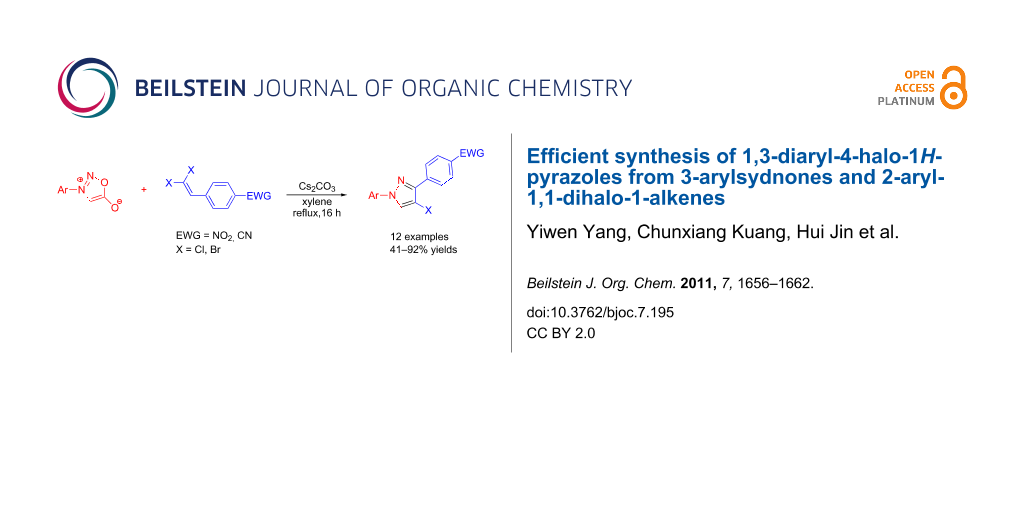
Abstract
An efficient synthesis of 1,3-diaryl-4-halo-1H-pyrazoles was achieved. The synthesis involves the [3 + 2] dipolar cycloaddition of 3-arylsydnones and 2-aryl-1,1-dihalo-1-alkenes. The process proceeds smoothly in moderate to excellent yields. 1,3-Diaryl-4-halo-1H-pyrazoles are found to be important intermediates that can easily be converted into 1,2,5-triaryl-substituted pyrazoles via Pd-catalyzed C–H bond activation.

Graphical Abstract
Introduction
Over the past decade, pyrazoles as key motifs in biologically active compounds have received increasing attention from the synthetic community. Diazoles can be employed as a central building block in the synthesis of compound libraries in the pharmaceutical [1] and agrochemical [2] industries. Pyrazole and its derivatives are an important class of heterocyclic compounds. As medicines, many of them display anti-inflammatory [3], antimicrobial [3], antiplatelet [4], antiallergenic [5], antifungal [6], MAP Kinase inhibitor [7], and anticancer activities [8]. As pesticides, they are used as insecticides [9] and fungicides [10], and as well as antiviral [11] and antibacterial agents [12]. Pyrazoles are gaining interest as ligands for transition metals, and in the field of materials chemistry [13,14].
Pyrazole and its derivatives can be synthesized by several methods [15]. The most common approach is based on the condensation of hydrazines with 1,3-dicarbonyl compounds or their equivalents. However, the 1,3-dipolar cycloaddition offers a more convenient synthetic route. Sydnones are easily accessible aromatic compounds and versatile synthetic intermediates. They can be used as unusual, alternative cycloaddition substrates for pyrazole synthesis [16,17]. These dipolar compounds are readily prepared in two steps from N-functionalized amino acids, and are readily stored and handled. Methods have been disclosed for the [3 + 2] dipolar cycloaddition of sydnones with alkenyl silanes [18] and stannanes [18], alkenyl arenes [19], 1,3-dienes [20,21], α,β-unsaturated esters [19,22] and nitriles [23], phosphane oxides [24] or with alkynyl silanes [18], stannanes [18,25,26], arenes [27,28], esters [29-33], boronic esters [34,35]. However, the cycloaddition of sydnones with 1,1-dihaloalkenes is unknown, as is the direct formation of 4-halopyrazoles through the [3 + 2] dipolar cycloaddition of sydnones.
In the present study, a convenient and efficient synthesis of a series of new 1,3-diaryl-4-halo-1H-pyrazoles 3 in moderate to excellent yields is reported. The route employed involves 1,3-dipolar cycloaddition between 3-arylsydnones 1 and 2-aryl-1,1-dihalo-1-alkenes 2 (Scheme 1). 1,3-Diaryl-4-halo-1H-pyrazoles were found to be important intermediates that could easily be converted into 1,2,4-triaryl- or 1,2,5-triaryl-substituted pyrazoles via a Pd-catalyzed C–C coupling reaction. To the best of our knowledge, this synthesis of 1,3-diaryl-4-halo-1H-pyrazoles has not yet been reported.
![[1860-5397-7-195-i1]](/bjoc/content/inline/1860-5397-7-195-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Access to 1,3-diaryl-4-halo-1H-pyrazoles from 3-arylsydnones and 2-aryl-1,1-dihalo-1-alkenes.
Scheme 1: Access to 1,3-diaryl-4-halo-1H-pyrazoles from 3-arylsydnones and 2-aryl-1,1-dihalo-1-alkenes.
Results and Discussion
To determine the optimal reaction conditions, 3-phenylsydnone (1a) and 1-(2,2-dibromovinyl)-4-nitrobenzene (2a) were used as the model substrates. A mixture of 1a, 2a, Cs2CO3 and xylene was then stirred in the dark in a sealed tube maintained at 140 °C in an oil bath. After 16 h, the product 3a was isolated in 72% yield.
The effects of different bases, molar ratios of 1a to 2a, solvents, and temperatures on the formation of 3a were investigated. The optimization of the 1,3-dipolar cycloaddition process between 1a and 2a is summarized in Table 1. Several bases were examined. When 1a reacted with 2a in the presence of Cs2CO3 as a base in xylene (140 °C, 16 h) in the dark, the reaction proceeded smoothly to generate the desired product 3a in 72% yield. Changing the base to K2CO3 decreased the yield to 54%. When Et3N, DBU, or no base was used, product 3a was not obtained (Table 1, entries 1–5). Changing the solvent to DMSO and DMF led to traces of 3a or no product, respectively (Table 1, entries 6–7). Various molar ratios of 1a to 2a were also studied. When the molar ratio of 1a to 2a was 1:1, the yield (48%) was much lower than that obtained with a ratio of 1:2 (72%) (Table 1, entries 1 and 8). Changing the quantity of Cs2CO3 to 2.0 equiv decreased the yield to 56%, but increasing the amount of Cs2CO3 to 4.5 equiv led to only a slightly higher yield (73%) (Table 1, entries 9 and 10).
Table 1: Screening for optimal reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-195-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | base (3 equiv) | solvent | T (°C) | yield of 3a (%)b |
|---|---|---|---|---|
| 1 | Cs2CO3 | xylene | 140 | 72 |
| 2 | K2CO3 | xylene | 140 | 54 |
| 3 | Et3N | xylene | 140 | 0 |
| 4 | DBU | xylene | 140 | 0 |
| 5 | none | xylene | 140 | 0 |
| 6 | Cs2CO3 | DMSO | 140 | trace |
| 7 | Cs2CO3 | DMF | 140 | 0 |
| 8c | Cs2CO3 | xylene | 140 | 48 |
| 9d | Cs2CO3 | xylene | 140 | 56 |
| 10e | Cs2CO3 | xylene | 140 | 73 |
| 11 | Cs2CO3 | xylene | 160 | 80 |
| 12 | Cs2CO3 | xylene | 120 | 63 |
| 13 | Cs2CO3 | xylene | 90 | 31 |
aReaction conditions: 1.0 equiv of 1a and 2.0 equiv of 2a were stirred in the dark for 16 h. bIsolated yield. c1.0 equiv of 1a and 1.0 equiv of 2a. d2.0 equiv of Cs2CO3. e4.5 equiv of Cs2CO3.
The effects of the reaction temperature on the formation of 3a were also remarkable. At 120 °C and 90 °C, the yields were decreased to 63% and 31%, respectively (Table 1, entries 12–13). At 160 °C, product 3a was formed in 80% yield (Table 1, entry 11). Ultimately, the optimal reaction conditions were determined as 1:2 molar ratio of 1a to 2a, 3.0 equiv Cs2CO3 base, xylene solvent, 160 °C, and 16 h in the dark (Table 1, entry 11).
Under the optimized conditions, a series of 3-arylsydnones 1 and 2-aryl-1,1-dihalo-1-alkenes 2 substrates were examined. Table 2 shows that in most cases, the desired pyrazoles 3 were smoothly generated in high yields (Table 2, entries 1, 4, 5, and 7–9). In cases with the aromatic portion of 3-arylsydnones 1 carrying either an electron-withdrawing group, such as in chlorine 1d, or an electron-donating substituent, as in methyl 1b and methoxyl 1c, the reactions all proceeded smoothly in moderate to excellent yields. Higher yields were obtained when the aromatic portion of 3-arylsydnones 1 carried an electron-donating group. The presence of a strong electron-donating group in the aromatic portion of 3-arylsydnones 1c greatly increased the reaction yield (Table 2, entries 7–9). On the other hand, the presence of an electron-withdrawing substituent on the aromatic portion of 3-arylsydnones 1d lead to the reactions providing pyrazoles 3 in relatively low yields (Table 2, entries 10–12). The effects of 2-aryl-1,1-dihalo-1-alkenes 2 on the formation of pyrazoles 3 were also remarkable. In the cases of the aromatic portion of 2-aryl-1,1-dihalo-1-alkenes 2 carrying nitryl 2a or cyano 2c groups all reactions proceeded smoothly in moderate to excellent yields. However, the stronger electron-withdrawing nitryl group provided pyrazoles 3 in relatively high yields. Under the same conditions, the reactivity of 1-(2,2-dibromovinyl)-4-nitrobenzene 2a was higher than that of 1-(2,2-dichlorovinyl)-4-nitrobenzene 2b (Table 2, entries 1, 2, 4, 5, 7, 8, 10 and 11). Generally speaking, where present the electron-donating groups of the substrates 1 exhibited stronger electron-donating effects, relative to substrates 2, leading to higher yields. We also used 1-(2,2-dibromovinyl)-4-methylbenzene (2d) as a dipolarophile to react with 3-phenylsydnone. In the process, two isomers, 4-bromo-1-phenyl-3-p-tolyl-1H-pyrazole (3m) and 3-bromo-1-phenyl-4-p-tolyl-1H-pyrazole (3n) were found, and they were generated in 35% and 17% yields, respectively. The experimental results indicate that the electron-donating group, i.e., methyl on the aromatic portion of 2-aryl-1,1-dihalo-1-alkenes, reduced the yields and regioselectivity of the reaction. Products 3 (except 3m and 3n) were generated with excellent regiocontrol. It may be that the large substituent (aryl) on the alkyne and the strong electron-withdrawing group, i.e., nitryl or cyano, are of great benefit to the regioselectivity of the reaction.
Table 2: Synthesis of 1,3-diaryl-4-halo-1H-pyrazoles 3.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-195-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | sydnones 1 | 1,1-dihaloalkenes 2 | pyrazoles 3 | yield of 3 (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-195-i6.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-195-i7.svg?max-width=637&scale=1.0)
2a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-195-i8.svg?max-width=637&scale=1.0)
3a |
80 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-195-i9.svg?max-width=637&scale=1.0)
1a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-195-i10.svg?max-width=637&scale=1.0)
2b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-195-i11.svg?max-width=637&scale=1.0)
3b |
65 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-195-i12.svg?max-width=637&scale=1.0)
1a |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-195-i13.svg?max-width=637&scale=1.0)
2c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-195-i14.svg?max-width=637&scale=1.0)
3c |
56 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-195-i15.svg?max-width=637&scale=1.0)
1b |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-195-i16.svg?max-width=637&scale=1.0)
2a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-195-i17.svg?max-width=637&scale=1.0)
3d |
84 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-195-i18.svg?max-width=637&scale=1.0)
1b |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-195-i19.svg?max-width=637&scale=1.0)
2b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-195-i20.svg?max-width=637&scale=1.0)
3e |
73 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-195-i21.svg?max-width=637&scale=1.0)
1b |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-195-i22.svg?max-width=637&scale=1.0)
2c |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-195-i23.svg?max-width=637&scale=1.0)
3f |
63 |
| 7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-195-i24.svg?max-width=637&scale=1.0)
1c |
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-195-i25.svg?max-width=637&scale=1.0)
2a |
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-195-i26.svg?max-width=637&scale=1.0)
3g |
92 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-195-i27.svg?max-width=637&scale=1.0)
1c |
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-195-i28.svg?max-width=637&scale=1.0)
2b |
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-195-i29.svg?max-width=637&scale=1.0)
3h |
75 |
| 9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-7-195-i30.svg?max-width=637&scale=1.0)
1c |
![[Graphic 28]](/bjoc/content/inline/1860-5397-7-195-i31.svg?max-width=637&scale=1.0)
2c |
![[Graphic 29]](/bjoc/content/inline/1860-5397-7-195-i32.svg?max-width=637&scale=1.0)
3i |
72 |
| 10 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-7-195-i33.svg?max-width=637&scale=1.0)
1d |
![[Graphic 31]](/bjoc/content/inline/1860-5397-7-195-i34.svg?max-width=637&scale=1.0)
2a |
![[Graphic 32]](/bjoc/content/inline/1860-5397-7-195-i35.svg?max-width=637&scale=1.0)
3j |
66 |
| 11 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-7-195-i36.svg?max-width=637&scale=1.0)
1d |
![[Graphic 34]](/bjoc/content/inline/1860-5397-7-195-i37.svg?max-width=637&scale=1.0)
2b |
![[Graphic 35]](/bjoc/content/inline/1860-5397-7-195-i38.svg?max-width=637&scale=1.0)
3k |
49 |
| 12 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-7-195-i39.svg?max-width=637&scale=1.0)
1d |
![[Graphic 37]](/bjoc/content/inline/1860-5397-7-195-i40.svg?max-width=637&scale=1.0)
2c |
![[Graphic 38]](/bjoc/content/inline/1860-5397-7-195-i41.svg?max-width=637&scale=1.0)
3l |
41 |
aReaction conditions: A mixture of 1 (0.3 mmol), 2 (0.6 mmol), and Cs2CO3 (0.9 mmol) was stirred in 3 mL of xylene in a sealed tube at 160 °C for 16 h in the dark. bIsolated yield.
The polysubstituted pyrazoles 3 were characterized by NMR, IR and HRMS. The structure of pyrazole 3g was further confirmed by single-crystal X-ray diffraction studies (Figure 1).
![[1860-5397-7-195-1]](/bjoc/content/figures/1860-5397-7-195-1.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of pyrazole 3g.
Figure 1: Crystal structure of pyrazole 3g.
In order to elucidate the probable reaction mechanism, a mixture of 1a, 2a and xylene was stirred in the dark at 160 °C. After 16 h, no product was found. The results indicate that it was difficult for 2-aryl-1,1-dihalo-1-alkenes to react with 3-arylsydnone in the absence of a base. The steric hindrance of the two halogen atoms on the alkenes is possibly the key factor. Thus a plausible mechanism for the synthesis of pyrazoles 3 could be as follows (Scheme 2): Initially, a haloalkyne is obtained by the elimination of a hydrogen halide from 2-aryl-1,1-dihalo-1-alkenes 2 with Cs2CO3 as a base [36]. Then, haloalkyne reacts with 3-arylsydnone 1 in a 1,3-dipolar cycloaddition reaction. Finally, CO2 is lost [18,34] and pyrazole 3 is generated.
![[1860-5397-7-195-i2]](/bjoc/content/inline/1860-5397-7-195-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the synthesis of 3.
Scheme 2: Proposed mechanism for the synthesis of 3.
Subsequently, the arylation reaction of pyrazoles 3 with iodobenzene or phenylboronic acid was investigated (Scheme 3).
![[1860-5397-7-195-i3]](/bjoc/content/inline/1860-5397-7-195-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Arylation reactions of pyrazoles (3) with iodobenzene or phenylboronic acid.
Scheme 3: Arylation reactions of pyrazoles (3) with iodobenzene or phenylboronic acid.
Compounds 3b, 3h, and 3g were chosen as the model substrates. When compounds 3b and 3h were treated with 2 equiv each of iodobenzene and potassium carbonate in DMF with Pd(OAc)2 as a catalyst, the desired 5-aryl substituted products 4a and 4b were generated in 51% and 82% yields, respectively. When pyrazole 3g was treated with 2 equiv each of phenylboronic acid and potassium carbonate in DMF with Pd(OAc)2 as a catalyst, the desired 4-aryl substituted product 5 was formed in 86% yield. The three reactions mentioned above indicate that pyrazoles 3 are important intermediates that can easily be converted into 1,2,4-triaryl- or 1,2,5-triaryl-substituted pyrazoles. In addition, 1-(4-methoxyphenyl)-1H-pyrazoles (such as compounds 3g, 3h and 3i) are also important intermediates, as they can react with cerium(IV) ammonium nitrate (CAN), leading to N-dearylation followed by the generation of the parent NH-pyrazole and p-benzoquinone [37].
Conclusion
In summary, a series of novel 1,3-diaryl-4-halo-1H-pyrazoles was synthesized in moderate to excellent yields by using 3-arylsydnones and 2-aryl-1,1-dihalo-1-alkenes in the presence of the mild base Cs2CO3. The synthesis of a series of new 1,3,4-trisubstituted pyrazoles, which are important heterocycle compounds in medical and pesticide research, was convenient and efficient.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data for all compounds. | ||
| Format: PDF | Size: 904.3 KB | Download |
Acknowledgements
The work was supported by the Natural Science Foundation of China (No. 30873153), the Key Projects of Shanghai in Biomedical (No.08431902700) and the Scientific Research Foundation of State Education Ministry for the Returned Overseas Chinese Scholars. We would like to thank the Center for Instrumental Analysis, Tongji University, China.
References
-
Singh, P.; Paul, K.; Holzer, W. Bioorg. Med. Chem. 2006, 14, 5061. doi:10.1016/j.bmc.2006.02.046
And references cited therein.
Return to citation in text: [1] -
Lamberth, C. Heterocycles 2007, 71, 1467. doi:10.3987/REV-07-613
Return to citation in text: [1] -
Bekhit, A. A.; Abdel-Aziem, T. Bioorg. Med. Chem. 2004, 12, 1935. doi:10.1016/j.bmc.2004.01.037
Return to citation in text: [1] [2] -
Selwood, D. L.; Brummell, D. G.; Budworth, J.; Burtin, G. E.; Campbell, R. O.; Chana, S. S.; Charles, I. G.; Fernandez, P. A.; Glen, R. C.; Goggin, M. C.; Hobbs, A. J.; Kling, M. R.; Liu, Q.; Madge, D. J.; Meillerais, S.; Powell, K. L.; Reynolds, K.; Spacey, G. D.; Stables, J. N.; Tatlock, M. A.; Wheeler, K. A.; Wishart, G.; Woo, C.-K. J. Med. Chem. 2001, 44, 78. doi:10.1021/jm001034k
Return to citation in text: [1] -
Lien, J. C.; Huang, L. J.; Wang, J. P.; Teng, C. M.; Lee, K. H.; Kuo, S. C. Bioorg. Med. Chem. 1997, 5, 2111. doi:10.1016/S0968-0896(97)00133-8
Return to citation in text: [1] -
Ryu, C. K.; Song, E. H.; Shim, J. Y.; You, H. J.; Choi, K. U.; Choi, I. H.; Lee, E. Y.; Chae, M. J. Bioorg. Med. Chem. Lett. 2003, 13, 17. doi:10.1016/S0960-894X(02)00856-9
Return to citation in text: [1] -
Nobuyoshi, M.; Michitaka, S.; Koichi, H.; Norio, Y.; Katsuyuki, K.; Teruaki, M.; Arihiro, K.; Shuji, O.; Takahisa, S.; Shuichiro, S.; Akira, A.; Satoshi, D.; Motohiro, K.; Jun, S. Substituted Pyrazole Compounds. WO Patent 0075131A1, Dec 14, 2000.
Return to citation in text: [1] -
Tandon, V. K.; Yadav, D. B.; Chaturvedi, A. K.; Shukla, P. K. Bioorg. Med. Chem. Lett. 2005, 15, 3288. doi:10.1016/j.bmcl.2005.04.066
Return to citation in text: [1] -
Kenji, H.; Takamasa, F. Production of Arylpyruvic Acid. JP Patent 62116541A, May 28, 1987.
Return to citation in text: [1] -
Ma, Z.; Yan, G.; Yang, G.; Zhu, S.; Guan, L. Chin. Pharm. J. (Beijing, China) 2009, 44, 630.
Return to citation in text: [1] -
Zhang, H.; Song, B. A. Chin. J. Pestic. 2002, 41, 6.
Return to citation in text: [1] -
Li, M.; Wang, Z.; Song, B.; Hu, D. Agrochemicals 2008, 47, 391.
Return to citation in text: [1] -
Trofimenko, S. The Coordination Chemistry of Pyrazole-Derived Ligands. In Progress in Inorganic Chemistry; Lippard, S. J., Ed.; John Wiley and Sons: Hoboken, NJ, 1986; Vol. 34, pp 115–210. doi:10.1002/9780470166352.ch3
Return to citation in text: [1] -
Danel, A.; He, Z.; Milburn, G. H. W.; Tomasik, P. J. Mater. Chem. 1999, 9, 339. doi:10.1039/a808784i
Return to citation in text: [1] -
Zhang, Z.; Zhang, Y.; Hui, X.; Xu, P.; Shen, D. Chin. J. Org. Chem. 2004, 24, 1348.
Return to citation in text: [1] -
Gribble, G. W. In Synthetic Applications of Dipolar Cycloaddition Chemistry towards Heterocycles and Natural Product; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons: New York, 2002; pp 681–754.
Return to citation in text: [1] -
Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 553. doi:10.1016/j.tet.2009.10.085
Return to citation in text: [1] -
González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026
Return to citation in text: [1] [2] [3] [4] [5] -
Larsen, S. D.; Martinborough, E. Tetrahedron Lett. 1989, 30, 4625. doi:10.1016/S0040-4039(01)80759-0
Return to citation in text: [1] [2] -
Haneda, A.; Imagawa, T.; Kawanisi, M. Bull. Chem. Soc. Jpn. 1976, 49, 748. doi:10.1246/bcsj.49.748
Return to citation in text: [1] -
Lin, S.-T.; Tien, H.-J.; Ding, M.-F.; Chien, J.-S. J. Chin. Chem. Soc. 2006, 53, 1557.
Return to citation in text: [1] -
Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1206. doi:10.1002/cber.19791120415
Return to citation in text: [1] -
de la Hoz, A.; Díaz-Ortiz, A.; Elguero, J.; Martínez, L. J.; Moreno, A.; Sánchez-Migallón, A. Tetrahedron 2001, 57, 4397. doi:10.1016/S0040-4020(01)00340-4
Return to citation in text: [1] -
Gonzalez-Nogal, A. M.; Cuadrado, P.; Sarmentero, M. A. Tetrahedron 2010, 66, 9610. doi:10.1016/j.tet.2010.10.016
Return to citation in text: [1] -
Nicolaou, K. C.; Pratt, B. A.; Arseniyadis, S.; Wartmann, M.; O'Brate, A.; Giannakakou, P. ChemMedChem 2006, 1, 41. doi:10.1002/cmdc.200500056
Return to citation in text: [1] -
Sakamoto, T.; Shiga, F.; Uchiyama, D.; Kondo, Y.; Yamanaka, H. Heterocycles 1992, 33, 813. doi:10.3987/COM-91-S87
Return to citation in text: [1] -
Foster, R. S.; Jakobi, H.; Harrity, J. P. A. Tetrahedron Lett. 2011, 52, 1506. doi:10.1016/j.tetlet.2011.01.115
Return to citation in text: [1] -
Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2010, 75, 984. doi:10.1021/jo902514v
Return to citation in text: [1] -
Chang, E.; Huang, S.; Lee, C.; Lin, H.; Chen, C.; Huang, Y.; Lin, S.; Wong, F. Aust. J. Chem. 2009, 62, 1355. doi:10.1071/CH08344
Return to citation in text: [1] -
Delaunay, T.; Genix, P.; Es-Sayed, M.; Vors, J.; Monteiro, N.; Balme, G. Org. Lett. 2010, 12, 3328. doi:10.1021/ol101087j
Return to citation in text: [1] -
Rodriguez, A.; Moran, W. J. Synthesis 2009, 650. doi:10.1055/s-0028-1083344
Return to citation in text: [1] -
Chang, E.; Chen, T.; Wong, F. F.; Chang, E.; Yeh, M. Synlett 2006, 901. doi:10.1055/s-2006-939041
Return to citation in text: [1] -
Huisgen, R.; Grashey, R.; Gotthardt, H.; Schmidt, R. Angew. Chem. 1962, 74, 29. doi:10.1002/ange.19620740109
Return to citation in text: [1] -
Browne, D. L.; Vivat, J. F.; Plant, A.; Gomez-Bengoa, E.; Harrity, J. P. A. J. Am. Chem. Soc. 2009, 131, 7762. doi:10.1021/ja902460n
Return to citation in text: [1] [2] -
Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2009, 74, 396. doi:10.1021/jo802240e
Return to citation in text: [1] -
Huh, D. H.; Jeong, J. S.; Lee, H. B.; Ryu, H.; Kim, Y. G. Tetrahedron 2002, 58, 9925. doi:10.1016/S0040-4020(02)01324-8
Return to citation in text: [1] -
Butler, R. N.; Hanniffy, J. M.; Stephens, J. C.; Burke, L. A. J. Org. Chem. 2008, 73, 1354. doi:10.1021/jo702423z
Return to citation in text: [1]
| 34. | Browne, D. L.; Vivat, J. F.; Plant, A.; Gomez-Bengoa, E.; Harrity, J. P. A. J. Am. Chem. Soc. 2009, 131, 7762. doi:10.1021/ja902460n |
| 35. | Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2009, 74, 396. doi:10.1021/jo802240e |
| 36. | Huh, D. H.; Jeong, J. S.; Lee, H. B.; Ryu, H.; Kim, Y. G. Tetrahedron 2002, 58, 9925. doi:10.1016/S0040-4020(02)01324-8 |
| 18. | González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026 |
| 34. | Browne, D. L.; Vivat, J. F.; Plant, A.; Gomez-Bengoa, E.; Harrity, J. P. A. J. Am. Chem. Soc. 2009, 131, 7762. doi:10.1021/ja902460n |
| 1. |
Singh, P.; Paul, K.; Holzer, W. Bioorg. Med. Chem. 2006, 14, 5061. doi:10.1016/j.bmc.2006.02.046
And references cited therein. |
| 4. | Selwood, D. L.; Brummell, D. G.; Budworth, J.; Burtin, G. E.; Campbell, R. O.; Chana, S. S.; Charles, I. G.; Fernandez, P. A.; Glen, R. C.; Goggin, M. C.; Hobbs, A. J.; Kling, M. R.; Liu, Q.; Madge, D. J.; Meillerais, S.; Powell, K. L.; Reynolds, K.; Spacey, G. D.; Stables, J. N.; Tatlock, M. A.; Wheeler, K. A.; Wishart, G.; Woo, C.-K. J. Med. Chem. 2001, 44, 78. doi:10.1021/jm001034k |
| 15. | Zhang, Z.; Zhang, Y.; Hui, X.; Xu, P.; Shen, D. Chin. J. Org. Chem. 2004, 24, 1348. |
| 3. | Bekhit, A. A.; Abdel-Aziem, T. Bioorg. Med. Chem. 2004, 12, 1935. doi:10.1016/j.bmc.2004.01.037 |
| 16. | Gribble, G. W. In Synthetic Applications of Dipolar Cycloaddition Chemistry towards Heterocycles and Natural Product; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons: New York, 2002; pp 681–754. |
| 17. | Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 553. doi:10.1016/j.tet.2009.10.085 |
| 3. | Bekhit, A. A.; Abdel-Aziem, T. Bioorg. Med. Chem. 2004, 12, 1935. doi:10.1016/j.bmc.2004.01.037 |
| 13. | Trofimenko, S. The Coordination Chemistry of Pyrazole-Derived Ligands. In Progress in Inorganic Chemistry; Lippard, S. J., Ed.; John Wiley and Sons: Hoboken, NJ, 1986; Vol. 34, pp 115–210. doi:10.1002/9780470166352.ch3 |
| 14. | Danel, A.; He, Z.; Milburn, G. H. W.; Tomasik, P. J. Mater. Chem. 1999, 9, 339. doi:10.1039/a808784i |
| 8. | Tandon, V. K.; Yadav, D. B.; Chaturvedi, A. K.; Shukla, P. K. Bioorg. Med. Chem. Lett. 2005, 15, 3288. doi:10.1016/j.bmcl.2005.04.066 |
| 10. | Ma, Z.; Yan, G.; Yang, G.; Zhu, S.; Guan, L. Chin. Pharm. J. (Beijing, China) 2009, 44, 630. |
| 7. | Nobuyoshi, M.; Michitaka, S.; Koichi, H.; Norio, Y.; Katsuyuki, K.; Teruaki, M.; Arihiro, K.; Shuji, O.; Takahisa, S.; Shuichiro, S.; Akira, A.; Satoshi, D.; Motohiro, K.; Jun, S. Substituted Pyrazole Compounds. WO Patent 0075131A1, Dec 14, 2000. |
| 6. | Ryu, C. K.; Song, E. H.; Shim, J. Y.; You, H. J.; Choi, K. U.; Choi, I. H.; Lee, E. Y.; Chae, M. J. Bioorg. Med. Chem. Lett. 2003, 13, 17. doi:10.1016/S0960-894X(02)00856-9 |
| 37. | Butler, R. N.; Hanniffy, J. M.; Stephens, J. C.; Burke, L. A. J. Org. Chem. 2008, 73, 1354. doi:10.1021/jo702423z |
| 5. | Lien, J. C.; Huang, L. J.; Wang, J. P.; Teng, C. M.; Lee, K. H.; Kuo, S. C. Bioorg. Med. Chem. 1997, 5, 2111. doi:10.1016/S0968-0896(97)00133-8 |
| 9. | Kenji, H.; Takamasa, F. Production of Arylpyruvic Acid. JP Patent 62116541A, May 28, 1987. |
| 19. | Larsen, S. D.; Martinborough, E. Tetrahedron Lett. 1989, 30, 4625. doi:10.1016/S0040-4039(01)80759-0 |
| 18. | González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026 |
| 18. | González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026 |
| 27. | Foster, R. S.; Jakobi, H.; Harrity, J. P. A. Tetrahedron Lett. 2011, 52, 1506. doi:10.1016/j.tetlet.2011.01.115 |
| 28. | Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2010, 75, 984. doi:10.1021/jo902514v |
| 29. | Chang, E.; Huang, S.; Lee, C.; Lin, H.; Chen, C.; Huang, Y.; Lin, S.; Wong, F. Aust. J. Chem. 2009, 62, 1355. doi:10.1071/CH08344 |
| 30. | Delaunay, T.; Genix, P.; Es-Sayed, M.; Vors, J.; Monteiro, N.; Balme, G. Org. Lett. 2010, 12, 3328. doi:10.1021/ol101087j |
| 31. | Rodriguez, A.; Moran, W. J. Synthesis 2009, 650. doi:10.1055/s-0028-1083344 |
| 32. | Chang, E.; Chen, T.; Wong, F. F.; Chang, E.; Yeh, M. Synlett 2006, 901. doi:10.1055/s-2006-939041 |
| 33. | Huisgen, R.; Grashey, R.; Gotthardt, H.; Schmidt, R. Angew. Chem. 1962, 74, 29. doi:10.1002/ange.19620740109 |
| 18. | González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026 |
| 18. | González-Nogal, A. M.; Calle, M.; Cuadrado, P.; Valero, R. Tetrahedron 2007, 63, 224. doi:10.1016/j.tet.2006.10.026 |
| 25. | Nicolaou, K. C.; Pratt, B. A.; Arseniyadis, S.; Wartmann, M.; O'Brate, A.; Giannakakou, P. ChemMedChem 2006, 1, 41. doi:10.1002/cmdc.200500056 |
| 26. | Sakamoto, T.; Shiga, F.; Uchiyama, D.; Kondo, Y.; Yamanaka, H. Heterocycles 1992, 33, 813. doi:10.3987/COM-91-S87 |
| 23. | de la Hoz, A.; Díaz-Ortiz, A.; Elguero, J.; Martínez, L. J.; Moreno, A.; Sánchez-Migallón, A. Tetrahedron 2001, 57, 4397. doi:10.1016/S0040-4020(01)00340-4 |
| 24. | Gonzalez-Nogal, A. M.; Cuadrado, P.; Sarmentero, M. A. Tetrahedron 2010, 66, 9610. doi:10.1016/j.tet.2010.10.016 |
| 20. | Haneda, A.; Imagawa, T.; Kawanisi, M. Bull. Chem. Soc. Jpn. 1976, 49, 748. doi:10.1246/bcsj.49.748 |
| 21. | Lin, S.-T.; Tien, H.-J.; Ding, M.-F.; Chien, J.-S. J. Chin. Chem. Soc. 2006, 53, 1557. |
| 19. | Larsen, S. D.; Martinborough, E. Tetrahedron Lett. 1989, 30, 4625. doi:10.1016/S0040-4039(01)80759-0 |
| 22. | Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1206. doi:10.1002/cber.19791120415 |
© 2011 Yang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)