Abstract
A series of 6,8-diiodocoumarin-3-N-carboxamides (4–11) were prepared. Treatment of ethyl 6,8-diiodocoumarin-3-carboxylate (1) with ethyl cyanoacetate/NH4OAc gave ethyl 2-(3-carbamoyl-6,8-diiodocoumarin-4-yl)-2-cyanoacetate (12) and 2-amino-4-hydroxy-7,9-diiodocoumarino[3,4-c]pyridine-1-carbonitrile (13), and treatment with acetone in the presence of NH4OAc or methylamine gave the ethyl 4-oxo-2,6-methano-2-methyl-3,4,5,6-tetrahydro-8,10-diiodobenzo[2,1-g]-2H-1,3-oxazocine-5-carboxylate derivatives 14a,b. All compounds were evaluated for their antimicrobial activity and the compounds 12–14a,b exhibited a pronounced effect on all tested microorganisms.

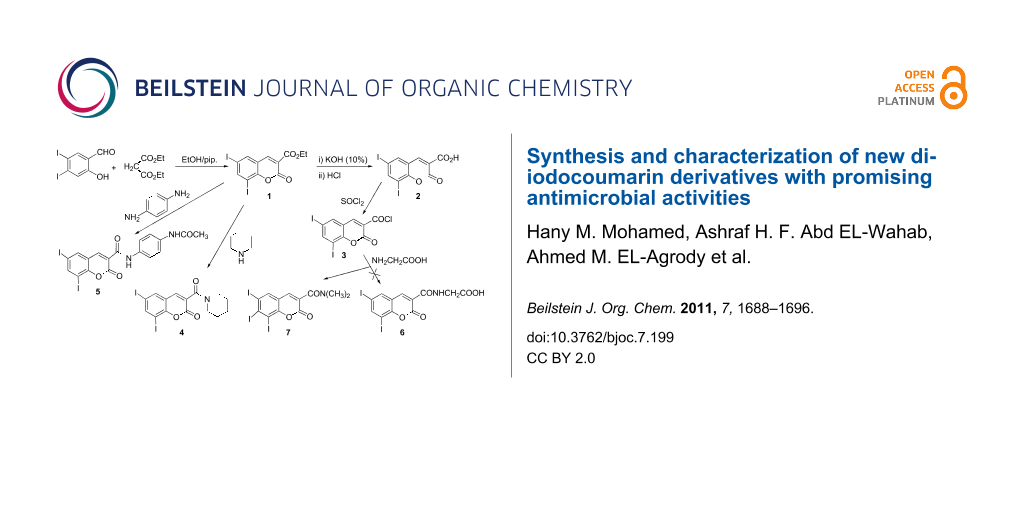
Graphical Abstract
Introduction
Coumarins and their derivatives are biologically and pharmaceutically interesting compounds known for their use as additives in food, perfumes, cosmetics, pharmaceuticals, platelet aggregation and agrochemicals [1,2]. Coumarins have also been reported to exhibit several biological activities, such as antimicrobial, anticancer, antifungal, anti-HIV and antioxidant properties [3-6], and they also served as versatile precursors for many organic transformations in the synthesis of a number of drug-like molecules [7,8]. Moreover, coumarin-based dyes and pigments are organic fluorescent materials exhibiting unique photochemical and photophysical properties, which render them useful in a variety of applications such as dye lasers, anion sensors, organic light-emitting diodes and solar cells [9,10].
Iodo-organic derivatives have been widely used as diagnostic-imaging drugs (such as diatrizoate meglumine, diatrizoic acid, iodipamide, iodixanol, iohexol, iomeprol and iopamidol) and as amebicides [11,12]. The benzoxazocine derivatives have received considerable attention due to their pharmacological properties, such as their antidepressant, antithrombotic, antipsychotic (for the central nervous system, CNS) and anti-breast-cancer activities [13].
In view of the important biological properties of the diiodocoumarin derivatives and iodo-organic compounds as medical agents, we planned to synthesize some new diiodocoumarin derivatives bearing side chains with different structures, as such derivatives could possess interesting and useful biological properties.
Results and Discussion
Interaction of 3,5-diiodosalicylaldehyde with diethyl malonate according to the literature procedure [14,15] afforded ethyl 6,8-diiodocoumarin-3-carboxylate (1). Treatment of 1 with hot ethanolic KOH (10%) followed by acidification with HCl gave the corresponding 6,8-diiodocoumarin-3-carboxylic acid (2), which on treatment with SOCl2 gave the 6,8-diiodocoumarin-3-carbonyl chloride (3). Treatment of 1 with piperidine in boiling ethanol or with p-phenylenediamine in boiling AcOH afforded the 6,8-diiodocoumarin-3-carboxamide derivatives 4 and 5, respectively. Interaction of 3 with glycine in dry benzene under reflux gave the new 6,8-diiodocoumarin-3-N,N-dimethylcarboxamide (7) instead of 6,8-diiodocoumarin-3-ylcarbonylglycine (6). The formation of compound 7 suggests that two glycine molecules react with 1 followed by the loss of ammonia and decarboxylation, furnishing the observed product (Scheme 1).
![[1860-5397-7-199-i1]](/bjoc/content/inline/1860-5397-7-199-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 6,8-diiodocoumarin derivatives 1–7.
Scheme 1: Synthesis of 6,8-diiodocoumarin derivatives 1–7.
The structures of compounds 3–5 and 7 were confirmed by IR, 1H NMR, 13C NMR and MS. The IR spectra for compound 3 showed 1774, 1718 cm−1 (2 CO); for compound 4 1713, 1631 cm−1 (2 CO); for compound 5 1718 cm−1 (CO); and for compound 7 1722, 1635 cm−1 (2 CO). 1H NMR for compounds 3–5 and 7 showed δ at 7.64–8.70 ppm (s, 1H, H-4), and 13C NMR for compounds 3 and 5 showed δ at 143.2 and 147.2 ppm (C-4), respectively. The mass spectra of compounds 3 and 7 showed the corresponding molecular ion peaks at m/z 460 (M+, 2.6%) and m/z 469 (M+, 18.5%). The fragmentation pattern of compounds 3 and 7 are illustrated in Scheme 2.
![[1860-5397-7-199-i2]](/bjoc/content/inline/1860-5397-7-199-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed fragmentation pathways for the EI ions of the substituted 6,8-diiodocoumarins 3 and 7.
Scheme 2: Proposed fragmentation pathways for the EI ions of the substituted 6,8-diiodocoumarins 3 and 7.
Reactions of 3 with 4-aminophenylethanol or p-aminophenol, or with potentially bifunctional amino acids (anthranilic acid and p-aminophenylacetic acid), was successful, and the corresponding 6,8-diiodocoumarin-3-carboxamide derivatives 8–11 were obtained (Scheme 3).
![[1860-5397-7-199-i3]](/bjoc/content/inline/1860-5397-7-199-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 6,8-diiodocoumarin-3-N-carboxamide derivatives 8–11.
Scheme 3: Synthesis of 6,8-diiodocoumarin-3-N-carboxamide derivatives 8–11.
The structures of compounds 8–11 were established by IR, 1H NMR, 13C NMR and MS. The IR spectra of compound 8 showed 3287 cm–1 (OH, NH) and 1719 cm–1 (CO) and for compound 9 3217 cm–1 (NH, OH) and 1720 cm–1 (CO). 1H NMR for 8 showed δ at 3.01 (t, J = 7.0 Hz, 2H, Ar-CH2), 3.56 (s, 1H, OH), 3.83 (t, J = 7.0 Hz, 2H, CH2–OH), and 10.49 ppm (s, 1H, NH), and for compound 11 at 3.55 (s, 2H, CH2), 8.71 (s, 1H, H-4), 10.10 (brs, 1H, NH), and 10.49 (s, 1H, OH). The 13C NMR for 11 showed δ at 160 (CO δ lactone), 163.4 (CONH), and 176.5 ppm (COOH). The mass spectra of compounds 8–11 provided additional evidence for the proposed structures.
As the C3–C4 olefinic bond in ethyl 6,8-diiodocoumarin-3-carboxylate (1) is activated by conjugation with electron-withdrawing carbonyl groups, the behavior of 1 towards activated methylene compounds under Michael reaction conditions was investigated. Thus, treatment of 1 with ethyl cyanoacetate/NH4OAc in boiling ethanol afforded two reaction products. The insoluble reaction product was identified as ethyl 2-(3-carbamoyl-6,8-diiodocoumarin-4-yl)-2-cyanoacetate (12) and the soluble reaction product was identified as 2-amino-4-hydroxy-7,9-diiodocoumarino[3,4-c]pyridine-1-carbonitrile (13), which probably formed as a result of amide formation, dehydration and intramolecular cyclization (Scheme 4).
![[1860-5397-7-199-i4]](/bjoc/content/inline/1860-5397-7-199-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of ethyl cyanoacetate and pyridine derivatives 12 and 13.
Scheme 4: Synthesis of ethyl cyanoacetate and pyridine derivatives 12 and 13.
The structures of compounds 12 and 13 were established by IR, 1H NMR, 13C NMR and MS. The IR spectra of compound 12 showed 3309, 3277 cm–1 (NH2), 2206 cm–1 (CN), and 1643 cm–1 (CO), while the 1H NMR for compound 13 showed δ at 7.89 (brs, 2H, NH2), and 9.06 (brs, 1H, OH). The spectral data of compound 13 confirmed its enol structure.
Reaction of 1 with acetone in the presence of NH4OAc or methylamine at room temperature for 7 days gave 1,3-oxazocine-5-carboxylate derivatives (14a,b) [16-18] (Scheme 5). The formation of 14 indicates that the activated methylene compounds attack at the C3–C4 olefinic bond in 1 under Michael reaction conditions to yield a cyclic Michael adduct, which underwent hydrolysis by NH3 or MeNH2 and cyclization through the elimination of H2O (Scheme 5).
![[1860-5397-7-199-i5]](/bjoc/content/inline/1860-5397-7-199-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 1,3-oxazocine derivatives 14a,b.
Scheme 5: Synthesis of 1,3-oxazocine derivatives 14a,b.
The structure of compound 14a was established by 13C NMR, which showed δ at 42.5 (CH2(c)), 168.4 cm–1 (CONH), and 170 cm–1 (CO). The structures of all newly synthesized compounds were confirmed by IR, 1H NMR, 13C NMR and mass spectrometry.
The inhibitory effects of the synthetic compounds against these organisms are given in Table 1, Figure 1 and Figure 2. Among the series tested, compounds 12–14a,b exhibited excellent antibacterial activity, better than the standard ampicillin, against two species of Gram-positive bacteria, Staphylococcus aureus (NCTC-7447), Bacillus cereus (ATCC-14579) and two Gram-negative bacteria, Escherichia coli (NCTC-10410) and Serratia marcescens (IMRU-70), while the same compounds showed moderate antifungal activity against the tested organisms. Compounds 9–11 exhibited comparable activity to ampicillin against the tested bacteria and moderate to weak antifungal activity against the tested organisms. Furthermore, compounds 1–8 showed moderate to weak activities against all the tested bacteria and fungi, compared with the standards ampicillin and calforan. In addition, compounds 2 and 5 in the series were found to be inactive against Escherichia coli (NCTC-10410), while compound 11 was inactive against Serratia marcescens (IMRU-70). An investigation of the structure–activity relationship (SAR) revealed that the activity is considerably affected by the presence of the diiodocoumarino[3,4-c]pyridine, 2-methyl-8,10-diiodobenzo[2,1-g]-2H-1,3-oxazocine, diiodocoumarin-3-carboxamide or 2,3-dimethyl-8,10-diiodobenzo[2,1-g]-2H-1,3-oxazocine, and slightly decreases with the presence of different amide groups at position C-3 of the diiodocoumarin moiety or with the presence of ester, acid or acid chloride at position C-3 of the diiodocoumarin moiety.
Table 1: Biological activity of the newly synthesized compounds.
|
Compound
no.a |
Inhibition-zone diameter (mm/mg sample) | |||||
|---|---|---|---|---|---|---|
| Gram-positive | Gram-negative | Fungi | ||||
|
Staphylococcus
aureus (NCTC-7447) |
Bacillus
cereus (ATCC-14579) |
Escherichia
coli (NCTC-10410) |
Serratia
marcescens (IMRU-70) |
Aspergillus fumigatus
(MTCC-3008) |
Candida albicans
(MTCC-227) |
|
| 1 | 10 | 11 | 15 | 10 | 9 | – |
| 2 | 13 | 10 | – | 13 | – | 10 |
| 3 | 16 | 15 | 10 | 12 | 10 | 10 |
| 4 | 15 | 14 | 12 | 10 | – | – |
| 5 | 10 | 12 | – | 15 | 10 | – |
| 7 | 10 | 10 | – | 15 | 11 | – |
| 8 | 20 | 18 | 14 | 10 | 16 | 15 |
| 9 | 22 | 22 | 22 | 17 | 14 | 13 |
| 10 | 22 | 15 | 22 | 15 | 17 | 11 |
| 11 | 20 | 22 | 20 | – | 15 | 12 |
| 12 | 26 | 27 | 28 | 26 | 16 | 18 |
| 13 | 27 | 28 | 28 | 26 | 17 | 17 |
| 14a | 26 | 28 | 27 | 28 | 15 | 14 |
| 14b | 25 | 26 | 25 | 27 | 18 | 15 |
| Ampicillin | 22 | 22 | 22 | 22 | – | – |
| Calforan | – | – | – | – | 20 | 20 |
ac = 1 mg mL–1 in DMF.
![[1860-5397-7-199-1]](/bjoc/content/figures/1860-5397-7-199-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Graphical representation of the antibacterial activity of tested compounds compared to ampicillin.
Figure 1: Graphical representation of the antibacterial activity of tested compounds compared to ampicillin.
![[1860-5397-7-199-2]](/bjoc/content/figures/1860-5397-7-199-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Graphical representation of antifungal activity of tested compounds, compared to calforan.
Figure 2: Graphical representation of antifungal activity of tested compounds, compared to calforan.
Experimental
General methods
Melting points were determined on a Stuart melting point apparatus and are uncorrected; IR spectra were recorded in KBr on a FT-IR 5300 spectrometer and Perkin Elmer spectrum RXIFT-IR system (ν, cm−1). The 1H NMR spectra at 300 MHz and 13C NMR spectra at 75 MHz were recorded in CDCl3 or DMSO-d6 on a Varian Mercury VX-300 NMR spectrometer. Chemical shifts (δ) are related to that of the solvent. Mass spectra were measured on a Shimadzu GMMS-QP-1000 EX mass spectrometer at 70 eV. The elemental analyses were performed at the Microanalytical Center, Cairo University, Cairo (Egypt).
Ethyl 6,8-diiodocoumarin-3-carboxylate (1). Ethyl 6,8-diiodocoumarin-3-carboxylate (1) was prepared by the interaction of 3,5-diiodosalicylaldehyde with diethyl malonate according to the literature procedures [19-21].
6,8-Diiodocoumarin-3-carboxylic acid (2). A solution of compound 1 (0.47 g, 10 mmol) in absolute ethanol (20 mL) was mixed with ethanolic solution of KOH (10%), which was then refluxed for 10 min. The reaction mixture was poured onto ice, acidified with HCl and recrystallized from ethanol [22].
6,8-Diiodocoumarin-3-carbonyl chloride (3). Compound 2 (0.44 g, 10 mmol) was dissolved in dry benzene (40 mL), 2 mL of thionyl chloride was added and the solution was refluxed for 1 h. A few drops of formic acid were added to eliminate the unreacted thionyl chloride, and the solvent was removed under reduced pressure. The solid obtained was recrystallized from benzene. Yellow crystals: Yield 92%; mp 180 °C; Anal. calcd for C10H3ClI2O3: C, 26.10; H, 0.65; found: C, 26.11; H, 0.67; IR (KBr, cm–1): 3055 (C–H aromatic), 1774, 1718 (2 CO); 1H NMR (300 MHz, CDCl3, δ/ppm) 8.01 (d, J = 1.8 Hz, 1H, Ar-H-7), 8.44 (d, J = 1.8 Hz, 1H, Ar-H-5), 8.58 (s, 1H, H-4); 13C NMR (75 MHz, CDCl3, δ/ppm) 86.0 (C-6), 89.1 (C-8), 118.6 (C-3), 120.3 (C-4a), 138.2 (C-5), 147.2 (C-4), 149.4 (C-7), 153.6 (C-8a), 155.0 (CO δ lactone), 162.9 (CO); MS m/z (% relative intensity): 460 (M+, 2.6), 459 (M – 1, 30.4), 425 (83.4), 341 (19.3), 214 (10.9), 87 (100).
6,8-Diiodo-3-(piperidine-1-carbonyl)coumarin (4). A solution of compound 1 (0.47 g, 10 mmol) in absolute ethanol (30 mL) was refluxed with piperidine (0.9 g, 10 mmol) for 1 h. After cooling, the solid formed was filtered off, washed with ethanol and dried under vacuum. The solid obtained was recrystallized from benzene. Colorless crystals: Yield 80%; mp 230 °C; Anal. calcd for C15H13I2NO3: C, 35.37; H, 2.55; N, 2.75; found: C, 35.36; H, 2.53; N, 2.76; IR (KBr, cm–1): 3040 (C–H aromatic), 2935, 2854 (C–H aliphatic), 1713, 1631 (CO); 1H NMR (300 MHz, CDCl3, δ/ppm) 1.59, 1.67, 3.29, 3.69 (m, 10H, (CH2)5), 7.64 (s, 1H, H-4), 7.78 (d, J = 2.1 Hz, 1H, Ar-H-7), 8.28 (d, J = 2.1 Hz, 1H, Ar-H-5); 13C NMR (75 MHz, CDCl3, δ/ppm) 24.30, 25.40, 48.03 (CH2 piperidine), 86.0 (C-8), 89.1 (C-6), 118.6 (C-3), 120.3 (C-4a), 138.2 (C-5), 147.2 (C-4), 149.4 (C-7), 153.6 (C-8a), 155.0 (CO δ lactone), 162.9 (CO-amide); MS m/z (% relative intensity): 509 (M+, 0.3), 424 (3.4), 341 (2.7), 214 (1.5), 84 (100).
N-(4-Acetamidophenyl)-6,8-diiodocoumarin-3-carboxamide (5). A solution of compound 1 (0.47 g, 10 mmol) in glacial acetic acid (30 mL) was refluxed with p-phenylenediamine (1.10 g, 10 mmol) for 2 h. After cooling, the solid formed was filtered off, washed with ethanol, dried under vacuum and recrystallized from benzene. Colorless crystals: Yield 87%; mp 319 °C; Anal. calcd for C18H12I2N2O4: C, 37.64; H, 2.90; N, 4.88; found: C, 37.65; H, 2.92; N, 4.90; IR (KBr, cm–1): 3285 (NH), 1718 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 2.40 (s, 3H, CH3), 7.28, 7.66 (2d, 4H, J = 8.4 Hz, AB-q, Ar-H), 8.37 (d , J = 1.8 Hz, 1H, Ar-H-7), 8.46 (d , J = 1.8 Hz, 1H, Ar-H-5), 8.70 (s, 1H, H-4), 8.90 (s, 1H, CH3CONH), 10.12 (brs, 1H, NH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 23.1 (CH3), 89.0 (C-6), 90.1 (C-8), 121.5, 128.0 (C-2', 3', 5', 6'), 114.3, 133.5, 135.6 (C-3, 1', 4'), 125.3 (C-4a), 136.2 (C-5), 143.2 (C-4), 146.4 (C-7), 148.6 (C-8a), 160 (CO δ lactone), 163.7 (CO-amide), 170.0 (COCH3); MS m/z (% relative intensity): 574 (M+, 3), 532 (M – CH2=C=O, 38.7), 424 (18.2), 341 (33.8), 298 (6.5), 171 (9.3), 107 (100).
6,8-Diiodocoumarin-3-N,N-dimethylcarboxamide (7). A solution of compound 3 (0.46 g, 1 mmol) in dry benzene (50 mL) was refluxed with glycine (0.75 g, 10 mmol) for 2 h. After cooling, the solid formed was filtered off, washed with ethanol, dried under vacuum, and recrystallized from dioxane. Colorless crystals: Yield 83%; mp 302 °C; Anal. calcd for C12H9I2NO3: C, 30.70; H, 1.92; N, 2.98; found: C, 30.72; H, 1.94; N, 3.00; IR (KBr, cm–1): 2931 (C–H aliphatic), 1722 and 1635 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 2.93 (s, 3H, N-CH3), 2.97 (s, 3H, N-CH3), 8.02 (s, 1H, H-4), 8.13 (d, J = 2.1 Hz, 1H, Ar-H-7), 8.38 (d, J = 2.1 Hz, 1H, Ar-H-5); MS m/z (% relative intensity): 469 (M+, 18.5), 425 (83.4), 397 (16.1), 341 (19.3) and 72 (100).
General procedure for the synthesis of 6,8-diiodocoumarin-3-carboxamide derivatives 8–11
To a well-stirred solution of 3 (0.46 g, 1 mmol) in dry dichloromethane (DCM) containing a few drops of triethylamine (TEA) an equivalent amount of an ambient nucleophile [4-aminophenylethanol, p-aminophenol, anthranilic acid and p-aminophenylacetic acid (1.2 mmol)] was added. The reaction mixture was stirred at room temperature under dry conditions for 3 h. DCM was removed under reduced pressure until dryness, the obtained solid was then washed with 10% HCl and the remaining solid recrystallized from dioxane.
N-(4-(2-Hydroxyethyl)phenyl)-6,8-diiodocoumarin-3-carboxamide (8). Yellow crystals: Yield 87%; mp 291 °C; Anal. calcd for C18H13I2NO4: C, 38.51; H, 2.32; N, 2.50; found: C, 38.52; H, 2.34; N, 2.51; IR (KBr, cm–1): 3287 (OH, NH), 3049 (Ar-H), 2958, 2928 (aliphatic-H), 1719 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 3.01 (t, J = 7.0 Hz, 2H, Ar-CH2), 3.56 (s, 1H, OH), 3.83 (t, J = 7.0 Hz, 2H, CH2-OH), 7.29, 7.63 (2d, J = 8.2 Hz, 4H, AB-q, Ar-H), 8.37 (d, J = 2.0 Hz, 1H, H-7), 8.46 (d, J = 2.0 Hz, 1H, H-5), 8.71 (s, 1H, H-4), 10.49 (s, 1H, NH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 38.4 (Ar-CH2), 62.2 (CH2-OH), 111.2, 114.0 (C-6,8), 114.5, 119.8 (C-3',2',5',6'), 129.3, 132.1 (C-5,7), 135.4, 135.9, 136.0 (C-3,1',4'), 148.3 (C-4), 156.4, 159.9 (C4a,8a), 161.4 (CO δ lactone), 163.9 (CO-amide); MS m/z (% relative intensity): 561 (M+, 0), 543 (M – H2O, 3), 530 (67), 425 (M – NH-C6H4-CH2CH2OH, 100), 341 (6), 107 (36), 128 (23), 127 (14), 87 (36).
N-(4-Hydroxyphenyl)-6,8-diiodocoumarin-3-carboxamide (9). Yellow crystal: Yield 85%; mp 303 °C; Anal. calcd for C16H9I2NO4: C, 36.03; H, 1.69; N, 2.63; found: C, 36.05; H, 1.68; N, 2.63; IR (KBr, cm–1): 3217 (NH, OH), 1720 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 7.25–7.85 (m, 4H, Ar-H), 8.35 (d, J = 1.8 Hz, 1H, Ar-H-7), 8.43 (d, J = 1.8 Hz, 1H, Ar-H-5), 8.70 (s, 1H, H-4), 10.12 (brs, 1H, OH), 11.9 (brs, 1H, NH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 89.5 (C-6), 92.0 (C-8), 130.5, 124.4, 134.3, 121.5 (C-3',4',5',6'), 114.3, 140.8, 116.0 (C-3,1',2'), 125.5 (C-4a), 134.2 (C-5), 139.5 (C-4), 144.5 (C-7), 146.6 (C-8a), 160 (CO δ lactone), 163.2 (CONH), 170 (COOH); MS m/z (% relative intensity): 533 (M+, 45), 425 (M+ – NH-C6H4-OH, 100), 341 (16), 214 (10), 171 (17), 87 (63).
2-(6,8-Diiodocoumarin-3-carboxamido)benzoic acid (10). Yellow crystals: Yield 91%; mp 315 °C; Anal. calcd for C17H9I2NO5: C, 36.37; H, 1.60; N, 2.50; found: C, 36.39; H, 36.39; N, 2.52; IR (KBr, cm–1): 3271 (OH, NH), 3055 (Ar-H), 1751, 1651 (CO, CONH); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 7.27, 7.64 (2d, J = 8.4 Hz, 4H, AB-q, Ar-H), 8.38 (d, J = 1.8 Hz, 1H, Ar-H-7), 8.47 (d, J = 1.8 Hz, 1H, Ar-H-5), 8.71 (s, 1H, H-4), 10.10 (brs, 1H, NH), 10.49 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 90.0 (C-6), 92.1 (C-8), 123.0, 130.0 (C-2',3',5',6'), 114.3, 135.0, 130.5 (C-3,1',4'), 125.5 (C-4a), 133.2 (C-5), 138.5 (C-4), 144.2 (C-7), 146.1 (C-8a), 160 (CO δ lactone), 163.4 (CO-amide), 176.5 (COOH); MS m/z (% relative intensity): 561 (M+, 7.1), 560 (M – 1, 40.8), 517 (M+ – CO2, 2.7), 516 (M – CO2H, 24.5), 425 (M – NHC6H4-2-CO2H, 11.5), 424 (100), 341 (17), 171 (14.5) and 87 (58.2).
2-(4-(6,8-Diiodocoumarin-3-carboxamido)phenyl)acetic acid (11). Yellow crystals: Yield 93%; mp 285 °C; Anal. calcd for C18H11I2NO5: C, 37.57; H, 1.91; N, 2.44; found: C, 37.59; H, 1.92; N, 2.46; IR (KBr, cm–1): 3286 (OH, NH), 3047 (Ar-H), 2916 (aliphatic-H), 1720 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 3.55 (s, 2H, CH2), 7.27, 7.64 (2d, J = 8.4 Hz, 4H, AB-q, Ar-H), 8.38 (d, J = 1.8 Hz, 1H, Ar-H-7), 8.47 (d, J = 1.8 Hz, 1H, Ar-H-5), 8.71 (s, 1H, H-4), 10.10 (brs, 1H, NH), 10.49 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 90.0 (C-6), 92.1 (C-8), 123.0, 130.0 (C2',3',5',6'), 114.3, 135.0, 130.5 (C-3,1',4'), 125.5 (C-4a), 133.2 (C-5), 138.5 (C-4), 144.2 (C-7), 146.1 (C-8a), 160 (CO δ lactone), 163.4 (CO-amide), 176.5 (COOH); MS m/z (% relative intensity): 575 (M+, 12.4), 574 (M – 1, 68.9), 531 (M – CO2, 7.3), 425 (M – NH-C6H4-4-CH2COOH, 13.5), 424 (100), 341 (18.9), 171 (27.2), 106 (94.6) and 87 (59.5).
General procedure for the synthesis of ethyl cyanoacetate and pyridine derivatives 12 and 13
Ethanolic solution of ethyl 6,8-diiodocoumarin-3-carboxylate (1) (0.47 g, 10 mmol, 30 mL) was refluxed with ethyl cyanoacetate (1.13 g, 10 mmol) for 6 h. The solid precipitated was filtered off while hot, washed with ethanol and dried under vacuum, and was identified as compound 12. The filtrate evaporated under reduced pressure to produce a solid identified as compound 13. Compound 12 crystallized from dioxane, whereas compound 13 crystallized from chloroform.
Ethyl 2-(3-carbamoyl-6,8-diiodocoumarin-4-yl)-2-cyanoacetate (12). Pale yellow crystal: Yield 82%; mp 310 °C; Anal. calcd for C15H10I2N2O5: C, 32.62; H, 1.81; N, 5.07; found: C, 32.64; H, 1.79; N, 5.05; IR (KBr, cm–1): 3309, 3277 (NH2), 2206 (CN), 1643 (CO); 1H NMR (300 MHz, CDCl3, δ/ppm) 1.50 (t, J = 7.2 Hz, 3H, CH3), 4.44 (q, J = 7.2 Hz, 2H, CH2), 5.05 (s, 1H, CH), 8.00 (d, J = 1.8 Hz, 1H, Ar-H-7), 8.40 (d, J = 1.8 Hz, 1H, Ar-H-5), 8.70 (brs, 2H, NH2, exchangeable with D2O); MS m/z (% relative intensity): 552 (M+, 2), 388 (8.0), 313 (30.0), 264 (4.0), 236 (35.0).
2-Amino-4-hydroxy-7,9-diiodocoumarino[3,4-c]pyridine-1-carbonitrile (13). Pale yellow crystals: Yield 84%; mp 340 °C; Anal. calcd for C13H5I2N3O3: C, 30.90; H, 0.99; N, 8.32; found C, 30.92; H, 1.00; N, 8.34; IR (KBr, cm-1): 3374 (OH), 3277, 3228 (NH2), 2207 (CN), 1707, 1662 (CO); 1H NMR (300 MHz, CDCl3, δ/pm) 9.06 (brs, 1H, OH, exchangeable with D2O), 8.20 (s, 1H, H-8), 7.97 (s, 1H, H-10), 7.89 (brs, 2H, NH2, exchangeable with D2O); MS m/z (% relative intensity): 505 (M+, 100), 477 (M – CO, 18.9), 397 (20.8), 341 (18.9), 171 (25.2), 106 (32.6) and 87 (35.5).
General procedure for the synthesis of 1,3-oxazocine-5-carboxylate derivatives 14a,b
A mixture of compound 1 (2.35 g, 5 mmol), acetone (30 mL) and (a) ammonium acetate (0.4 g, 5 mmol) or (b) methylamine (0.16 g, 5 mmol) was stirred at room temperature for 7 days. In both cases a colorless solid formed after the solvent had evaporated under reduced pressure, and the products were identified as compounds 14a and 14b. The crude products were crystallized from benzene.
Ethyl 4-oxo-2,6-methano-2-methyl-3,4,5,6-tetrahydro-8,10-diiodobenzo[2,1-g]-2H-1,3-oxazocine-5-carboxylate (14a). Colorless: Yield 72%; mp 222 °C; Anal. calcd for C15H15I2NO4: C, 34.16; H, 2.85; N, 2.66; found: C, 34.18; H, 2.87; N, 2.68; IR (KBr, cm–1): 3217 (NH), 2977 (aliphatic-H), 1728, 1689 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 0.87 (t, J = 6.9 Hz, 3H, CH3 (e)), 1.64–1.95 (m, 5H, CH2(c), CH3(f)), 3.80 (q, J = 4.5 Hz, 2H, CH2 (d)), 3.72–3.97 (m, 2H, H(a) + H(b)), 7.16 (d, J = 1.8 Hz, 1H, Ar-H-9), 7.92 (d, J = 2.1 Hz, 1H, Ar-H-7), 8.88 (brs, 1H, NH); 13C NMR (75 MHz, DMSO-d6, δ/ppm) 14.1 (CH3(e)), 24.7 (CH3(f)), 42.5 (CH2(c)), 56.0, 57.3 (CH(a)-CH(b)), 61.2 (CH2(d)), 130, 138, 141.9, 154 (C-2,3,5), 87.1, 87.5 (C-4,6),168.4 (CONH), 170 (CO); MS m/z (% relative intensity): 527 (M+, 4.3) 454 (M – CO2C2H5, 42.5), 182 (100), 136 (44.8), 57 (13.4).
Ethyl 3-methyl-4-oxo-2,6-methano-2,3-dimethyl-3,4,5,6-tetrahydro-8,10-diiodobenzo[2,1-g]-2H-1,3-oxazocine-5-carboxylate (14b). Colorless: Yield 70%; mp 198 °C; Anal. calcd for C16H17I2NO4: C, 35.50; H, 3.14; N, 2.59; found: C, 35.51; H, 3.16; N, 2.61; IR (KBr, cm–1): 3051 (Ar-H), 2985.6 (aliphatic-H), 1735, 1651 (CO); 1H NMR (300 MHz, DMSO-d6, δ/ppm) 1.23 (t, J = 7.2 Hz, 3H, CH3(e)), 1.78 (s, 3H, CH3(f)), 2.83 (s, 3H, NCH3), 2.38–2.42 (m, 2H, CH2(b)), 3.4–3.57 (m, 2H, H(a) + H(b)), 4.18 (q, J = 7.2 Hz, 2H, CH2(d)), 7.66 (d, 1H, Ar-H-9), 7.95 (d, J = 1.8 Hz, 1H, Ar-H-7); MS m/z (% relative intensity): 541 (M+, 3.4), 196 (59.2), 150 (27.6), 56 (100).
Antimicrobial assays
The newly synthesized compounds were screened for their antimicrobial activities in vitro against two species of Gram-positive bacteria, namely Staphylococcus aureus (NCTC-7447), Bacillus cereus (ATCC-14579), and two Gram-negative bacteria, namely Escherichia coli (NCTC-10410), Serratia marcescens (IMRU-70); and against two species of fungi, namely Aspergillus fumigatus (MTCC-3008) and Candida albicans (MTCC-227). The tested microorganisms were obtained from the Regional Center for Mycology & Biotechnology (RCMP), Al-Azhar University.
The activities of these compounds were tested by using the disc-diffusion method [23] for bacteria and the paper-disk-diffusion method [24] for fungi. The area of zone inhibition was measured with ampicillin (30 µg mL−1) as the standard antibiotic reference for antibacterial activity, and calforan (30 µg mL−1) was used as a reference antifungal activity. The tested compounds were dissolved in N,N-dimethylformamide (DMF) to give a solution of 1 mg mL−1. The inhibition zones (diameter of the hole) were measured in millimeters (6 mm) at the end of an incubation period of 48 h at 28 °C; N,N-dimethylformamide showed no inhibition zone.
Conclusion
It was interesting to note that four of the new compounds (12, 13 and 14a,b) were found to have an antimicrobial activity greater than that of the standard antibiotic ampicillin or the standard antifungal claforan, while compounds 1–11 were either inactive or only weakly active against the tested microorganisms. The presence of fused diiodocoumarino[3,4-c]pyridine and diiodobenzo[2,1-g]-2H-1,3-oxazocine nucleus increased the antimicrobial activity, whereas the presence of diiodocoumarin-3-carboxamides decreased the antimicrobial activity.
References
-
O’Kennedy, R.; Thornes, R. D. Coumarins: Biology, Applications and Mode of Action; Wiley & Sons: Chichester, UK, 1997.
Return to citation in text: [1] -
Zahradnik, M. The Production and Application of Fluorescent Brightening Agents; Wiley & Sons, 1992.
Return to citation in text: [1] -
Bailly, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.-P.; Hildebrand, M.-P.; Peyrot, V.; Wattez, N. J. Med. Chem. 2003, 46, 5437–5444. doi:10.1021/jm030903d
Return to citation in text: [1] -
Yeh, J.-Y.; Coumar, M. S.; Horng, J.-T.; Shiao, H.-Y.; Kuo, F.-M.; Lee, H.-L.; Chen, I.-C.; Chang, C.-W.; Tang, W.-F.; Tseng, S.-N. J. Med. Chem. 2010, 53, 1519–1533. doi:10.1021/jm901570x
Return to citation in text: [1] -
Pierson, J.-T.; Dumètre, A.; Hutter, S.; Delmas, F.; Laget, M.; Finet, J.-P.; Azas, N.; Combes, S. Eur. J. Med. Chem. 2010, 45, 864–869. doi:10.1016/j.ejmech.2009.10.022
Return to citation in text: [1] -
Basile, A.; Sorbo, S.; Spadaro, V.; Bruno, M.; Maggio, A.; Faraone, N.; Rosselli, S. Molecules 2009, 14, 939–952. doi:10.3390/molecules14030939
Return to citation in text: [1] -
Zhao, P.-L.; Wang, L.; Zhu, X.-L.; Huang, X.; Zhan, C.-G.; Wu, J.-W.; Yang, G.-F. J. Am. Chem. Soc. 2010, 132, 185–194. doi:10.1021/ja905756c
Return to citation in text: [1] -
Teichert, J. F.; Feringa, B. L. Chem. Commun. 2011, 47, 2679–2681. doi:10.1039/c0cc05160h
Return to citation in text: [1] -
Key, J. A.; Kho, S.; Timerghazin, Q. K.; Brown, A.; Cairo, C. W. Dyes Pigm. 2009, 82, 196–203. doi:10.1016/j.dyepig.2009.01.001
Return to citation in text: [1] -
Zhou, S.; Jia, J.; Gao, J.; Han, L.; Li, Y.; Sheng, W. Dyes Pigm. 2010, 86, 123–128. doi:10.1016/j.dyepig.2009.12.005
Return to citation in text: [1] -
Craven, P. A.; Pfanstiel, J.; DeRubertis, F. R. J. Clin. Invest. 1986, 77, 850–859. doi:10.1172/JCI112382
Return to citation in text: [1] -
Rathbone, D. L.; Su, D.; Wang, Y.; Billington, D. C. Tetrahedron Lett. 2000, 41, 123–126. doi:10.1016/S0040-4039(99)02027-4
Return to citation in text: [1] -
Mishra, J. K.; Samanta, K.; Jain, M.; Dikshit, M.; Panda, G. Med. Chem. Lett. 2010, 20, 244–247. doi:10.1016/j.bmcl.2009.10.126
Return to citation in text: [1] -
Burton, H. J. Chem. Soc. 1945, 280, 280–283. doi:10.1039/JR9450000280
Return to citation in text: [1] -
Bonsignore, L.; Cottiglia, F.; Maccioni, A. M.; Sacci, D.; Lavagna, S. M. J. Heterocycl. Chem. 1995, 32, 573–577. doi:10.1002/jhet.5570320234
Return to citation in text: [1] -
Koelsch, C. F. J. Am. Chem. Soc. 1945, 67, 569–574. doi:10.1021/ja01220a023
Return to citation in text: [1] -
Koelsch, C. F.; Freerks, M. C. J. Org. Chem. 1953, 18, 1538–1545. doi:10.1021/jo50017a013
Return to citation in text: [1] -
Bedair, A. H.; Aly, F. M.; El-Agrody, A. M.; El-Assy, R. K. M. Acta Pharm. 1986, 36, 363–369.
Return to citation in text: [1] -
Kadnikov, D. V.; Larock, R. C. J. Organomet. Chem. 2003, 687, 425–435. doi:10.1016/S0022-328X(03)00786-1
Return to citation in text: [1] -
Potdar, M. K.; Mohile, S. S.; Salunkhe, M. M. Tetrahedron Lett. 2001, 42, 9285–9287. doi:10.1016/S0040-4039(01)02041-X
Return to citation in text: [1] -
Yavari, I.; Adib, M.; Hojabri, L. Tetrahedron 2002, 58, 6895–6899. doi:10.1016/S0040-4020(02)00758-5
Return to citation in text: [1] -
Creaven, B. S.; Egan, D. A.; Kavanagh, K.; McCann, M.; Noble, A.; Thati, B.; Walsh, M. Inorg. Chim. Acta 2006, 359, 3976–3984. doi:10.1016/j.ica.2006.04.006
Return to citation in text: [1] -
European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Clin. Microbiol. Infect. 2000, 6, 509–515.
Return to citation in text: [1] -
National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 5th ed; Approved Standard M7-A5.Wayne, PA: NCCLS, 2000.
Return to citation in text: [1]
| 1. | O’Kennedy, R.; Thornes, R. D. Coumarins: Biology, Applications and Mode of Action; Wiley & Sons: Chichester, UK, 1997. |
| 2. | Zahradnik, M. The Production and Application of Fluorescent Brightening Agents; Wiley & Sons, 1992. |
| 11. | Craven, P. A.; Pfanstiel, J.; DeRubertis, F. R. J. Clin. Invest. 1986, 77, 850–859. doi:10.1172/JCI112382 |
| 12. | Rathbone, D. L.; Su, D.; Wang, Y.; Billington, D. C. Tetrahedron Lett. 2000, 41, 123–126. doi:10.1016/S0040-4039(99)02027-4 |
| 9. | Key, J. A.; Kho, S.; Timerghazin, Q. K.; Brown, A.; Cairo, C. W. Dyes Pigm. 2009, 82, 196–203. doi:10.1016/j.dyepig.2009.01.001 |
| 10. | Zhou, S.; Jia, J.; Gao, J.; Han, L.; Li, Y.; Sheng, W. Dyes Pigm. 2010, 86, 123–128. doi:10.1016/j.dyepig.2009.12.005 |
| 7. | Zhao, P.-L.; Wang, L.; Zhu, X.-L.; Huang, X.; Zhan, C.-G.; Wu, J.-W.; Yang, G.-F. J. Am. Chem. Soc. 2010, 132, 185–194. doi:10.1021/ja905756c |
| 8. | Teichert, J. F.; Feringa, B. L. Chem. Commun. 2011, 47, 2679–2681. doi:10.1039/c0cc05160h |
| 3. | Bailly, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.-P.; Hildebrand, M.-P.; Peyrot, V.; Wattez, N. J. Med. Chem. 2003, 46, 5437–5444. doi:10.1021/jm030903d |
| 4. | Yeh, J.-Y.; Coumar, M. S.; Horng, J.-T.; Shiao, H.-Y.; Kuo, F.-M.; Lee, H.-L.; Chen, I.-C.; Chang, C.-W.; Tang, W.-F.; Tseng, S.-N. J. Med. Chem. 2010, 53, 1519–1533. doi:10.1021/jm901570x |
| 5. | Pierson, J.-T.; Dumètre, A.; Hutter, S.; Delmas, F.; Laget, M.; Finet, J.-P.; Azas, N.; Combes, S. Eur. J. Med. Chem. 2010, 45, 864–869. doi:10.1016/j.ejmech.2009.10.022 |
| 6. | Basile, A.; Sorbo, S.; Spadaro, V.; Bruno, M.; Maggio, A.; Faraone, N.; Rosselli, S. Molecules 2009, 14, 939–952. doi:10.3390/molecules14030939 |
| 19. | Kadnikov, D. V.; Larock, R. C. J. Organomet. Chem. 2003, 687, 425–435. doi:10.1016/S0022-328X(03)00786-1 |
| 20. | Potdar, M. K.; Mohile, S. S.; Salunkhe, M. M. Tetrahedron Lett. 2001, 42, 9285–9287. doi:10.1016/S0040-4039(01)02041-X |
| 21. | Yavari, I.; Adib, M.; Hojabri, L. Tetrahedron 2002, 58, 6895–6899. doi:10.1016/S0040-4020(02)00758-5 |
| 23. | European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Clin. Microbiol. Infect. 2000, 6, 509–515. |
| 16. | Koelsch, C. F. J. Am. Chem. Soc. 1945, 67, 569–574. doi:10.1021/ja01220a023 |
| 17. | Koelsch, C. F.; Freerks, M. C. J. Org. Chem. 1953, 18, 1538–1545. doi:10.1021/jo50017a013 |
| 18. | Bedair, A. H.; Aly, F. M.; El-Agrody, A. M.; El-Assy, R. K. M. Acta Pharm. 1986, 36, 363–369. |
| 24. | National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 5th ed; Approved Standard M7-A5.Wayne, PA: NCCLS, 2000. |
| 14. | Burton, H. J. Chem. Soc. 1945, 280, 280–283. doi:10.1039/JR9450000280 |
| 15. | Bonsignore, L.; Cottiglia, F.; Maccioni, A. M.; Sacci, D.; Lavagna, S. M. J. Heterocycl. Chem. 1995, 32, 573–577. doi:10.1002/jhet.5570320234 |
| 13. | Mishra, J. K.; Samanta, K.; Jain, M.; Dikshit, M.; Panda, G. Med. Chem. Lett. 2010, 20, 244–247. doi:10.1016/j.bmcl.2009.10.126 |
| 22. | Creaven, B. S.; Egan, D. A.; Kavanagh, K.; McCann, M.; Noble, A.; Thati, B.; Walsh, M. Inorg. Chim. Acta 2006, 359, 3976–3984. doi:10.1016/j.ica.2006.04.006 |
© 2011 Mohamed et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)