Abstract
In the last few years, palladium-mediated three-component synthesis has emerged as an important synthetic methodology to gain access to nitrogen-containing structures. The latest developments in this area are discussed in this review.

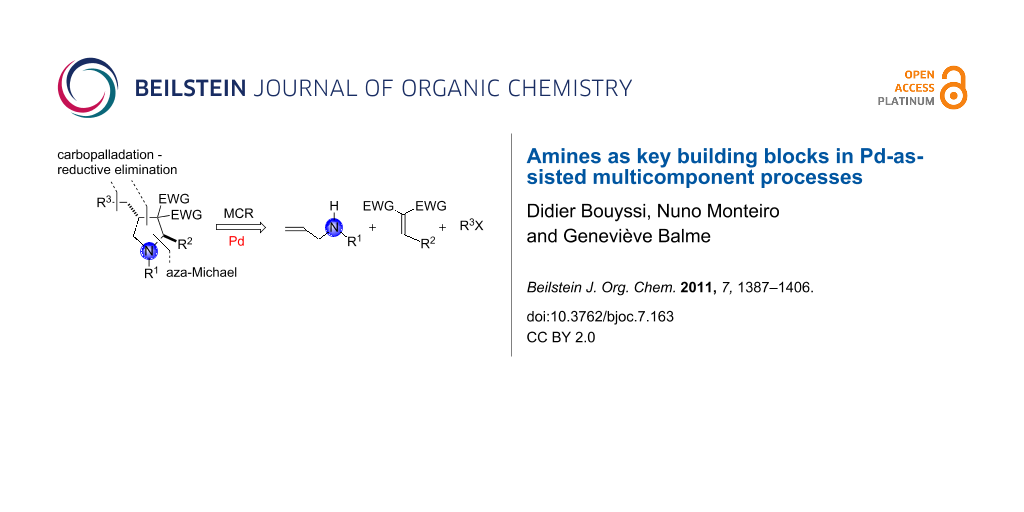
Graphical Abstract
Introduction
Nitrogen-containing structures are present in numerous bioactive natural and synthetic products. The development of new methodologies to prepare these useful frameworks has attracted great attention from organic chemists. Among these developments, multicomponent strategies offer significant advantages over stepwise procedures since several bonds are formed in a one-pot operation, minimizing the formation of waste and competitive reactions [1]. In line with this, remarkable new strategies have been developed based on palladium-mediated coupling process. The purpose of this review is to discuss recent achievements in the design of palladium-catalyzed multicomponent preparation of nitrogen-containing structures and this article is divided into sections relating to the introduction of the amine functionality.
Review
Imines as electrophilic partners
The imine function plays an important role in the development of multicomponent approaches to polyfunctionalized nitrogen acyclic or cyclic compounds due to the ease of their in situ preparation. Many strategies have been developed based on this concept, the imines being either directly used as starting building blocks or generated in situ as part of the multicomponent process.
Arndtsen and coworkers elaborated a three-component process allowing the synthesis of α-substituted amides. This methodology relied on the oxidative addition of an N-acyliminium species, generated in situ from an imine and an acid chloride, to a Pd(0) complex, furnishing a stable chelated palladium adduct 1, which was isolated and fully characterized. When vinyltributyltin was added as a third component in the reaction medium, a transmetallation step occurred, followed by a reductive elimination step, furnishing amides 2 in good to excellent yields [2]. This reaction tolerated various functional groups on the imine moiety, such as ether, thioether and ester groups, although enolizable alkylimines were not suitable under these conditions (Scheme 1).
![[1860-5397-7-163-i1]](/bjoc/content/inline/1860-5397-7-163-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of substituted amides.
Scheme 1: Synthesis of substituted amides.
Replacement of the acid chloride with a chloroformate under 1 atmosphere of carbon monoxide as a fourth component led to ketocarbamates 3 in a single operation through a carbonylative coupling [3]. Various chloroformates and imines can participate in this reaction, stannanes being limited to aryl, benzyl or ethyl ones. When vinylstannane was used, the transmetallation step was more rapid than the CO insertion, giving instead substituted carbamates. After removal of the solvents in vacuo, the addition of acetic acid and 15 equivalents of ammonium acetate to the crude mixture resulted in a postcyclization leading to imidazolones 4, with spontaneous elimination of the initial chloroformate substituent (Scheme 2).
![[1860-5397-7-163-i2]](/bjoc/content/inline/1860-5397-7-163-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of ketocarbamates and imidazolones.
Scheme 2: Synthesis of ketocarbamates and imidazolones.
Arndtsen also demonstrated that 3-amido-substituted β-lactams 7 can be obtained, based on a similar strategy, through the assembly of four components, namely, imines, acid chloride and carbon monoxide. The process is thought to begin with formation of a münchnone 5, resulting from oxidative addition of an acyliminium species to Pd(0), followed by CO insertion and β-hydride elimination. This münchnone is in equilibrium with its ketene isomeric form 6, and a formal [2 + 2] cycloaddition with a second equivalent of imine generates the lactam (Scheme 3). The authors pointed out that the trapping of HCl by a sterically hindered base (NEtiPr2) is the key point in this methodology to enable access to this heterocycle and to avoid formation of imidazolinium salts [4].
![[1860-5397-7-163-i3]](/bjoc/content/inline/1860-5397-7-163-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
To increase the structural diversity of the final lactam, a modified process was developed that allows introduction of two distinct imines in this reaction. This coupling process was catalyzed by palladacycle 8 and led to the mesoionic compound 5 under these conditions. Subsequent addition of a second, different imine produced β-lactams 9 in good yields, after heating at 55 °C for 24 h (Scheme 4) [4].
![[1860-5397-7-163-i4]](/bjoc/content/inline/1860-5397-7-163-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Access to β-lactams with increased structural diversity.
Scheme 4: Access to β-lactams with increased structural diversity.
As mentioned above, imidazolinium salts 12 can be obtained by a dipolar cycloaddition of münchnone intermediates with imines. Arndtsen and coworkers developed a new highly active palladium catalyst to improve previous results in this area. Moreover, this strategy allows the selective incorporation of two different imines leading to polysubstituted imidazoliniums 13. After a large screening of palladium precatalysts and ligands, the palladacycle 10 in combination with the di-tert-butyl-2-biphenylphosphine (11) furnished the best results in terms of reaction time and yield. A large variety of imines and acid chlorides can be used in this reaction, with only enolizable imines and those bearing bulky nitrogen substituents being incompatible. In order to have four independent tunable substrates, the authors added a base (NEtiPr2) to the reaction medium that favors formation of the münchnone intermediate. The second imine was added after 16 h of heating at 45 °C, together with PhSO3H, which catalyzed the dipolar cycloaddition and avoided formation of a β-lactam as shown before (Scheme 5) [5].
![[1860-5397-7-163-i5]](/bjoc/content/inline/1860-5397-7-163-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of imidazolinium salts.
Scheme 5: Synthesis of imidazolinium salts.
The palladium-catalyzed trans-addition-alkylative cyclization (anti-Wacker cyclization) of o-ethynylbenzaldehyde with organoboron reagents in the presence of secondary amines was accomplished by Tsukamoto and coworkers [6]. This novel strategy, dedicated to the synthesis of indenamines 14, involves addition of an electron-rich palladium/phosphine complex to a triple bond, followed by nucleophilic addition to an iminium ion generated in situ by addition of a secondary amine to an aldehyde. Transmetallation of the resulting species with a boronic acid or triethylborane, followed by a reductive elimination, afforded the indenamine core in good to excellent yields. However, this Pd-catalyzed cyclization was only effective for aldehydes since ketones did not participate in the process (Scheme 6).
![[1860-5397-7-163-i6]](/bjoc/content/inline/1860-5397-7-163-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Tsukamato extended this methodology further to the cyclization of alkynyl- and allenyliminiums in order to access 1,4-disubstituted-1,2,3,6-tetrahydropyridines 15 or 16 following the same strategy [7]. For alkynyliminiums, two different catalytic systems were developed according to the nature of the aryl- or heteroarylboronic acids used. For neutral or electron-rich acids, Pd(PPh3)4 as catalyst gave excellent results, whereas it was necessary to use PdCp(η3-C3H5) in the presence of PPh(c-C6H11)2 as a ligand for those bearing electron-withdrawing groups (Scheme 7).
![[1860-5397-7-163-i7]](/bjoc/content/inline/1860-5397-7-163-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of substituted tetrahydropyridines.
Scheme 7: Synthesis of substituted tetrahydropyridines.
The proposed mechanism involves addition of Pd(0) onto the triple bond, followed by nucleophilic attack on the iminium generated in situ. The resulting vinylpalladium species reacts with the boron or alkynyl compound as previously shown. Allenylamines are also compatible with this anti-Wacker process, leading to more substituted tetrahydropyridines 16 in good to excellent yields (Scheme 8).
![[1860-5397-7-163-i8]](/bjoc/content/inline/1860-5397-7-163-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of more substituted tetrahydropyridines.
Scheme 8: Synthesis of more substituted tetrahydropyridines.
Another strategy allowing access to pyridine derivatives was developed by Katsumura and coworkers. They showed that chiral 2,4-disubstituted 1,2,5,6-tetrahydropyridines 17 can be obtained through a one pot imine synthesis, Stille coupling, 6π-azaelectrocyclization and aminoacetal formation. The chiral auxilliary can be removed by further treatment with DIBAL-H and Pb(OAc)4 (Scheme 9) [8].
![[1860-5397-7-163-i9]](/bjoc/content/inline/1860-5397-7-163-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of chiral tetrahydropyridines.
Scheme 9: Synthesis of chiral tetrahydropyridines.
The Strecker reaction, employing aldehydes or ketones and a cyanide source, is a very useful route for the preparation of α-aminonitriles 19 or 20. A general and efficient three-component method was reported by Jung and coworkers who used a new catalytic system based on a NHC–amidate palladium(II) complex 18. This complex acts as a Lewis acid to favor addition of cyanide to the imine generated in situ. This methodology employs smooth conditions and works with aldehydes as well as ketones, giving good to excellent yields (Scheme 10) [9].
![[1860-5397-7-163-i10]](/bjoc/content/inline/1860-5397-7-163-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Preparation of α-aminonitrile by a catalyzed Strecker reaction.
Scheme 10: Preparation of α-aminonitrile by a catalyzed Strecker reaction.
Barluenga and coworkers reported a synthesis of spiroacetals 22, through a Pd(II)-catalyzed three-component cascade reaction, starting from an alkynol, an aldehyde and a primary amine. The authors suggested that the first step of the reaction was the attack of the hydroxyl group onto the triple bond activated by a Pd(II) cationic complex, followed by a protodemetalation, which afforded the methylidenefuran 21. This reacts with the imine activated by the Pd(II) species through a Mannich-type process. Finally, addition of the phenol to the oxonium can lead to spiroacetal 22. One major drawback of this MCR is the formation of an equimolar amount of two diastereomers, which can be circumvented by further treatment of the crude mixture with 5 equivalents of MgClO4 and 1.6 equivalents of HClO4 in CH2Cl2/MeCN at room temperature. Under these acidic conditions, one diastereomer was cleanly and completely transformed into the other one (Scheme 11) [10].
![[1860-5397-7-163-i11]](/bjoc/content/inline/1860-5397-7-163-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Hallberg and coworkers developed a one-pot strategy towards the synthesis of masked 3-aminoindan-1-ones 23. This process was initiated by Heck addition of an aryl triflate to a vinyl ether, leading to an α-arylation product, followed by iminium formation in the presence of a secondary amine and subsequent tandem cyclization. The authors showed the importance of the ratio of the diverse reactants, notably that the amount of amine should remain low to avoid formation of aminal derivatives that would block the ring closure (Scheme 12) [11].
![[1860-5397-7-163-i12]](/bjoc/content/inline/1860-5397-7-163-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of masked 3-aminoindan-1-ones.
Scheme 12: Synthesis of masked 3-aminoindan-1-ones.
Homoallylic amines and α-aminoesters 24 were prepared by Malinakova and coworkers, by a palladium(II)-catalyzed coupling of boronic acids, 1,2-nonadiene, and aliphatic, aromatic or heteroaromatic imines [12]. The authors postulated a transmetalation step between the Pd(II) complex and a boronic acid activated by CsF, followed by insertion of the resulting σ-arylpalladium(II) into the allenic moiety leading to a π-allyl intermediate. This can undergo a nucleophilic allyl transfer to the imine, generating an amino-Pd(II) complex, which can subsequently add to another allenic unit. After a new transmetalation step with the boronic acid, the active catalytic species can be released and entered into a new catalytic cycle (Scheme 13).
![[1860-5397-7-163-i13]](/bjoc/content/inline/1860-5397-7-163-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of homoallylic amines and α-aminoesters.
Scheme 13: Synthesis of homoallylic amines and α-aminoesters.
Imine as a nucleophilic partner
A tandem four-component reaction allowing access to 1,2-dihydroisoquinolin-1-ylphosphonates 26 was reported by Wu and coworkers. Initial Sonogashira coupling was effected between a 2-bromobenzaldehyde and an alkyne in the presence of catalytic amounts of PdCl2(PPh3)2 and CuI. After complete conversion of the aldehyde into the coupling product 25 (TLC control), a primary amine and diethylphosphite were added to the reaction medium with concomitant addition of 10 mol % of Cu(OTf)2 necessary to complete the cyclization step. The proposed mechanism involves formation of an imine intermediate, which attacks the triple bond activated by the copper(II) complex. The resulting iminium was finally trapped by addition of diethylphosphite. Moderate to good yields were obtained depending on the nature of the various components (Scheme 14) [13].
![[1860-5397-7-163-i14]](/bjoc/content/inline/1860-5397-7-163-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Preparation of 1,2-dihydroisoquinolin-1-ylphosphonates.
Scheme 14: Preparation of 1,2-dihydroisoquinolin-1-ylphosphonates.
Amines as hetero-Michael donors
Many multicomponent approaches to nitrogen heterocycles have been developed based on the reaction of nitrogen-centered nucleophiles with α,β-unsaturated ketones generated in situ by Pd-catalyzed Sonogashira cross-coupling reactions. For instance, by building on their expertise in this area [14] Müller and coworkers recently developed a very effective and modular three-component strategy to assemble a series of 3,5-bis(hetero)aromatic pyrazoles in a consecutive fashion from terminal alkynes, acid chlorides, and hydrazine derivatives. Classical approaches to these valuable compounds are notably based on the cyclocondensation of hydrazine derivatives with 1,3-disubstituted three-carbon units, including α,β-unsaturated ketones, and particularly alkynones. In situ generation of the latter is an interesting means of overcoming the poor commercial availability of these compounds and also offers the flexibility needed for library production (Scheme 15). Thus various (hetero)aryl acid chlorides and terminal alkynes were heated in THF in the presence of Et3N and catalytic amounts of PdCl2(PPh3)2 and CuI. The resulting ynones 27 were then treated in situ with diversely substituted hydrazine derivatives to produce, upon microwave heating, a series of pyrazoles 28–30 (Scheme 15). As previously established for this type of cycloaddition, one of the two possible regioisomers was obtained preferentially depending on the hydrazine derivatives used, N-alkyl- and N-arylhydrazines giving opposite regioselectivities [15].
![[1860-5397-7-163-i15]](/bjoc/content/inline/1860-5397-7-163-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Pyrazole elaboration by cycloaddition of hydrazines with alkynones generated in situ.
Scheme 15: Pyrazole elaboration by cycloaddition of hydrazines with alkynones generated in situ.
The carbonylative coupling of terminal alkynes with aryl (and heteroaryl) halides was proposed by Mori and coworkers as a different approach to α,β-alkynyl ketone derivatives as pyrazole precursors. They established a four-component domino process combining various organic halides, terminal alkynes, hydrazines, and carbon monoxide at room temperature. In this case, all components are mixed at the very beginning of the process, in aqueous THF, under ambient pressure of CO and in the presence of 1 mol % PdCl2(PPh3)2 as the sole catalyst. However, one drawback of this approach is that it is, so far, limited to simple hydrazine and N-methylhydrazine (Scheme 16). From a mechanistic point of view, it is interesting to note that the intermediacy of α,β-alkynyl ketones in the four-component process could not be confirmed (TLC). In addition, their reaction with hydrazines was shown to be ineffective under the present solvent system in the presence or absence of palladium catalyst. This may suggest that if α,β-alkynyl ketones are formed, they immediately react with hydrazine to form pyrazole by a specific rate acceleration in the one-pot process [16].
![[1860-5397-7-163-i16]](/bjoc/content/inline/1860-5397-7-163-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: An alternative approach to pyrazoles involving hydrazine cycloaddition.
Scheme 16: An alternative approach to pyrazoles involving hydrazine cycloaddition.
The Sonogashira cross-coupling of acid chlorides with terminal alkynes has also been demonstrated as a valuable tool to generate, in situ, ynones bearing a pendant amine group 31, which will undergo addition–intramolecular cyclocondensation processes leading to the formation of pyrrole derivatives. For instance, a series of (hetero)aryl-, alkynyl-, and cycloalkyl acid chlorides were cross-coupled with N-Boc-protected propargylamine at room temperature, and the resulting ynones were then treated in situ with sodium iodide and PTSA to yield 2-substituted N-Boc-4-iodopyrroles 32 in good overall yields. Interestingly, this product may be further transformed in situ into the corresponding N-Boc-4-alkynylpyrroles 33 by a further Sonogashira coupling that makes use of the still-operative palladium complex. To do so, a terminal alkyne and caesium carbonate were added to the reaction mixture containing the newly formed 4-iodopyrrole, and the reaction temperature was increased to 70 °C (Scheme 17) [17].
![[1860-5397-7-163-i17]](/bjoc/content/inline/1860-5397-7-163-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of pyrroles by cyclization of propargyl amines.
Scheme 17: Synthesis of pyrroles by cyclization of propargyl amines.
Grigg and coworkers reported a three-component cascade process for the synthesis of isoindolones and phthalazones starting from ortho-halogenated cinnamates 34 and related compounds in the presence of hydrazine derivatives and carbon monoxide. The process is thought to begin with carbonylation of the starting aryl iodide to give an acylpalladium species 35, which is intercepted by the hydrazine nucleophile to give an acylhydrazide intermediate 36. The latter undergoes intramolecular Michael addition to give either N-aminoisoindolones 37 or mono-N- and di-N,N′-phthalazones 38, depending essentially on whether a monosubstituted or 1,2-disubstituted hydrazine derivative is used. A proper choice of catalyst and reaction conditions is also needed to improve the efficiency of each reaction (Scheme 18) [18].
![[1860-5397-7-163-i18]](/bjoc/content/inline/1860-5397-7-163-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Isoindolone and phthalazone synthesis by cyclization of acylhydrazides.
Scheme 18: Isoindolone and phthalazone synthesis by cyclization of acylhydrazides.
Consecutive one-pot transformations initiated by Heck reaction and terminated by intramolecular aza–Michael addition were developed by Hanson and coworkers to access a series of benzo-fused sultams. A range of α-bromobenzenesulfonyl chlorides 40 were first coupled with various amines in DMF at room temperature in the presence of Et3N to generate intermediate sulfonamides 41. Subsequent in situ addition of a Michael acceptor in large excess together with Et3N, Bu4NCl, and catalytic Pd2(dba)3·CHCl3 led to the production of the desired sultams 42 upon heating at 110 °C. A series of sultam derivatives of bioactive, related isoindol-1-one amides 43 were also prepared by entering acrylic acid into the Heck–aza–Michael process and coupling a second amine derivative (after removal of excess acrylic acid) with the aid of an oligomeric alkyl carbodiimide 44 (Scheme 19) [19].
![[1860-5397-7-163-i19]](/bjoc/content/inline/1860-5397-7-163-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Sultam synthesis by cyclization of sulfonamides.
Scheme 19: Sultam synthesis by cyclization of sulfonamides.
Interestingly, Willis and coworkers have shown that aryl N-aminosulfonamides may be accessed by three-component coupling of aryl iodides, hydrazines, and DABCO·(SO2)2 as a convenient source of sulfur dioxide. However, this Pd-catalyzed aminosulfonylation process proved inefficient with primary amines (Scheme 20) [20].
![[1860-5397-7-163-i20]](/bjoc/content/inline/1860-5397-7-163-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of sulfonamides by aminosulfonylation of aryl iodides.
Scheme 20: Synthesis of sulfonamides by aminosulfonylation of aryl iodides.
Multicomponent synthesis of nitrogen-containing heterocycles may also be initiated by an aza-Michael addition and terminated by a palladium-catalyzed ring-closure process [21]. For instance, Balme and coworkers reported a Pd-catalyzed three-component assembly of highly functionalized 4-benzyl- and allyl-pyrrolidines 46 based on a combination of allylamines (in situ transformed to their sodium salts by treatment with NaH), gem-diactivated alkenes 45 as Michael acceptors, and unsaturated halides (or triflate). Equal amounts of each of the three partners were reacted at room temperature in the presence of a catalytic quantity of a palladium(0) catalyst generated in situ by reduction of PdCl2(PPh3)2 with n-butyllithium. The key step in this one-pot transformation is the Pd-mediated cyclofunctionalization of the allyl moiety by carbopalladation/reductive elimination [22]. It is interesting to note that 3-sulfonylpyrolidin-2-ones (γ-lactams) 48 may also be accessed in high yield as single trans-diastereomers upon simple treatment of N-allyl- or N-methylpyrrolidines with 2-mercaptobenzoic acid in boiling MeCN. Acid-promoted formation of a ring-opened iminium salt intermediate 47, followed by hydrolysis and subsequent intramolecular attack of the released secondary amine onto the ester group, would account for the formation of the γ-lactams [23]. This unexpected transformation was observed during attempted Pd-catalyzed deallylation of N-allyl-3-sulfonylpyrrolidines in the presence of 2-mercaptobenzoic acid according to the procedure developed by Genêt and coworkers [24] (Scheme 21).
![[1860-5397-7-163-i21]](/bjoc/content/inline/1860-5397-7-163-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Pyrrolidine synthesis by carbopalladation of allylamines.
Scheme 21: Pyrrolidine synthesis by carbopalladation of allylamines.
Amines as coupling partners through hydroamination of alkyne derivatives
Many synthetic methods for the preparation of indole derivatives have been reported because they occur in numerous natural products and bioactive compounds. Among these different strategies, those involving a palladium-catalyzed coupling reaction have received much attention [25] and one of the most commonly used procedures involves a one-pot two-step reaction with, first, a Sonogashira coupling of o-haloanilines with terminal alkynes, followed by a cyclization reaction of the resulting 2-alkynyaniline derivatives [26,27]. A strategy for the preparation of indoles through a three-component reaction consisted of generating the terminal alkyne precursor 49 in situ through a Pd/Cu mediated coupling reaction between (trimethylsilyl)acetylene (TMSA) with an aryl iodide, followed by a desilylation reaction. The subsequent addition of the third partner, an o-iodoanilide derivative, allowed a Pd/Cu tandem C–C/C–N-bond-forming reaction. The main advantage of this multicomponent reaction is to suppress the isolation of the pure form of the arylalkyne derivatives, which often represents a problem due to their ability to dimerize. This one-pot four-step reaction proceeded well with a series of electron-rich and electron-poor aryl iodide derivatives, and the best results were obtained when Pd/C–PPh3 was used as the catalyst system (Scheme 22).
![[1860-5397-7-163-i22]](/bjoc/content/inline/1860-5397-7-163-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of indoles through a sequential C–C coupling/desilylation–coupling/cyclization reaction.
Scheme 22: Synthesis of indoles through a sequential C–C coupling/desilylation–coupling/cyclization reaction.
Another attractive palladium-mediated multicomponent approach towards the synthesis of indole derivatives involving the cyclization of a 2-alkynylaniline intermediate is based on a sequential, site-selective Pd-catalyzed cross-coupling approach starting from 1-chloro-2-iodobenzenes, phenylacetylene and a variety of primary amines [28,29]. The sequential three-component reaction was performed with the aid of an N-heterocyclic carbene-palladium complex generated in situ, derived from imidazolium salt 50 and Pd(OAc)2, and with CuI as the catalyst system. A first Sonogashira coupling reaction occurred, in the presence of Cs2CO3 as base, leading to ortho-alkynylchloroarene intermediates 51. A subsequent amination was possible due to the high catalytic activity of this palladiumcarbene complex in the coupling of aryl chlorides. This was followed by an intramolecular alkyne–hydroamination (addition of an N–H bond across a carbon–carbon multiple bond) leading to the corresponding indole derivatives 52. The amination/alkyne–hydroamination sequence requires the addition of 1.5 equiv of t-BuOK to reach completion. A variety of amines were involved in this one-pot sequential three-component reaction allowing the introduction of different protecting groups of the indole moiety. This site-selective, Pd/Cu-catalyzed cross-coupling approach was also performed on 1-chloro-2-iodo-4-(trifluoromethyl)benzene as o-dihaloarene partner and the corresponding polysubstituted indoles were isolated in good yields as single regioisomers (Scheme 23).
![[1860-5397-7-163-i23]](/bjoc/content/inline/1860-5397-7-163-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of indoles by a site selective Pd/C catalyzed cross-coupling approach.
Scheme 23: Synthesis of indoles by a site selective Pd/C catalyzed cross-coupling approach.
Based on this concept, Alper and coworkers reported the synthesis of isoindolin-1-one derivatives 53 through a four-component reaction starting from ortho-dihaloarenes and conducted in phosphonium salt-based ionic liquids (PSILs) with PdCl2(PPh3)2/CuI/DBU as the catalyst system [30]. In this case, the palladium-mediated Sonogashira coupling reaction leading to 1-halo-2-alkynylbenzene derivatives is followed by a carboxyamidation in the presence of carbon monoxide and primary amines [31]. This is followed by an in situ intramolecular hydroamination of the resulting amide on the triple bond, leading to substituted 3-methyleneisoindolin-1-ones in high selectivities in favor of the (Z)-isomers (Scheme 24).
![[1860-5397-7-163-i24]](/bjoc/content/inline/1860-5397-7-163-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of isoindolin-1-one derivatives through a sequential Sonogashira coupling/carbonylation/hydroamination reaction.
Scheme 24: Synthesis of isoindolin-1-one derivatives through a sequential Sonogashira coupling/carbonylation/h...
A palladium-mediated three-component process for the preparation of substituted pyrroles involving a dihalogeno substrate and a sequential Sonogashira coupling followed by an hydroamination was developed by Duchêne and Parrain [32]. In this one-pot sequence, the first reaction is an allylic amination between the 3,4-diiodobut-2-enoic acid (54) and a primary amine, which can be in competition with the intramolecular lactonization reaction. The best yields of the expected pyrroles were obtained when the three-component reaction was conducted, with five equivalents of the amine partner, at room temperature in DMF, with PdCl2(PPh3)2/CuI as the catalyst system. The initial C–N allylic amination, followed by a Sonogashira cross-coupling and an intramolecular hydroamination furnished a dihydroexoalkylidene pyrrole 55, which rearranges into pyrrole 56. This Pd/Cu-mediated three-component approach is influenced by the nature of the nitrogen nucleophile, and the reaction failed with tosylamine and benzylcarbamate, whereas aryl-, alkyl- and benzylamines were used successfully in this reaction (Scheme 25).
![[1860-5397-7-163-i25]](/bjoc/content/inline/1860-5397-7-163-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis of pyrroles through an allylic amination/Sonogashira coupling/hydroamination reaction.
Scheme 25: Synthesis of pyrroles through an allylic amination/Sonogashira coupling/hydroamination reaction.
A three-component reaction involving in the first step a Sonogashira coupling of o-haloanilines 57 with terminal alkynes and leading to o-alkynylaniline intermediates 58 was developed by Lu and co-workers [33]. This one-pot reaction is based on a stepwise synthesis of indole derivatives reported by Cachi’s group, and involves, in the last step, a palladium-mediated cyclization of o-alkynylaniline derivatives in the presence of aryl halides [34]. In this process, oxidative addition of the aryl halide to the Pd(0) catalyst generates an organopalladium reagent, which activates the alkyne moiety towards nucleophilic attack of the amino group. A reductive elimination generates the indole derivatives 59.
In this one-pot three-component reaction, the same palladium complex catalyzes the Sonogashira coupling and the cyclofunctionalization reaction. However, the presence of a strong electron-withdrawing substituent on the amino group is needed for the intramolecular cyclization reaction. Therefore, a protocol for a copper-free Sonogashira coupling was developed in order to suppress the concurrent formation of 2-substituted indoles 60 by direct cyclization of o-alkynylaniline intermediates under the classical Sonogashira reaction conditions. Interestingly, aryl bromides were used as a third partner and may be added at the beginning of this one-pot reaction since no competition between the Sonogashira coupling with these substrates and iodoanilides is observed. A variety of 2,3-disubstituted indoles 59 were obtained under mild conditions in good yields (Scheme 26).
![[1860-5397-7-163-i26]](/bjoc/content/inline/1860-5397-7-163-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Synthesis of indoles through a Sonogashira coupling/cyclofunctionalization reaction.
Scheme 26: Synthesis of indoles through a Sonogashira coupling/cyclofunctionalization reaction.
A similar three-component reaction was further developed under microwave irradiation by Larock and coworkers [35]. In this case, N,N-dimethyl-2-iodoanilines, terminal alkynes and various aryl iodides were involved in the reaction due to the high nucleophilicity of the N,N-dialkylamino moiety. Here, the reaction needs to be performed in two steps, the aryl iodide in acetonitrile being added after the completion of the first Sonogashira coupling reaction. Regarding the mechanism of the reaction, the intramolecular attack of the amino nucleophile affords here indolium species 61. Removal of a methyl group by the iodide anion generated in situ, followed by reductive elimination allows the preparation of various 2,3-disubstituted indole derivatives 62 (Scheme 27).
![[1860-5397-7-163-i27]](/bjoc/content/inline/1860-5397-7-163-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of indoles through a one-pot two-step Sonogashira coupling/cyclofunctionalization reaction.
Scheme 27: Synthesis of indoles through a one-pot two-step Sonogashira coupling/cyclofunctionalization reactio...
Amines as coupling partners through Buchwald–Hartwig amination
Other strategies used for the palladium-mediated three-component preparation of substituted indole derivatives involve an efficient Buchwald–Hartwig amination as the key step. Xi and co-workers developed an elegant one-pot synthesis of 2-alkynylindoles 64 involving o-bromo-(2,2-dibromovinyl)benzenes 63, arylamines and terminal alkynes as starting partners [36]. It should be noted that the three components are present at the same time in the reaction system and the best results for this Pd-catalyzed tandem Sonogashira/double C–N coupling reaction were obtained when Pd(OAc)2 was used as the catalyst along with a bulky bidentate phosphine ligand such as Xantphos in the presence of Cs2CO3 as base. Most likely, the reaction proceeds through a Pd-catalyzed Sonogashira coupling leading to a mono-alkynylated product, followed by an intermolecular Buchwald–Hartwig amination and a subsequent intramolecular amination. This Pd-catalyzed tandem coupling reaction allows the preparation of a variety of 2-alkynylindoles 64 (Scheme 28).
![[1860-5397-7-163-i28]](/bjoc/content/inline/1860-5397-7-163-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of α-alkynylindoles through a Pd-catalyzed Sonogashira/double C–N coupling reaction.
Scheme 28: Synthesis of α-alkynylindoles through a Pd-catalyzed Sonogashira/double C–N coupling reaction.
An elegant three-component process based on a Pd-catalyzed cascade sequence, involving an alkenyl amination, a C-arylation and a subsequent intramolecular N-arylation, was developed by Barluenga and coworkers for the preparation of indole derivatives 68 [37]. Here, equimolecular amounts of haloalkene 65, o-dihaloarene 66, and amines are mixed at the start of the reaction. The higher reactivity of the haloalkene toward oxidative addition with palladium, when compared to the haloarene, allowed the unique formation of the imine intermediate 67. This was followed by the formation of the corresponding aza-allylic anion by deprotonation in basic media. A subsequent Pd-mediated intermolecular alkylation with the dihalogeno substrate followed by an intramolecular N-arylation furnished 2-substituted indoles 68. In this cascade reaction, the palladium catalyst intervenes in three different coupling reactions: Intermolecular N-alkenylation, C-arylation and intramolecular N-arylation (Scheme 29).
![[1860-5397-7-163-i29]](/bjoc/content/inline/1860-5397-7-163-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Synthesis of indoles through a Pd-catalyzed sequential alkenyl amination/C-arylation/N-arylation.
Scheme 29: Synthesis of indoles through a Pd-catalyzed sequential alkenyl amination/C-arylation/N-arylation.
The palladium-mediated amination reaction coupled with a nitrogen–carbon bond-forming reaction was also used for the stereoselective synthesis of N-aryl-2-benzylpyrrolidines 71 starting from linear 4-pentenylamine and its derivatives [38]. In this tandem reaction, two different aryl bromides are sequentially added to the primary aliphatic amine in the presence of a palladium(0) catalyst. The first selective, Pd-catalyzed mono-N-arylation leading to the corresponding γ-(N-arylamino)alkenes 69 is followed by a carboamination reaction, developed by the same group, after addition of the second aryl bromide [39]. A plausible mechanism for this cyclization/coupling reaction involves formation of intermediate 70 by reaction of the organopalladium complex with the newly formed γ-(N-arylamino)alkene 69. A syn-insertion of the alkene into the Pd–N bond in 70 followed by reductive elimination furnishes N-aryl-2-benzylpyrrolidine derivatives 71. In this process, both reactions are catalyzed by zerovalent palladium and the choice of the phosphine ligand for the N-arylation of amines and the carboamination reactions is of great significance and an in situ modification of the catalyst by phosphine ligand exchange was necessary to achieve the selective diarylation in good yields (Scheme 30).
![[1860-5397-7-163-i30]](/bjoc/content/inline/1860-5397-7-163-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Synthesis of N-aryl-2-benzylpyrrolidines through a sequential N-arylation/carboamination reaction.
Scheme 30: Synthesis of N-aryl-2-benzylpyrrolidines through a sequential N-arylation/carboamination reaction.
A three-component reaction involving a palladium-catalyzed double N-arylation in combination with a S-arylation in a single operation was developed for the preparation of phenothiazine derivatives 72 starting from primary amines, 2-bromothiophenol and substituted 1-bromo-2-iodobenzenes [40]. Ferrocene ligands, such as dppf, and Pd2dba3 as the palladium source were found to be the most suitable and efficient catalyst systems for the preparation of a series of phenotiazine derivatives. This one-pot procedure worked with a wide variety of primary amines including allyl-, benzyl-, alkyl- and arylamines, and antipsychotic promazine as well as some analogues 74 were synthesized when 3-(dimethylamino)-1-propylamine (73) was used as the amine component (Scheme 31).
![[1860-5397-7-163-i31]](/bjoc/content/inline/1860-5397-7-163-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Synthesis of phenothiazine derivatives through a one-pot palladium-catalyzed double C–N arylation in combination with a S-arylation.
Scheme 31: Synthesis of phenothiazine derivatives through a one-pot palladium-catalyzed double C–N arylation i...
Amines as coupling partners through a Pd-mediated allylic amination
The allene carbopalladation process with organic halides is known to generate a π-allylpalladium intermediate, which can be trapped by intermolecular carbo- or heteronucleophiles to produce the corresponding three-component adduct. This strategy was used by Ma and coworkers for the selective preparation of five-membered nitrogen heterocycles starting from allene-bearing nucleophilic centers [41]. In this context, the same authors developed a new synthesis of substituted imidazolidinones 75 through a palladium-catalyzed, three-component reaction of 2,3-allenylamines, organic halides and isocyanates [42]. In this process, there is first a carbopalladation of the functionalized allene with the aryl iodide, followed by reaction of the internal aza-nucleophile with the highly electrophilic isocyanate derivative, before premature trapping of the initially formed π-allylpalladium intermediate that would lead to 2,5-dihydropyrrole or vinylic azacyclopropane derivatives. This is followed by a five-membered ring cyclization leading to polysubstituted imidazolidinones 75 in rather good yields and excellent selectivity (Scheme 32).
![[1860-5397-7-163-i32]](/bjoc/content/inline/1860-5397-7-163-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of substituted imidazolidinones through a palladium-catalyzed three-component reaction of 2,3-allenyl amines, organic halides and isocyanates.
Scheme 32: Synthesis of substituted imidazolidinones through a palladium-catalyzed three-component reaction of...
A conceptually related strategy was developed by Yoshida, Itami and Tonogaki [43]. In this case, the palladium-catalyzed allenation with an aryl iodide is performed on the allenylboronate pinacol ester 76 in the presence of benzylamine to afford the functionalized alkenylboronate 77 in quantitative yields and with complete regio- and stereoselectivity. A four-component reaction was further developed through an in situ post C–B arylation by adding a second aryl iodide, with Cs2CO3 and water, to the newly formed alkenylboronate 78. The subsequent Suzuki–Miyaura coupling led to the formation of 2,3-diarylated amines 79 and the best results were obtained with secondary amines, the remaining N–H functionality interfering with the C–B arylation step with primary amines as coupling partners (Scheme 33).
![[1860-5397-7-163-i33]](/bjoc/content/inline/1860-5397-7-163-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Synthesis of 2,3-diarylated amines through a palladium-catalyzed four-component reaction involving an allenylboronate pinacol ester.
Scheme 33: Synthesis of 2,3-diarylated amines through a palladium-catalyzed four-component reaction involving ...
This palladium-catalyzed three-component coupling was applied to the synthesis of rolipram, which is a phosphodiesterase-4 inhibitor. In this process, the Pd-mediated three-component reaction that gives access to the alkenylboronate 80 was followed by a palladium-mediated carbonylative cyclization reaction. Hydrogenation of the resulting unsaturated lactam 81 and removal of the N-benzyl group afforded rolipram (Scheme 34).
![[1860-5397-7-163-i34]](/bjoc/content/inline/1860-5397-7-163-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Synthesis of rolipram involving a Pd-catalyzed three-component reaction.
Scheme 34: Synthesis of rolipram involving a Pd-catalyzed three-component reaction.
Alper and coworkers developed several multicomponent approaches for the synthesis of nitrogen-containing heterocycles based on a palladium-mediated carbonylation reaction [44]. An interesting, related strategy for the preparation of unsaturated seven-membered ring lactams 84, starting from a Baylis–Hillman adduct bearing an aryl bromide moiety 82, with primary amines and carbon monoxide, was developed by the same group [45]. The sequence involves first a selective palladium(0)-catalyzed amination on the Baylis–Hillman acetates with primary amines leading to allylic amines 83. This is followed by oxidative addition of the palladium species to the aryl bromide, which undergoes CO insertion to form the corresponding acylpalladium, which in turn is intercepted by the allylamine to give, after reductive elimination, the seven-membered ring lactams 84 in good to excellent yields. A wide range of amine components are compatible with this one-pot procedure (Scheme 35).
![[1860-5397-7-163-i35]](/bjoc/content/inline/1860-5397-7-163-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Synthesis of seven-membered ring lactams through a Pd-catalyzed amination/intramolecular cyclocarbonylation.
Scheme 35: Synthesis of seven-membered ring lactams through a Pd-catalyzed amination/intramolecular cyclocarbo...
Conclusion
In summary, this review highlights the usefulness of amines as key building blocks in the development of Pd-mediated multicomponent approaches to polyfunctionalized nitrogen acyclic or cyclic compounds. Amines may be involved in several bond-forming transformations, including aza-Michael additions, hydroaminations of alkynes, Buchwald–Hartwig aminations, and allylic aminations, thereby allowing the creation of several covalent bonds in a single operation. Imine derivatives are also of high synthetic value as they may act either as electrophilic or nucleophilic partners. It is expected that further useful, new multicomponent processes in which amines play a central role will be developed in the near future.
References
-
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Davis, J. L.; Dhawan, R.; Arndtsen, B. A. Angew. Chem., Int. Ed. 2004, 43, 590–594. doi:10.1002/anie.200352123
Return to citation in text: [1] -
Siamaki, A. R.; Black, D. A.; Arndtsen, B. A. J. Org. Chem. 2008, 73, 1135–1138. doi:10.1021/jo701875b
Return to citation in text: [1] -
Dhawan, R.; Dghaym, R. D.; Cyr, D. J. S.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. doi:10.1021/ol061308j
Return to citation in text: [1] [2] -
Worrall, K.; Xu, B.; Bontemps, S.; Arndtsen, B. A. J. Org. Chem. 2011, 76, 170–180. doi:10.1021/jo101858d
Return to citation in text: [1] -
Tsukamoto, H.; Ueno, T.; Kondo, Y. Org. Lett. 2007, 9, 3033–3036. doi:10.1021/ol071107v
Return to citation in text: [1] -
Tsukamoto, H.; Kondo, Y. Angew. Chem., Int. Ed. 2008, 47, 4851–4854. doi:10.1002/anie.200800823
Return to citation in text: [1] -
Kobayashi, T.; Nakashima, M.; Hakogi, T.; Tanaka, K.; Katsumura, S. Org. Lett. 2006, 8, 3809–3812. doi:10.1021/ol061405c
Return to citation in text: [1] -
Jarusiewicz, J.; Choe, Y.; Yoo, K. S.; Park, C. P.; Jung, K. W. J. Org. Chem. 2009, 74, 2873–2876. doi:10.1021/jo900163w
Return to citation in text: [1] -
Barluenga, J.; Mendoza, A.; Rodríguez, F.; Fañanás, F. J. Angew. Chem., Int. Ed. 2009, 48, 1644–1647. doi:10.1002/anie.200805519
Return to citation in text: [1] -
Arefalk, A.; Larhed, M.; Hallberg, A. J. Org. Chem. 2005, 70, 938–942. doi:10.1021/jo048187q
Return to citation in text: [1] -
Hopkins, C. D.; Malinakova, H. C. Org. Lett. 2006, 8, 5971–5974. doi:10.1021/ol0624528
Return to citation in text: [1] -
Zhou, H.; Jin, H.; Ye, S.; He, X.; Wu, J. Tetrahedron Lett. 2009, 50, 4616–4618. doi:10.1016/j.tetlet.2009.05.106
Return to citation in text: [1] -
Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609
And references cited therein.
Return to citation in text: [1] -
Willy, B.; Müller, T. J. J. Eur. J. Org. Chem. 2008, 4157–4168. doi:10.1002/ejoc.200800444
Return to citation in text: [1] -
Mohamed Ahmed, M. S.; Kobayashi, K.; Mori, A. Org. Lett. 2005, 7, 4487–4489. doi:10.1021/ol051841j
Return to citation in text: [1] -
Merkul, E.; Boersch, C.; Frank, W.; Müller, T. J. J. Org. Lett. 2009, 11, 2269–2272. doi:10.1021/ol900581a
Return to citation in text: [1] -
Grigg, R.; Sridharan, V.; Shah, M.; Mutton, S.; Kilner, C.; MacPherson, D.; Milner, P. J. Org. Chem. 2008, 73, 8352–8356. doi:10.1021/jo800822p
Return to citation in text: [1] -
Rolfe, A.; Young, K.; Hanson, P. R. Eur. J. Org. Chem. 2008, 5254–5262. doi:10.1002/ejoc.200800651
Return to citation in text: [1] -
Nguyen, B.; Emmett, E. J.; Willis, M. C. J. Am. Chem. Soc. 2010, 132, 16372–16373. doi:10.1021/ja1081124
Return to citation in text: [1] -
Balme, G.; Bouyssi, D.; Lomberget, T.; Monteiro, N. Synthesis 2003, 2115–2134. doi:10.1055/s-2003-42082
Return to citation in text: [1] -
Martinon, L.; Azoulay, S.; Monteiro, N.; Kündig, E. P.; Balme, G. J. Organomet. Chem. 2004, 689, 3831–3836. doi:10.1016/j.jorganchem.2004.07.039
Return to citation in text: [1] -
Martinon, L. Ph.D. Thesis, University of Lyon/University of Geneva, France/Switzerland, 2006.
Return to citation in text: [1] -
Lemaire-Audoire, S.; Savignac, M.; Genêt, J.-P.; Bernard, J.-M. Tetrahedron Lett. 1995, 36, 1267–1270. doi:10.1016/0040-4039(95)00003-U
Return to citation in text: [1] -
Li, J. J.; Gribble, G. W. Metal-Catalyzed Cross-Coupling Reactions for Indoles. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble, G. W., Ed.; Topics in Heterocyclic Chemistry, Vol. 26; Springer: Berlin, 2011; pp 193–234. doi:10.1007/7081_2010_36
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873–2920. doi:10.1021/cr040639b
Return to citation in text: [1] -
Layek, M.; Lakshmi, U.; Kalita, D.; Barange, D. K.; Islam, A.; Mukkanti, K.; Pal, M. Beilstein J. Org. Chem. 2009, 5, No. 46. doi:10.3762/bjoc.5.46
Return to citation in text: [1] -
Ackermann, L. Org. Lett. 2005, 7, 439–442. doi:10.1021/ol047649j
Return to citation in text: [1] -
Kaspar, L. T.; Ackermann, L. Tetrahedron 2005, 61, 11311–11316. doi:10.1016/j.tet.2005.09.095
Return to citation in text: [1] -
Bradaric, C. J.; Downard, A.; Kennedy, C.; Robertson, A. J.; Zhou, Y. Green Chem. 2003, 5, 143–152. doi:10.1039/b209734f
Return to citation in text: [1] -
Cao, H.; McNamee, L.; Alper, H. Org. Lett. 2008, 10, 5281–5284. doi:10.1021/ol8021403
Return to citation in text: [1] -
Lamandé-Langle, S.; Abarbri, M.; Thibonnet, J.; Duchêne, A.; Parrain, J.-L. Chem. Commun. 2010, 46, 5157–5159. doi:10.1039/C0CC00500B
Return to citation in text: [1] -
Lu, B. Z.; Zhao, W.; Wei, H.-X.; Dufour, M.; Farina, V.; Senanayake, C. H. Org. Lett. 2006, 8, 3271–3274. doi:10.1021/ol061136q
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Marinelli, F. Tetrahedron Lett. 1992, 33, 3915–3918. doi:10.1016/S0040-4039(00)74818-0
Return to citation in text: [1] -
Chen, Y.; Markina, N. A.; Larock, R. C. Tetrahedron 2009, 65, 8908–8915. doi:10.1016/j.tet.2009.07.075
Return to citation in text: [1] -
Liang, Y.; Meng, T.; Zhang, H.-J.; Xi, Z. Synlett 2011, 911–914. doi:10.1055/s-0030-1259699
Return to citation in text: [1] -
Barluenga, J.; Jiménez-Aquino, A.; Valdés, C.; Aznar, F. Angew. Chem., Int. Ed. 2007, 46, 1529–1532. doi:10.1002/anie.200604407
Return to citation in text: [1] -
Yang, Q.; Ney, J. E.; Wolfe, J. P. Org. Lett. 2005, 7, 2575–2578. doi:10.1021/ol050647u
Return to citation in text: [1] -
Ney, J. E.; Wolfe, J. P. Angew. Chem., Int. Ed. 2004, 43, 3605–3608. doi:10.1002/anie.200460060
Return to citation in text: [1] -
Dahl, T.; Tornøe, C. W.; Bang-Andersen, B.; Nielsen, P.; Jorgensen, M. Angew. Chem., Int. Ed. 2008, 47, 1726–1728. doi:10.1002/anie.200705209
Return to citation in text: [1] -
Ma, S.; Jiao, N. Angew. Chem., Int. Ed. 2002, 41, 4737–4740. doi:10.1002/anie.200290033
For a three component strategy involving carbopalladation of γ-allenic malonates.
Return to citation in text: [1] -
Shu, W.; Yu, Q.; Jia, G.; Ma, S. Chem.–Eur. J. 2011, 17, 4720–4723. doi:10.1002/chem.201003611
Return to citation in text: [1] -
Tonogaki, K.; Itami, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2006, 128, 1464–1465. doi:10.1021/ja057778a
Return to citation in text: [1] -
Zeng, F.; Alper, H. Org. Lett. 2010, 12, 3642–3644. doi:10.1021/ol101428v
Return to citation in text: [1] -
Cao, H.; Vieira, T. O.; Alper, H. Org. Lett. 2011, 13, 11–13. doi:10.1021/ol102699a
Return to citation in text: [1]
| 28. | Ackermann, L. Org. Lett. 2005, 7, 439–442. doi:10.1021/ol047649j |
| 29. | Kaspar, L. T.; Ackermann, L. Tetrahedron 2005, 61, 11311–11316. doi:10.1016/j.tet.2005.09.095 |
| 30. | Bradaric, C. J.; Downard, A.; Kennedy, C.; Robertson, A. J.; Zhou, Y. Green Chem. 2003, 5, 143–152. doi:10.1039/b209734f |
| 31. | Cao, H.; McNamee, L.; Alper, H. Org. Lett. 2008, 10, 5281–5284. doi:10.1021/ol8021403 |
| 1. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. |
| 4. | Dhawan, R.; Dghaym, R. D.; Cyr, D. J. S.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. doi:10.1021/ol061308j |
| 14. |
Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609
And references cited therein. |
| 38. | Yang, Q.; Ney, J. E.; Wolfe, J. P. Org. Lett. 2005, 7, 2575–2578. doi:10.1021/ol050647u |
| 4. | Dhawan, R.; Dghaym, R. D.; Cyr, D. J. S.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. doi:10.1021/ol061308j |
| 15. | Willy, B.; Müller, T. J. J. Eur. J. Org. Chem. 2008, 4157–4168. doi:10.1002/ejoc.200800444 |
| 39. | Ney, J. E.; Wolfe, J. P. Angew. Chem., Int. Ed. 2004, 43, 3605–3608. doi:10.1002/anie.200460060 |
| 3. | Siamaki, A. R.; Black, D. A.; Arndtsen, B. A. J. Org. Chem. 2008, 73, 1135–1138. doi:10.1021/jo701875b |
| 12. | Hopkins, C. D.; Malinakova, H. C. Org. Lett. 2006, 8, 5971–5974. doi:10.1021/ol0624528 |
| 36. | Liang, Y.; Meng, T.; Zhang, H.-J.; Xi, Z. Synlett 2011, 911–914. doi:10.1055/s-0030-1259699 |
| 2. | Davis, J. L.; Dhawan, R.; Arndtsen, B. A. Angew. Chem., Int. Ed. 2004, 43, 590–594. doi:10.1002/anie.200352123 |
| 13. | Zhou, H.; Jin, H.; Ye, S.; He, X.; Wu, J. Tetrahedron Lett. 2009, 50, 4616–4618. doi:10.1016/j.tetlet.2009.05.106 |
| 37. | Barluenga, J.; Jiménez-Aquino, A.; Valdés, C.; Aznar, F. Angew. Chem., Int. Ed. 2007, 46, 1529–1532. doi:10.1002/anie.200604407 |
| 8. | Kobayashi, T.; Nakashima, M.; Hakogi, T.; Tanaka, K.; Katsumura, S. Org. Lett. 2006, 8, 3809–3812. doi:10.1021/ol061405c |
| 10. | Barluenga, J.; Mendoza, A.; Rodríguez, F.; Fañanás, F. J. Angew. Chem., Int. Ed. 2009, 48, 1644–1647. doi:10.1002/anie.200805519 |
| 34. | Arcadi, A.; Cacchi, S.; Marinelli, F. Tetrahedron Lett. 1992, 33, 3915–3918. doi:10.1016/S0040-4039(00)74818-0 |
| 7. | Tsukamoto, H.; Kondo, Y. Angew. Chem., Int. Ed. 2008, 47, 4851–4854. doi:10.1002/anie.200800823 |
| 11. | Arefalk, A.; Larhed, M.; Hallberg, A. J. Org. Chem. 2005, 70, 938–942. doi:10.1021/jo048187q |
| 35. | Chen, Y.; Markina, N. A.; Larock, R. C. Tetrahedron 2009, 65, 8908–8915. doi:10.1016/j.tet.2009.07.075 |
| 6. | Tsukamoto, H.; Ueno, T.; Kondo, Y. Org. Lett. 2007, 9, 3033–3036. doi:10.1021/ol071107v |
| 32. | Lamandé-Langle, S.; Abarbri, M.; Thibonnet, J.; Duchêne, A.; Parrain, J.-L. Chem. Commun. 2010, 46, 5157–5159. doi:10.1039/C0CC00500B |
| 5. | Worrall, K.; Xu, B.; Bontemps, S.; Arndtsen, B. A. J. Org. Chem. 2011, 76, 170–180. doi:10.1021/jo101858d |
| 9. | Jarusiewicz, J.; Choe, Y.; Yoo, K. S.; Park, C. P.; Jung, K. W. J. Org. Chem. 2009, 74, 2873–2876. doi:10.1021/jo900163w |
| 33. | Lu, B. Z.; Zhao, W.; Wei, H.-X.; Dufour, M.; Farina, V.; Senanayake, C. H. Org. Lett. 2006, 8, 3271–3274. doi:10.1021/ol061136q |
| 18. | Grigg, R.; Sridharan, V.; Shah, M.; Mutton, S.; Kilner, C.; MacPherson, D.; Milner, P. J. Org. Chem. 2008, 73, 8352–8356. doi:10.1021/jo800822p |
| 16. | Mohamed Ahmed, M. S.; Kobayashi, K.; Mori, A. Org. Lett. 2005, 7, 4487–4489. doi:10.1021/ol051841j |
| 40. | Dahl, T.; Tornøe, C. W.; Bang-Andersen, B.; Nielsen, P.; Jorgensen, M. Angew. Chem., Int. Ed. 2008, 47, 1726–1728. doi:10.1002/anie.200705209 |
| 17. | Merkul, E.; Boersch, C.; Frank, W.; Müller, T. J. J. Org. Lett. 2009, 11, 2269–2272. doi:10.1021/ol900581a |
| 41. |
Ma, S.; Jiao, N. Angew. Chem., Int. Ed. 2002, 41, 4737–4740. doi:10.1002/anie.200290033
For a three component strategy involving carbopalladation of γ-allenic malonates. |
| 42. | Shu, W.; Yu, Q.; Jia, G.; Ma, S. Chem.–Eur. J. 2011, 17, 4720–4723. doi:10.1002/chem.201003611 |
| 25. | Li, J. J.; Gribble, G. W. Metal-Catalyzed Cross-Coupling Reactions for Indoles. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble, G. W., Ed.; Topics in Heterocyclic Chemistry, Vol. 26; Springer: Berlin, 2011; pp 193–234. doi:10.1007/7081_2010_36 |
| 26. | Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873–2920. doi:10.1021/cr040639b |
| 27. | Layek, M.; Lakshmi, U.; Kalita, D.; Barange, D. K.; Islam, A.; Mukkanti, K.; Pal, M. Beilstein J. Org. Chem. 2009, 5, No. 46. doi:10.3762/bjoc.5.46 |
| 23. | Martinon, L. Ph.D. Thesis, University of Lyon/University of Geneva, France/Switzerland, 2006. |
| 24. | Lemaire-Audoire, S.; Savignac, M.; Genêt, J.-P.; Bernard, J.-M. Tetrahedron Lett. 1995, 36, 1267–1270. doi:10.1016/0040-4039(95)00003-U |
| 21. | Balme, G.; Bouyssi, D.; Lomberget, T.; Monteiro, N. Synthesis 2003, 2115–2134. doi:10.1055/s-2003-42082 |
| 45. | Cao, H.; Vieira, T. O.; Alper, H. Org. Lett. 2011, 13, 11–13. doi:10.1021/ol102699a |
| 22. | Martinon, L.; Azoulay, S.; Monteiro, N.; Kündig, E. P.; Balme, G. J. Organomet. Chem. 2004, 689, 3831–3836. doi:10.1016/j.jorganchem.2004.07.039 |
| 19. | Rolfe, A.; Young, K.; Hanson, P. R. Eur. J. Org. Chem. 2008, 5254–5262. doi:10.1002/ejoc.200800651 |
| 43. | Tonogaki, K.; Itami, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2006, 128, 1464–1465. doi:10.1021/ja057778a |
| 20. | Nguyen, B.; Emmett, E. J.; Willis, M. C. J. Am. Chem. Soc. 2010, 132, 16372–16373. doi:10.1021/ja1081124 |
© 2011 Bouyssi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)