200801639
Return to citation in text: [1] [2] [3] -
Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2010, 5, 555. doi:10.1002/asia.200900230
Return to citation in text: [1] [2] [3] [4] -
Gale, P. A.; Hiscock, J. R.; Jie, C. Z.; Hursthouse, M. B.; Light, M. E. Chem. Sci. 2010, 1, 215. doi:10.1039/c0sc00202j
Return to citation in text: [1] [2] -
Gaussian 03, Revision B.3; Gaussian, Inc.: Pittsburgh PA, 2004.
Return to citation in text: [1] [2] -
Gaussian 09, Revision A.5; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] [2] -
Miertus, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117. doi:10.1016/0301-0104(81)85090-2
Return to citation in text: [1] -
Miertus, S.; Tomasi, J. Chem. Phys. 1982, 65, 239. doi:10.1016/0301-0104(82)85072-6
Return to citation in text: [1]
| 1. | Bianchi, A.; Bowman-James, K.; García-España, E., Eds. Supramolecular Chemistry of Anions; Wiley-VCH: New York, 1997. |
| 2. | Schmidtchen, F. P.; Berger, M. Chem. Rev. 1997, 97, 1609. doi:10.1021/cr9603845 |
| 3. | Beer, P. D. Acc. Chem. Res. 1998, 31, 71. doi:10.1021/ar9601555 |
| 4. | Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675. doi:10.1021/jo982382l |
| 5. | Beer, P. D.; Gale, P. A. Angew. Chem., Int. Ed. 2001, 40, 486. doi:10.1002/1521-3773(20010202)40:3<486::AID-ANIE486>3.0.CO;2-P |
| 6. | Wiskur, S. L.; Ait-Haddou, H.; Lavigne, J. J.; Anslyn, E. V. Acc. Chem. Res. 2001, 34, 963. doi:10.1021/ar9600796 |
| 7. | Vilar, R. Angew. Chem., Int. Ed. 2003, 42, 1460. doi:10.1002/anie.200200551 |
| 8. | Bowman-James, K. Acc. Chem. Res. 2005, 38, 671. doi:10.1021/ar040071t |
| 9. | Gale, P. A. Acc. Chem. Res. 2006, 39, 465. doi:10.1021/ar040237q |
| 10. | Sessler, J. L.; Gale, P. A.; Cho, W. S. Anion Receptor Chemistry; Royal Society of Chemistry: Cambridge, 2006. |
| 11. | Albrecht, M. Naturwissenschaften 2007, 94, 951. doi:10.1007/s00114-007-0282-7 |
| 12. | Lankshear, M. D.; Beer, P. D. Acc. Chem. Res. 2007, 40, 657. doi:10.1021/ar7000217 |
| 13. | Vilar, R. Eur. J. Inorg. Chem. 2008, 357. doi:10.1002/ejic.200701017 |
| 24. | Chmielewski, M. J.; Charon, M.; Jurczak, J. Org. Lett. 2004, 6, 3501. doi:10.1021/ol048661e |
| 25. | Piatek, P.; Lynch, V. M.; Sessler, J. L. J. Am. Chem. Soc. 2004, 126, 16073. doi:10.1021/ja045218q |
| 26. | Chang, K.-J.; Moon, D.; Lah, M. S.; Jeong, K.-S. Angew. Chem., Int. Ed. 2005, 44, 7926. doi:10.1002/anie.200503121 |
| 27. | Curiel, D.; Cowley, A.; Beer, P. D. Chem. Commun. 2005, 236. doi:10.1039/b412363h |
| 28. | Pfeffer, F. M.; Lim, K. F.; Sedgwick, K. J. Org. Biomol. Chem. 2007, 5, 1795. doi:10.1039/b702804k |
| 29. | Chmielewski, M. J.; Zhao, L. Y.; Brown, A.; Curiel, D.; Sambrook, M. R.; Thompson, A. L.; Santos, S. M.; Felix, V.; Davis, J. J.; Beer, P. D. Chem. Commun. 2008, 3154. doi:10.1039/b804941f |
| 30. | Suk, J.-m.; Chae, M. K.; Kim, N.-K.; Kim, U.-I.; Jeong, K.-S. Pure Appl. Chem. 2008, 80, 599. doi:10.1351/pac200880030599 |
| 31. | Zhao, L.; Mullen, K. M.; Chmielewski, M. J.; Brown, A.; Bampos, N.; Beer, P. D.; Davis, J. J. New J. Chem. 2009, 33, 760. doi:10.1039/b818854h |
| 42. | Miertus, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117. doi:10.1016/0301-0104(81)85090-2 |
| 43. | Miertus, S.; Tomasi, J. Chem. Phys. 1982, 65, 239. doi:10.1016/0301-0104(82)85072-6 |
| 16. | Bondy, C. R.; Loeb, S. J. Coord. Chem. Rev. 2003, 240, 77. doi:10.1016/S0010-8545(02)00304-1 |
| 17. | Gómez, D. E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. Org. Biomol. Chem. 2005, 3, 1495. doi:10.1039/b500123d |
| 18. | Kang, S. O.; Begum, R. A.; Bowman-James, K. Angew. Chem., Int. Ed. 2006, 45, 7882. doi:10.1002/anie.200602006 |
| 19. | Gale, P. A.; García-Garrido, S. E.; Garric, J. Chem. Soc. Rev. 2008, 37, 151. doi:10.1039/b715825d |
| 20. | Caltagirone, C.; Gale, P. A. Chem. Soc. Rev. 2009, 38, 520. doi:10.1039/b806422a |
| 21. | Gale, P. A. Chem. Commun. 2005, 3761. doi:10.1039/b504596g |
| 22. | Gale, P. A. Chem. Soc. Rev. 2010, 39, 3746. doi:10.1039/c001871f |
| 23. | Gale, P. A. Chem. Commun. 2011, 47, 82. doi:10.1039/c0cc00656d |
| 38. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2010, 5, 555. doi:10.1002/asia.200900230 |
| 36. | Caltagirone, C.; Gale, P. A.; Hiscock, J. R.; Brooks, S. J.; Hursthouse, M. B.; Light, M. E. Chem. Commun. 2008, 3007. doi:10.1039/b806238b |
| 37. | Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236. doi:10.1002/chem.200801639 |
| 38. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2010, 5, 555. doi:10.1002/asia.200900230 |
| 37. | Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236. doi:10.1002/chem.200801639 |
| 33. | Makuc, D.; Lenarčič, M.; Bates, G. W.; Gale, P. A.; Plavec, J. Org. Biomol. Chem. 2009, 7, 3505. doi:10.1039/b908947k |
| 34. | Makuc, D.; Triyanti; Albrecht, M.; Plavec, J.; Rissanen, K.; Valkonen, A.; Schalley, C. A. Eur. J. Org. Chem. 2009, 2009, 4854. doi:10.1002/ejoc.200900721 |
| 35. | Makuc, D.; Albrecht, M.; Plavec, J. Supramol. Chem. 2010, 22, 603. doi:10.1080/10610278.2010.506544 |
| 36. | Caltagirone, C.; Gale, P. A.; Hiscock, J. R.; Brooks, S. J.; Hursthouse, M. B.; Light, M. E. Chem. Commun. 2008, 3007. doi:10.1039/b806238b |
| 37. | Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236. doi:10.1002/chem.200801639 |
| 38. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2010, 5, 555. doi:10.1002/asia.200900230 |
| 39. | Gale, P. A.; Hiscock, J. R.; Jie, C. Z.; Hursthouse, M. B.; Light, M. E. Chem. Sci. 2010, 1, 215. doi:10.1039/c0sc00202j |
| 32. | Verschueren, K. H. G.; Seljée, F.; Rozeboom, H. J.; Kalk, K. H.; Dijkstra, B. W. Nature 1993, 363, 693. doi:10.1038/363693a0 |
| 38. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2010, 5, 555. doi:10.1002/asia.200900230 |
| 39. | Gale, P. A.; Hiscock, J. R.; Jie, C. Z.; Hursthouse, M. B.; Light, M. E. Chem. Sci. 2010, 1, 215. doi:10.1039/c0sc00202j |
© 2011 Makuc et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
