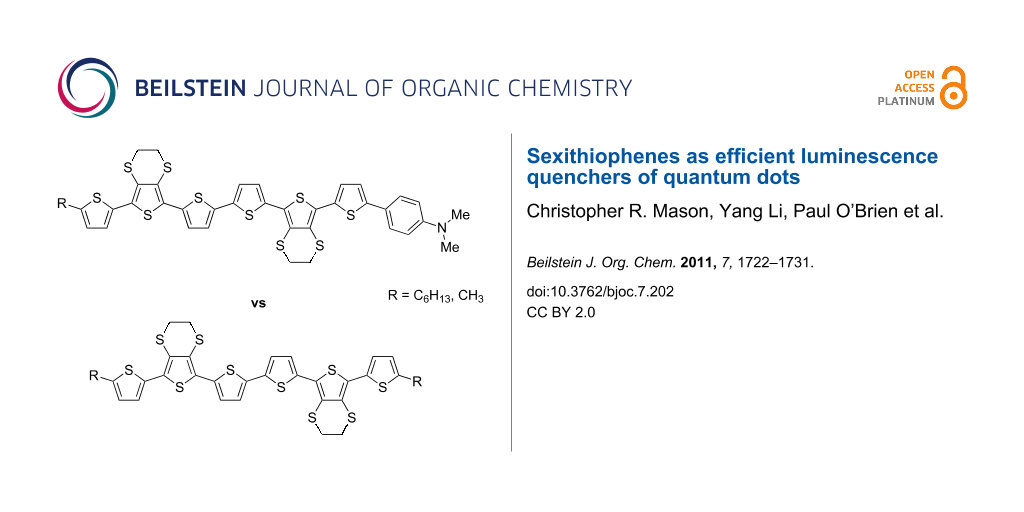
Abstract
Sexithiophenes 1a and 1b, in which a 4-(dimethylamino)phenyl unit is incorporated as an end-capping group, were synthesised and characterised by cyclic voltammetry, absorption spectroscopy and UV–vis spectroelectrochemistry. Additionally, their ability to function as effective luminescence quenchers for quantum dot emission was studied by photoluminescence spectroscopy and compared with the performance of alkyl end-capped sexithiophenes 2a and 2b.

Graphical Abstract
Introduction
Structurally well-defined oligothiophenes as functional organic materials have attracted significant interest due to the advantages they offer over polythiophenes, namely (i) monodispersity, (ii) a precise structure with no isomeric impurities, (iii) high chemical stability, (iv) good solubility, and (v) direct processability from solution or by vacuum deposition [1]. This has led to the application of oligothiophenes in numerous organic devices including solar cells [2-4], light-emitting diodes (LEDs) [5,6], field-effect transistors [7-9] and electrochromics [10]. Furthermore, end-capped oligothiophenes are particularly attractive materials for study due to their enhanced stability, and this can lead to improved performance in devices through enhanced intermolecular ordering [11]. Tailoring of the properties of oligothiophenes can be achieved by the appropriate choice of the end-capping functionality, for example, the incorporation of perfluoroalkyl groups for n-type semiconductors [12,13]. Previously, we reported the synthesis and properties of alkyl end-capped oligothiophenes 2 (Figure 1), which incorporate the ethylene dithiothiophene (EDTT) unit, including their performance as the electron donor material in a bilayer photovoltaic device [14]. In this article, the role of dimethylamino end-capping groups in coordinating to nanocrystalline particles is reported.
![[1860-5397-7-202-1]](/bjoc/content/figures/1860-5397-7-202-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Dimethylaminophenylene end-capped sexithiophenes 1a and 1b, and dialkyl end-capped sexithiophenes 2a and 2b.
Figure 1: Dimethylaminophenylene end-capped sexithiophenes 1a and 1b, and dialkyl end-capped sexithiophenes 2a...
Recently, there has been significant interest in small, nanocrystalline particles, or quantum dots, from both a fundamental point of view, and also with regards to their potential role in a range of device architectures [15,16]. Additionally, the formation of blends of conjugated oligomers and quantum dots can lead to attractive properties, taking advantage of facile charge transfer due to the high electron affinity of the quantum dot nanoparticles [17,18], as well as overcoming processing difficulties associated with devices containing quantum dots alone [15]. The complementary properties of conjugated oligomers/polymers and nanoparticles, and the possible photoinduced electron-transfer processes between them, have led to these materials being combined successfully in LEDs and photovoltaic cells [19-23]. Previously, we reported the synthesis and characterisation of CdS quantum dots in polystyrene beads, in which beads ranging in size from 100 nm to 500 μm were prepared and confocal microscopy showed an even distribution of CdS throughout the polymer, with retention of the photoluminescence behaviour of the quantum dot [24]. Herein, we present a comparative study on the photophysical properties of sexithiophenes 1 or 2 in varying concentrations, in the presence of a fixed concentration of CdSe(ZnS) core/shell quantum dots. Our choice of compounds represents molecules with (1a,b) and without (2a,b) a Lewis base functionality for quantum-dot surface coordination, with a view to determining whether structural complexity is required to achieve nanoparticle–sexithiophene electronic interactions.
Results and Discussion
Synthesis
The preparation of sexithiophenes 1a and 1b (Scheme 1) began in a similar fashion to our previously published methodology for the synthesis of compounds 2a and 2b [14]. After the formation of terthiophenes 3 [14], bromination with NBS under acidic conditions afforded the key intermediates 4a and 4b in high yields (97% and 88%, respectively). In parallel, dibrominated terthiophene 6 was prepared in an analogous fashion to 4a and 4b with 2.2 equiv of NBS. Subsequent Negishi coupling of compound 6 with organozinc intermediates of 4a and 4b, which were prepared by lithiation followed by reaction with zinc chloride, led to the isolation of 7a and 7b in modest yields (40% and 20%, respectively). Sexithiophenes 1a and 1b were subsequently isolated following Suzuki–Miyaura coupling with 4-(dimethylamino)phenylboronic acid (1a, 28%; 1b, 44%). As a comparison, nonfunctionalised sexithiophenes 2a and 2b were also synthesised in order to study the role of the dimethylaminophenyl group on the oligothiophene properties. The synthesis of 2a and 2b was completed by following our published procedure [14], with oxidative coupling of 3a or 3b with the aid of FeCl3 affording sexithiophenes 2a and 2b in moderate yields (40% and 50%, respectively).
![[1860-5397-7-202-i1]](/bjoc/content/inline/1860-5397-7-202-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of functionalised oligothiophenes 1a,b and 2a,b. Reagents and conditions: a) NBS, CH3CO2H, THF (1:1, v/v); 4a, 97%, 4b, 88%; b) 2.2 equiv NBS, CH3CO2H, THF (1:1, v/v); 6, 91%; c) (i) n-BuLi, (ii) ZnCl2, (iii) 4a or 4b, Pd(PPh3)4, THF; 7a, 40%, 7b, 20%; d) 4-(dimethylamino)phenylboronic acid, Pd(PPh3)4, toluene, EtOH, NaHCO3, H2O; 1a, 28%, 1b, 44%; e) FeCl3, CHCl3; 2a, 40%, 2b, 50%.
Scheme 1: The synthesis of functionalised oligothiophenes 1a,b and 2a,b. Reagents and conditions: a) NBS, CH3...
Characterisation of physical properties
A film of methyl-capped dimethylaminosexithiophene 1b on ITO glass was obtained by spin coating from a chloroform solution, and the redox properties were compared to those of nonfunctionalised analogue 2b (Figure 2; Table 1).
![[1860-5397-7-202-2]](/bjoc/content/figures/1860-5397-7-202-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Solid-state voltammograms of 1b and 2b, as spin-coated films on ITO glass, versus Ag/AgCl reference electrode, platinum wire as the counter electrode, TBAPF6 as the supporting electrolyte in CH3CN (0.1 M), scan rate 100 mV s−1.
Figure 2: Solid-state voltammograms of 1b and 2b, as spin-coated films on ITO glass, versus Ag/AgCl reference...
Table 1: Redox and peak separation potentials of 1b in the solid state compared to 2b. The HOMO–LUMO gap was determined from the difference in the onsets for the reduction and first-oxidation processes.
| Entry | E1½ (V) | ΔE1a–c (mV) | E2½ (V) | ΔE2a–c (mV) | HOMO–LUMO gap (eV) |
|---|---|---|---|---|---|
| 2b | +0.73 | 340 | +0.95 | 170 | 2.2 |
| 1b | +0.56 | 290 | +0.74 | 270 | 2.1 |
In the positive scan, the cyclic voltammogram for 1b shows two quasi-reversible redox waves consistent with the formation of a polaronic cation at the first step followed by the formation of the dication species at the second oxidation. Compared to 2b, the first oxidation peak of 1b is shifted to a less positive value, viz. from +0.90 V to +0.70 V. The corresponding reduction process is also shifted from +0.56 V to +0.41 V. The second oxidation potential for 1b experiences a shift to lower values (1.04 V compared to 0.89 V), but the reversibility is diminished somewhat; ΔE2a–c (mV) 2b, 170 mV; 1b, 270 mV. The decrease in oxidation potentials of 1b compared to those of 2b indicates a more effective stabilisation or accommodation of the positive polaron and bipolaron species in 1b, due to the addition of the electron-donating dimethylaminophenyl substituent. In the negative scan, similar reduction potentials were observed at −1.75 and −1.80 V for 1b and 2b, respectively. The electrochemical band gap for 1b in the solid state is approximately 2.1 eV, which is slightly lower than that of 2b (2.2 eV) due to the electronic contribution of the conjugated phenylamine.
Absorption studies (in dichloromethane) on functionalised sexithiophenes 1a and 1b revealed a bathochromic shift in the absorption maximum compared to the corresponding dialkyl end-capped sexithiophenes 2a and 2b (shifts of 10 and 12 nm for compounds 1a (λmax = 468 nm) and 1b (λmax = 469 nm), respectively (Figure 3; Table 2) [14]). These bathochromic shifts are consistent with the increase in π-electron delocalisation due to the addition of the conjugated phenyl ring. This increase in conjugation is also evidenced by a slight reduction in the optically determined HOMO–LUMO gap of 1a and 1b (2.2 eV for both, Table 2). The solid-state absorption spectra of 1a and 1b are very similar, but the optical HOMO–LUMO gaps in the solid state are red-shifted compared to those in solution (2.0 eV).
![[1860-5397-7-202-3]](/bjoc/content/figures/1860-5397-7-202-3.png?scale=2.1&max-width=1024&background=FFFFFF)
Figure 3: Absorption spectra in solution (dichloromethane) and solid state.
Figure 3: Absorption spectra in solution (dichloromethane) and solid state.
Table 2: Comparison of the optical properties of 1a, 1b, 2a and 2b in solution and solid state. The HOMO–LUMO gap was determined from the onset of the longest-wavelength absorption band.
| Entry | Absorption maxima (nm) | HOMO–LUMO gap (eV) |
|---|---|---|
| 1a | 468 | 2.2 |
| 2a | 458 | 2.3 |
| 1a solid | 469 | 2.0 |
| 1b | 469 | 2.2 |
| 2b | 457 | 2.3 |
| 1b solid | 467 | 2.0 |
UV–vis spectroelectrochemical measurements (SEC) were performed in acetonitrile on functionalised sexithiophene 1b and nonfunctionalised sexithiophene 2b as thin films drop-cast onto ITO glass (Figure 4). The methyl derivatives were used in this study since the hexyl analogues were found to dissolve partially, upon oxidation and reduction. In this context, it should be noted that the difference between the spectroelectrochemical behaviours of the methyl and hexyl sexithiophenes is expected to be negligible. During the experiments, no new peaks appeared in either sample until the second oxidation process. At this point, two new peaks emerge above +0.80 V at 668 and >1100 nm for 1b and are assigned to the dication, as are both new peaks in 2b (at 651 and >1100 nm). These continue to grow up to +1.50 V until a sharp decline in absorption is seen due to the degradation or dissolution of the films (Figure 4, compound 1b). Intermediate polaron peaks were not seen for either 1b or 2b. The difference between the first and second oxidation potentials (0.19 V) for 1b is slightly larger than that for 2b (0.13 V) but the peaks may still be sufficiently close enough together to prevent the experimental detection of the intermediate polaron (1b+·). The increase in wavelength of the high-energy absorption band of 1b2+ (668 nm) compared to that of 2b2+ (631 nm) indicates a more delocalised charged species. This increase is a consequence of the resonance effect of the phenyldimethylamine group.
![[1860-5397-7-202-4]](/bjoc/content/figures/1860-5397-7-202-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–visible spectroelectrochemical measurements of 1b (left) and 2b (right) drop-cast onto ITO glass.
Figure 4: UV–visible spectroelectrochemical measurements of 1b (left) and 2b (right) drop-cast onto ITO glass....
Sexithiophene/quantum dot composites in solution
A comparative study on the photophysical properties of sexithiophenes 1a,b and 2a,b, in varying concentrations, with a fixed concentration of CdSe(ZnS) core/shell quantum dots was conducted to determine the effect of the Lewis base group in sexithiophenes 1a and 1b. The quantum dot CdSe core was prepared by the method of Cumberland et al. [25], and the ZnS shell was added using the dithiocarbamate precursor Zn(S2CNMeHex)2 [26]. The resulting dots incorporate a hexadecylamine (HDA) capping group and were prepared so as to ensure that their fluorescence did not overlap with the absorption profile of the sexithiophenes under study. Thus, the photoluminescence maximum for these quantum dots under excitation of light at 590 nm is 634 nm in chloroform, while the high-energy absorption edge for the nanocrystals is 657 nm. Comparison with the absorption profiles for 1b and 2b confirms that no overlap occurs between the emission from the quantum dot and the absorption by the oligothiophene (Figure 5). As such, any luminescence quenching observed would not be a result of Förster energy transfer.
![[1860-5397-7-202-5]](/bjoc/content/figures/1860-5397-7-202-5.png?scale=2.3&max-width=1024&background=FFFFFF)
Figure 5: Absorption spectra for 1b and 2b, together with the absorption and emission profiles for the CdSe(ZnS) quantum dots, all in chloroform.
Figure 5: Absorption spectra for 1b and 2b, together with the absorption and emission profiles for the CdSe(Z...
The effect of each of the corresponding sexithiophenes with or without a Lewis base group (1 or 2) was then studied by preparing separate stock solutions of quantum dots and sexithiophenes 1b or 2b, of known concentration. A fixed amount of quantum dot solution was used combined with various amounts of sexithiophene 1b or 2b (additional pure solvent was added to maintain a fixed concentration of quantum dots). Each solution was allowed to equilibrate for 2 h, to ensure that any surface-exchange processes had reached a steady state, before the UV–vis absorption spectra were recorded (Figure 6). Although an increase in absorption was observed with increasing concentration of sexithiophene, no evidence for any ground-state interactions between the sexithiophene, 1b or 2b, and the CdSe(ZnS) quantum dots was observed. Next, in order to examine any possible transfer processes within the mixed solutions, photoluminescence quenching experiments were performed (Figure 7). The quantum dots were excited at 590 nm (outside the absorption region for oligothiophenes 1b and 2b) and the photoluminescence spectra were recorded at each concentration of 1b and 2b. The general trend observed was a decrease in the photoluminescence intensity with increasing sexithiophene concentration, irrespective of the presence (1b) or absence (2b) of a Lewis base (the anomaly at low concentrations of sexithiophenes is explained below). Since energy transfer can be excluded, it may be that hole transfer from the quantum dot (the HOMO of CdSe(ZnS) was reported as 6.5 eV [27]) to sexithiophenes occurs, leading to the extensive quenching observed here. A Stern–Volmer plot indicated that sexithiophenes 1b and 2b are equally effective (within experimental error) at quenching quantum dots (a plot of I0/I versus [Q] showed a slope of 1.120 ± 0.174 for 1b, and 1.101 ± 0.114 for 2b). As such, it is likely that both sexithiophenes 1b and 2b, regardless of the presence of a conventional Lewis base group, are effective in displacing HDA and quenching the luminescence of the quantum dots.
![[1860-5397-7-202-6]](/bjoc/content/figures/1860-5397-7-202-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The absorption spectra of increasing sexithiophene concentration with HDA capped CdSe(ZnS) quantum dots in chloroform; (i) 1b; (ii) 2b.
Figure 6: The absorption spectra of increasing sexithiophene concentration with HDA capped CdSe(ZnS) quantum ...
![[1860-5397-7-202-7]](/bjoc/content/figures/1860-5397-7-202-7.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 7: Photoluminescence quenching experiments; the effect of increasing sexithiophene concentration with HDA capped CdSe(ZnS) quantum dots, in chloroform. Excitation wavelength = 590 nm; (i) 1b; (ii) 2b.
Figure 7: Photoluminescence quenching experiments; the effect of increasing sexithiophene concentration with ...
As a further comparison, terthiophene 5 was also examined for photoluminescence quenching under identical conditions to those used for sexithiophenes 1b and 2b. Interestingly, the addition of terthiophene 5 has no quenching effect on the luminescence of the quantum dots, indicating that the HOMO and LUMO of terthiophene 5 are misaligned with respect to the quantum dot such that the quenching does not occur. However, a slight increase in the intensity of the quantum-dot photoluminescence was observed with increasing concentrations of terthiophene 5, indicating an effective passivation of the nonradiative decay sites [28]. At low concentration of sexithiophene 1b (0.2 × 10−5 M), a slight increase in luminescence intensity was observed, whereas the luminescence intensity at the same concentration of sexithiophene 2b remained constant. At these concentrations, the amount of sexithiophene bound to the quantum dot surface is at its lowest and a significant amount of HDA remains attached to the surface also. Since the photoluminescence is increased at these concentrations, we postulate that the sexithiophenes preferentially bind to defects on the surface of the quantum dots that otherwise act as intergap trap states for nonradiative emission.
Conclusion
Two novel sexithiophene families, 1a,b and 2a,b, were synthesised and investigated as luminescence quenchers of quantum dots. Their properties were investigated by cyclic voltammetry, UV–vis absorption and spectroelectrochemistry. The addition of the terminal 4-(dimethylamino)phenyl unit in 1a and 1b has a noticeable effect on the optical and electronic properties of the sexithiophene, compared to the nonfunctionalised systems 2a and 2b, notably lowering the band gap and red-shifting the absorbance due to the increased degree of conjugation that the functionalisation affords.
Composite solutions of CdSe(ZnS) quantum dots with various concentrations of 1b and 2b were prepared, and the excited state interactions between the components were studied by photoluminescence spectroscopy. Upon excitation of solely the quantum dot at 590 nm (no overlap of the sexithiophene absorption and quantum dot luminescence occurs at this wavelength), both sexithiophenes 1b and 2b proved to be effective luminescence quenchers across a range of concentrations. It is likely that a hole-transfer mechanism operates in this process. Thus, these results indicate that sexithiophenes based on the skeleton of compound 2 are effective luminescence quenchers even without functionalisation with a Lewis base group.
Experimental
General
Melting points were taken on a Stuart Scientific SMP1 Melting Point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Varian Unity Innova instrument at 300 and 75 MHz, respectively; chemical shifts are given in ppm. Peak multiplicities are denoted by s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet) or by a combination of these dd (doublet of doublets), dt (doublet of triplets) and td (triplet of doublets), with coupling constants (J) given in Hz. IR spectra were recorded on an ATI Mattson Genesis Series FTIR spectrometer. Electron-impact (EI) and chemical-ionisation (CI) mass spectra were recorded on a Micromass Trio 2000 spectrometer; high-resolution mass spectra were recorded on a Kratos Concept spectrometer. Elemental analyses were obtained on a Carlo Erba Instruments EA 1108 elemental analyser. Absorption spectra were measured on a Unicam UV 300 spectrophotometer.
All CV and spectroelectrochemical measurements were performed on a CH Instruments 660 A Electrochemical Workstation with iR compensation, with anhydrous CH2Cl2 or acetonitrile as the solvent, aqueous Ag/AgCl as the reference electrode and platinum wire and gold disk (or ITO glass for SEC) as the counter and working electrodes, respectively. All solutions were degassed (Ar) and, where relevant, were prepared so as to contain the substrate in concentrations of ca. 10−3 M, together with n-Bu4NPF6 (0.1 M) as the supporting electrolyte. Under these conditions, the redox potential for the FcH/FcH+ couple was +0.48 V (CH2Cl2, versus Ag/AgCl).
The photoluminescence properties of the oligothiophene and CdSe(ZnS) quantum dot blends were measured on a Spex FluoroMax instrument with a xenon lamp (150 W) and a 152 P photomultiplier tube as a detector. Spectra were obtained with the slits set at 5 nm and with an integration time of 1 s. The samples were placed in quartz cuvettes (1 cm path length). The wavelength of excitation is indicated in the text.
The compounds 2a, 2b and 3 were prepared according to our previously reported procedure [14].
5-(5-Bromothiophen-2-yl)-7-(5-hexylthiophen-2-yl)-2,3-dihydrothieno[3,4-b][1,4]dithiine (4a)
To a solution of 3a (0.62 g, 1.47 mmol) in tetrahydrofuran/glacial acetic acid (1:1 v/v, 40 mL), was added N-bromosuccinimide (0.26 g, 1.47 mmol) portionwise, in the dark under rapid stirring. The reaction mixture was stirred for 2 h after which water (40 mL) was added. The product was then extracted into dichloromethane (2 × 30 mL), washed with saturated sodium hydrogencarbonate (2 × 30 mL) and dried (MgSO4). The solvent was removed under reduced pressure to afford 4a as a dark green oil (0.71 g, 97%); 1H NMR (CDCl3) δ 7.14 (d, J = 3.7, 1H), 7.05 (s, 2H), 6.78 (d, J = 3.7, 1H), 3.30 (s, 4H), 2.85 (t, J = 6.6, 2H), 1.73 (q, 2H), 1.36 (m, 6H), 0.93 (t, J = 6.7, 3H); 13C NMR (CDCl3) δ 147.4, 136.7, 132.0, 130.4, 128.7, 126.5, 126.2, 124.8, 124.3, 123.1, 112.9, 31.8, 30.4, 29.1, 28.6, 28.5, 22.8, 14.4; MS (TOF, EI+) m/z: 519 (M + NH4+, 31%), 517 (M + NH4+, 28%), 503 (M + H+, 100%), 501 (M + H+, 95%); FTIR (10 scan) ν/cm−1: 2952, 2924, 2853, 1490, 1462, 1411, 1275, 1036; HRMS (TOF, EI+): calcd for C20H21BrS5 + H+, 500.9509; found, 500.9676.
5-(5-Bromothiophen-2-yl)-7-(5-methylthiophen-2-yl)-2,3-dihydrothieno[3,4-b][1,4]dithiine (4b)
Compound 4b was prepared as described for 4a from compound 3b (0.89 g, 2.50 mmol), in tetrahydrofuran/glacial acetic acid (1:1 v/v, 40 mL), and N-bromosuccinimide (0.45 g, 2.50 mmol) to afford 4b as a yellow solid (0.96 g, 88%); mp 93–96 °C; 1H NMR (CDCl3) δ 7.14 (d, J = 3.7, 1H), 7.06 (s, 2H), 6.78 (dd, J = 1.0 and 3.7, 1H), 3.31 (s, 4H), 2.56 (s, 3H); 13C NMR (CDCl3) δ 141.2, 136.6, 132.3, 130.4, 129.4, 128.5, 126.8, 126.2, 126.1, 124.3, 123.2, 112.9, 28.6, 28.5, 15.6; MS (EI) m/z: 432 (M+, 16%), 430 (M+, 13%), 352 (11%); MS (CI) m/z: 433 (M + H+, 41%), 431 (M + H+, 37%), 353 (100%); FTIR (KBr) ν/cm−1: 2945, 2908, 1655, 1489, 1412, 1276, 1217, 1056; Anal. calcd for C14H8O2S5: C, 41.76; H, 2.57; S, 37.16; Br, 18.52%; found: C, 41.62; H, 2.27; S, 37.20; Br, 18.30%.
4,5-Bis-(hydroxythiophene-2-yl-methyl)-[1,3]dithiole-2-thione (A) [29]
A solution of 1,3-dithiole-2-thione (4.00 g, 29.8 mmol) in dry tetrahydrofuran (250 mL) was cooled to −78 °C under dry nitrogen. Lithium diisopropylamide mono(tetrahydrofuran) (1.5 M in cyclohexanes, 21.9 mL, 32.9 mmol) was added and the mixture was stirred for 20 minutes after which time thiophene-2-carboxaldehyde (3.1 mL, 32.9 mmol) was added and the mixture stirred for a further 10 min. Another portion of lithium diisopropylamide mono(tetrahydrofuran) (1.5 M in cyclohexanes, 21.9 mL, 32.9 mmol) was added and the mixture was stirred for 15 min after which time thiophene-2-carboxaldehyde (3.1 mL, 32.9 mmol) was added and the mixture was stirred for a further 5 min. The reaction mixture was stirred for a further 1.5 h whilst being allowed to warm to room temperature. Saturated sodium hydrogencarbonate (150 mL) was added and the organic phase was removed. The aqueous phase was washed with dichloromethane (3 × 50 mL) and the combined organic extracts were dried (MgSO4). The solvent was removed under reduced pressure and the product was isolated by column chromatography (silica, petroleum ether (40–60 °C)/dichloromethane (1:1 v/v) to remove the starting materials and impurities, then ethyl acetate to remove the product) to afford A as a red oil (~10.1 g; product unstable, no data).
[5-(Thiophene-2-carbonyl)-2-thioxo-[1,3]dithiol-4-yl]thiophen-2-yl-methanone (B) [29]
To a solution of the diol A (10.1 g), in dichloromethane (250 mL), manganese dioxide (10× excess w/w, ~101 g) was added portionwise and the mixture was stirred for approximately 2 min. The mixture was filtered through a silica plug (eluted with dichloromethane) and the solvent removed under reduced pressure to afford B as a yellow solid (9.50 g, 90% from 1,3-dithiole-2-thione 3); mp 91–92 °C (lit. [29] mp 92 °C); 1H NMR (CDCl3) δ 7.76 (dd, J = 1.1 and 4.9, 2H), 7.72 (dd, J = 1.1 and 4.0, 2H), 7.14 (t, J = 4.4, 2H); MS (EI) m/z: 354 (M+, 75%), 278 (12%), 250 (13%); MS (CI) m/z: 372 (M + NH4+, 100%), 355 (M + H+, 58%), 281 (21%).
4,5-Bis(thiophene-2-carbonyl)[1,3]dithiole-2-one (C) [30]
To a solution of the diketone B (9.5 g, 26.8 mmol) in dichloromethane/glacial acetic acid (3:1 v/v, 200 mL), was added mercuric acetate (11.98 g, 37.6 mmol). The mixture was stirred at room temperature for 16 h and filtered through a silica plug (eluting with dichloromethane). The organic extract was washed with water (2 × 100 mL) and saturated sodium hydrogencarbonate (2 × 100 mL), and was dried (MgSO4). The solvent was removed under reduced pressure to afford C as an off-white solid (7.35 g, 81%); mp 130–131 °C (lit. [30] mp 130–132 °C); 1H NMR (CDCl3) δ 7.75 (dd, J = 1.1 and 4.9, 2H), 7.73 (dd, J = 1.1 and 4.0, 2H), 7.14 (t, J = 4.4, 2H); MS (EI) m/z: 338 (M+, 19%); MS (CI) m/z: 356 (M + NH4+, 100%), 339 (M + H+, 19%).
4,6-Di-thiophen-2-yl-thieno[3,4-d][1,3]dithiol-2-one (D) [30]
A mixture of the tricarbonyl C (3.68 g, 10.9 mmol), sodium hydrogencarbonate (4.57 g, 54.5 mmol) and phosphorus pentasulfide (24.2 g, 54.5 mmol) in 1,4-dioxane (150 mL) was stirred whilst the temperature was raised to 90 °C. This temperature was maintained for 3 h. The mixture was cooled, water (200 mL) was added portionwise (CAUTION! H2S and CO2 evolution) and the suspension was stirred overnight. After cooling, the mixture was filtered, washed with boiling water and dried in vacuo to afford the crude product. The crude product was dissolved in minimal hot chloroform, dried (MgSO4), stirred with decolourising charcoal for 30 min and then filtered through a silica plug (eluting with chloroform). The solvent was reduced in volume and the product isolated by precipitation with petroleum ether (40–60 °C), to afford D as an orange/yellow solid (2.17 g, 59%); mp 169–170 °C (lit. [30] mp 170–172 °C); 1H NMR (CDCl3) δ 7.42 (dd, J = 1.1 and 5.1, 2H), 7.28 (dd, J = 1.1 and 3.7, 2H), 7.15 (t, J = 4.4, 2H); MS (EI) m/z: 338 (M+, 51%), 310 (19%), 265 (19%); MS (CI) m/z: 356 (M + NH4+, 5%), 339 (M + H+, 100%).
5,7-Dithiophen-2-yl-2,3-dihydrothieno[3,4-b][1,4]dithiine (5) [30]
To a solution of the terthiophene D (2.17 g, 6.4 mmol) in dry tetrahydrofuran (150 mL), was added sodium ethoxide (0.2 M solution in ethanol, 70.6 mL, 14.1 mmol) under dry nitrogen. The reaction mixture was stirred for 30 min after which time 1,2-dibromoethane (0.55 mL, 6.4 mmol) was added and the mixture was stirred at room temperature for a further 16 h. The reaction mixture was filtered through a silica plug (eluting with tetrahydrofuran) and the solvent removed under reduced pressure to leave the crude product. The crude product was dissolved in chloroform, stirred with decolourising charcoal for 30 min and then filtered through a silica plug (eluting with chloroform). The solvents were removed under reduced pressure to afford 5 as a yellow solid (1.94 g, 66%); mp 140–141 °C (lit. [30] mp 140–142 °C); 1H NMR (CDCl3) δ 7.39 (dd, J = 1.2 and 5.1, 2H), 7.36 (dd, J = 1.2 and 3.7, 2H), 7.13 (t, J = 4.5, 2H), 3.34 (s, 4H); MS (EI) m/z: 338 (M+, 18%); MS (CI) m/z: 339 (M + H+, 100%).
5,7-Bis-(5-bromothiophen-2-yl)-2,3-dihydrothieno[3,4-b][1,4]dithiine (6)
To a solution of the terthiophene 5 (1.94 g, 5.2 mmol), in tetrahydrofuran/glacial acetic acid (1:1 v/v, 100 mL) was added N-bromosuccinimide (2.04 g, 11.5 mmol) portionwise, in the dark and under rapid stirring. The reaction mixture was stirred for 2 h after which water (150 mL) was added and the resulting precipitate was collected by filtration. The crude product was redissolved in chloroform and dried (MgSO4). The chloroform was reduced in volume and the product was isolated by precipitation with petroleum ether (40–60 °C) to afford 6 as a yellow solid (2.57 g, 91%); mp 156–157 °C; 1H NMR (CDCl3) δ 7.06 (s, 4H), 3.33 (s, 4H); MS (APCI+) m/z: 496 (MH+, 21%), 418 (100%); MS (APCI−) m/z: 468 ([M – C2H4]−, 100%); FTIR (KBr) ν/cm−1: 2926, 1655, 1560, 1508, 1476, 1419, 1272, 1217; Anal. calcd for C14H8Br2S5: C, 33.88; H, 1.62; S, 32.30; Br, 32.20%; found: C, 34.14; H, 1.42; S, 32.64; Br, 31.98%.
5-Bromo-5'''''-hexyl-3',3'''',4',4''''-ethylenedithio-2,2';5',2'';5'',2''';5''',2'''';5'''',2'''''-sexithiophene (7a)
To a solution of compound 4a (0.71 g, 1.42 mmol), in dry tetrahydrofuran (10 mL), under dry nitrogen and at −78 °C, was added n-butyllithium (2.5 M in hexanes, 0.74 mL, 1.85 mmol) and the mixture was stirred for 45 min. Then, to this a solution of zinc(II)chloride (0.25 g, 1.85 mmol) in dry tetrahydrofuran (20 mL) prepared under dry nitrogen was added by cannula, before being allowed to warm to room temperature. This was then added to a solution of compound 6 (2.10 g, 4.26 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.12 g, 0.099 mmol) in dry tetrahydrofuran (50 mL) under dry nitrogen, and heated under reflux for 16 h. The solution was allowed to cool and the crude product precipitated with petroleum ether (40–60 °C). The crude product was redissolved in a minimal amount of chloroform, reduced in volume and reprecipitated with petroleum ether (40–60 °C). The crude product was precipitated a number of times from chloroform to remove the majority of the excess of compound 6 used. The traces of 6 were removed by washing the solid (by using soxhlet extraction apparatus) with methanol. The crude product was then collected with chloroform and isolated by precipitation. The product was isolated by column chromatography (silica, toluene) and then precipitation to afford 7a as a red solid (0.47 g, 40%); mp 177 °C (by DSC); 1H NMR (CDCl3) δ 7.27 (m, 2H), 7.20 (dd, J = 0.8 and 3.9, 2H), 7.17 (d, J = 3.5, 1H), 7.08 (m, 2H), 6.80 (d, J = 3.7, 1H), 3.36 (s, 8H), 2.86 (t, J = 7.7, 2H), 1.74 (q, 2H), 1.37 (m, 6H), 0.94 (t, J = 6.9, 3H); MS (MALDI-TOF) m/z: 838 (M+, 100%); FTIR (KBr) ν/cm−1: 2924, 2855, 1634, 1266, 1161, 1040; Anal. calcd for C34H29BrS10: C, 48.72; H, 3.49; Br, 9.53%; found: C, 48.31; H, 3.22; Br, 9.05%.
5-Bromo-5'''''-methyl-3',3'''',4',4''''-ethylenedithio-2,2';5',2'';5'',2''';5''',2'''';5'''',2'''''-sexithiophene (7b)
Compound 7b was prepared as described for 7a from compound 4b (0.66 g, 1.5 mmol) in dry tetrahydrofuran (20 mL), n-butyllithium (2.5 M in hexanes, 0.80 mL, 2.0 mmol), zinc(II)chloride (0.27 g, 2.0 mmol) in dry tetrahydrofuran (20 mL), compound 6 (2.27 g, 4.60 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.13 g, 0.11 mmol) in dry tetrahydrofuran (60 mL). The product was isolated by column chromatography (silica, toluene) and then precipitation to afford 7b as a red solid (0.23 g, 20%); mp 195 °C (by DSC); 1H NMR (CDCl3) δ 7.26 (m, 2H), 7.20 (d, J = 4.0, 2H), 7.15 (d, J = 3.5, 1H), 7.08 (m, 2H), 6.80 (d, J = 3.5, 1H), 3.36 (s, 8H), 2.55 (s, 3H); MS (MALDI-TOF) m/z: 769 (M+, 68%), 767 (M+, 100%); FTIR (KBr) ν/cm−1: 2910, 1619, 1476, 1407, 1272, 1213, 1159, 1056; Anal. calcd for C29H19BrS10: C, 45.35; H, 2.49; Br, 10.40%; found: C, 45.08; H, 2.13; Br, 10.73%.
5-(4-[Dimethylamino]phenyl)-5'''''-hexyl-3',3'''',4',4''''-ethylenedithio-2,2';5',2'';5'',2''';5''',2'''';5'''',2'''''-sexithiophene (1a)
To a solution of 7a (136 mg, 0.16 mmol) and tetrakis(triphenylphosphine)palladium(0) (10 mg, 0.008 mmol) in toluene (20 mL) under dry nitrogen, was added, subsequently, a suspension of 4-(dimethylamino)phenylboronic acid (35 mg, 0.21 mmol) in ethanol (25 mL) and a solution of anhydrous sodium carbonate (45 mg, 0.42 mmol) in water (5 mL). The mixture was then heated under reflux for 16 h. Then, the solution was allowed to cool to room temperature and toluene (30 mL) was added to dilute the organic phase. The aqueous phase was removed and extracted with toluene (3 × 30 mL). The organic phases were combined and dried (MgSO4). The solvents were reduced in volume and the crude product was precipitated with petroleum ether (40–60 °C). The product was isolated by column chromatography (neutral alumina, toluene/ethyl acetate 9:1 (v/v) with gradual change to ethyl acetate). The solvents were reduced in volume and the product was precipitated with petroleum ether (40–60 °C) to afford 1a as a red solid (40 mg, 28%); mp 192–194 °C; 1H NMR (CDCl3) δ 7.55 (d, J = 8.6, 2H), 7.28 (m, 2H), 7.22 (m, 3H), 7.17 (d, J = 4.0, 2H), 6.78 (m, 3H), 3.36 (s, 8H), 3.04 (s, 6H), 2.86 (t, J = 7.5, 2H), 1.74 (q, 2H), 1.37 (m, 6H), 0.92 (t, J = 6.9, 3H); MS (APCI+) m/z: 895 (M + NH4+, 100%), 878 (M + H+, 53%); MS (APCI−) m/z: 848 ([M – 2CH3]−, 100%); FTIR (KBr) ν/cm−1: 3055, 2916, 2850, 1604, 1419, 1342, 1265, 1172, 1049; HRMS (TOF, EI+): calcd for C42H39NS10 + H+, 878.0362; found, 878.0372.
5-(4-[dimethylamino]phenyl)-5'''''-methyl-3',3'''',4',4''''-ethylenedithio-2,2';5',2'';5'',2''';5''',2'''';5'''',2'''''-sexithiophene (1b)
Compound 1b was prepared as described for 1a from compound 7b (78 mg, 0.10 mmol) in toluene (20 mL), tetrakis(triphenylphosphine)palladium(0) (6 mg, 0.005 mmol), 4-(dimethylamino)phenylboronic acid) (22 mg, 0.13 mmol) in ethanol (25 mL) and anhydrous sodium carbonate (28 mg, 0.26 mmol) in water (5 mL). The product was isolated by column chromatography (neutral alumina, toluene/ethyl acetate 9:1 (v/v) with a gradual change to ethyl acetate). The solvents were reduced in volume and the product precipitated with petroleum ether (40–60 °C) to afford 1b as a red solid (36 mg, 44%); mp 152–155 °C; 1H NMR (CDCl3) δ 7.55 (d, J = 8.78, 2H), 7.26 (m, 2H), 7.22 (m, 3H), 7.17 (d, J = 4.0, 1H), 7.15 (d, J = 4.0, 1H), 6.77 (m, 3H), 3.36 (s, 8H), 3.04 (s, 6H), 2.56 (s, 3H); MS (APCI+) m/z: 808 (MH+, 100%); FTIR (KBr) ν/cm−1: 3055, 2916, 1605, 1419, 1342, 1265, 1157, 1049; HRMS (TOF, EI+): calcd for C37H29NS10 + H+, 806.9502; found, 806.9494.
References
-
Mishra, A.; Ma, C.-Q.; Bäuerle, P. Chem. Rev. 2009, 109, 1141–1276. doi:10.1021/cr8004229
Return to citation in text: [1] -
Schulze, K.; Uhrich, C.; Schüppel, R.; Leo, K.; Pfeiffer, M.; Brier, E.; Reinold, E.; Bäuerle, P. Adv. Mater. 2006, 18, 2872–2875. doi:10.1002/adma.200600658
Return to citation in text: [1] -
Tamayo, A. B.; Walker, B.; Nguyen, T.-Q. J. Phys. Chem. C 2008, 112, 11545–11551. doi:10.1021/jp8031572
Return to citation in text: [1] -
Xia, P. F.; Lu, J.; Kwok, C. H.; Fukutani, H.; Wong, M. S.; Tao, Y. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 137–148. doi:10.1002/pola.23131
Return to citation in text: [1] -
Mazzeo, M.; Vitale, V.; Della Sala, F.; Pisignano, D.; Anni, M.; Barbarella, G.; Favaretto, L.; Zanelli, A.; Cingolani, R.; Gigli, G. Adv. Mater. 2003, 15, 2060–2063. doi:10.1002/adma.200305356
Return to citation in text: [1] -
Barbarella, G.; Favaretto, L.; Zanelli, A.; Gigli, G.; Mazzeo, M.; Anni, M.; Bongini, A. Adv. Funct. Mater. 2005, 15, 664–670. doi:10.1002/adfm.200400172
Return to citation in text: [1] -
Murphy, A. R.; Fréchet, J. M. J. Chem. Rev. 2007, 107, 1066–1096. doi:10.1021/cr0501386
Return to citation in text: [1] -
Ie, Y.; Hirose, T.; Aso, Y. J. Mater. Chem. 2009, 19, 8169–8175. doi:10.1039/b912744e
Return to citation in text: [1] -
Mei, J.; Graham, K. R.; Stalder, R.; Prakash Tiwari, S.; Cheun, H.; Shim, J.; Yoshio, M.; Nuckolls, C.; Kippelen, B.; Castellano, R. K.; Reynolds, J. R. Chem. Mater. 2011, 23, 2285–2288. doi:10.1021/cm1036869
Return to citation in text: [1] -
Yin, B.; Jiang, C.; Wang, Y.; La, M.; Liu, P.; Deng, W. Synth. Met. 2010, 160, 432–435. doi:10.1016/j.synthmet.2009.11.025
Return to citation in text: [1] -
Garnier, F.; Yassar, A.; Hajlaoui, R.; Horowitz, G.; Deloffre, F.; Servet, B.; Ries, S.; Alnot, P. J. Am. Chem. Soc. 1993, 115, 8716–8721. doi:10.1021/ja00072a026
Return to citation in text: [1] -
Facchetti, A.; Deng, Y.; Wang, A.; Koide, Y.; Sirringhaus, H.; Marks, T. J.; Friend, R. H. Angew. Chem., Int. Ed. 2000, 39, 4547–4551. doi:10.1002/1521-3773(20001215)39:24<4547::AID-ANIE4547>3.0.CO;2-J
Return to citation in text: [1] -
Facchetti, A.; Yoon, M.-H.; Stern, C. L.; Hutchinson, G. R.; Ratner, M. A.; Marks, T. J. J. Am. Chem. Soc. 2004, 126, 13480–13501. doi:10.1021/ja048988a
Return to citation in text: [1] -
Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Trindade, T.; O'Brien, P.; Pickett, N. L. Chem. Mater. 2001, 13, 3843–3858. doi:10.1021/cm000843p
Return to citation in text: [1] [2] -
Kamat, P. V. J. Phys. Chem. C 2008, 112, 18737–18753. doi:10.1021/jp806791s
Return to citation in text: [1] -
Greenham, N. C.; Peng, X.; Alivisatos, A. P. Phys. Rev. B 1996, 54, 17628–17637. doi:10.1103/PhysRevB.54.17628
Return to citation in text: [1] -
Ginger, D. S.; Greenham, N. C. Phys. Rev. B 1999, 59, 10622–10629. doi:10.1103/PhysRevB.59.10622
Return to citation in text: [1] -
Holder, E.; Tessler, N.; Rogach, A. L. J. Mater. Chem. 2008, 18, 1064–1078. doi:10.1039/b712176h
Return to citation in text: [1] -
Gaponik, N. P.; Talapin, D. V.; Rogach, A. L.; Eychmüller, A. J. Mater. Chem. 2000, 10, 2163–2166. doi:10.1039/b001958p
Return to citation in text: [1] -
Colvin, V. L.; Schlamp, M. C.; Alivisatos, A. P. Nature 1994, 370, 354–357. doi:10.1038/370354a0
Return to citation in text: [1] -
Liu, J.; Tanaka, T.; Sivula, K.; Alivisatos, A. P.; Fréchet, J. M. J. J. Am. Chem. Soc. 2004, 126, 6550–6551. doi:10.1021/ja0489184
Return to citation in text: [1] -
Reiss, P.; Couderc, E.; De Girolamo, J.; Pron, A. Nanoscale 2011, 3, 446–489. doi:10.1039/c0nr00403k
Return to citation in text: [1] -
Li, Y.; Liu, E. C. Y.; Pickett, N.; Skabara, P. J.; Cummins, S. S.; Ryley, S.; Sutherland, A. J.; O’Brien, P. J. Mater. Chem. 2005, 15, 1238–1243. doi:10.1039/B412317D
Return to citation in text: [1] -
Cumberland, S. L.; Hanif, K. M.; Javier, A.; Khitrov, G. A.; Strouse, G. F.; Woessner, S. M.; Yun, C. S. Chem. Mater. 2002, 14, 1576–1584. doi:10.1021/cm010709k
Return to citation in text: [1] -
Malik, M. A.; O'Brien, P.; Revaprasadu, N. Chem. Mater. 2002, 14, 2004–2010. doi:10.1021/cm011154w
Return to citation in text: [1] -
Hikmet, R. A. M.; Talapin, D. V.; Weller, H. J. Appl. Phys. 2003, 93, 3509–3514. doi:10.1063/1.1542940
Return to citation in text: [1] -
Milliron, D. J.; Alivisatos, A. P.; Pitios, C.; Edder, C.; Fréchet, J. M. J. Adv. Mater. 2003, 15, 58–61. doi:10.1002/adma.200390011
Return to citation in text: [1] -
Skabara, P. J.; Serebryakov, I. M.; Roberts, D. M.; Perepichka, I. F.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 1999, 64, 6418–6424. doi:10.1021/jo990198+
Return to citation in text: [1] [2] [3] -
Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h
Return to citation in text: [1] [2] [3] [4] [5] [6]
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 1. | Mishra, A.; Ma, C.-Q.; Bäuerle, P. Chem. Rev. 2009, 109, 1141–1276. doi:10.1021/cr8004229 |
| 10. | Yin, B.; Jiang, C.; Wang, Y.; La, M.; Liu, P.; Deng, W. Synth. Met. 2010, 160, 432–435. doi:10.1016/j.synthmet.2009.11.025 |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 7. | Murphy, A. R.; Fréchet, J. M. J. Chem. Rev. 2007, 107, 1066–1096. doi:10.1021/cr0501386 |
| 8. | Ie, Y.; Hirose, T.; Aso, Y. J. Mater. Chem. 2009, 19, 8169–8175. doi:10.1039/b912744e |
| 9. | Mei, J.; Graham, K. R.; Stalder, R.; Prakash Tiwari, S.; Cheun, H.; Shim, J.; Yoshio, M.; Nuckolls, C.; Kippelen, B.; Castellano, R. K.; Reynolds, J. R. Chem. Mater. 2011, 23, 2285–2288. doi:10.1021/cm1036869 |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 5. | Mazzeo, M.; Vitale, V.; Della Sala, F.; Pisignano, D.; Anni, M.; Barbarella, G.; Favaretto, L.; Zanelli, A.; Cingolani, R.; Gigli, G. Adv. Mater. 2003, 15, 2060–2063. doi:10.1002/adma.200305356 |
| 6. | Barbarella, G.; Favaretto, L.; Zanelli, A.; Gigli, G.; Mazzeo, M.; Anni, M.; Bongini, A. Adv. Funct. Mater. 2005, 15, 664–670. doi:10.1002/adfm.200400172 |
| 24. | Li, Y.; Liu, E. C. Y.; Pickett, N.; Skabara, P. J.; Cummins, S. S.; Ryley, S.; Sutherland, A. J.; O’Brien, P. J. Mater. Chem. 2005, 15, 1238–1243. doi:10.1039/B412317D |
| 2. | Schulze, K.; Uhrich, C.; Schüppel, R.; Leo, K.; Pfeiffer, M.; Brier, E.; Reinold, E.; Bäuerle, P. Adv. Mater. 2006, 18, 2872–2875. doi:10.1002/adma.200600658 |
| 3. | Tamayo, A. B.; Walker, B.; Nguyen, T.-Q. J. Phys. Chem. C 2008, 112, 11545–11551. doi:10.1021/jp8031572 |
| 4. | Xia, P. F.; Lu, J.; Kwok, C. H.; Fukutani, H.; Wong, M. S.; Tao, Y. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 137–148. doi:10.1002/pola.23131 |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 15. | Trindade, T.; O'Brien, P.; Pickett, N. L. Chem. Mater. 2001, 13, 3843–3858. doi:10.1021/cm000843p |
| 16. | Kamat, P. V. J. Phys. Chem. C 2008, 112, 18737–18753. doi:10.1021/jp806791s |
| 15. | Trindade, T.; O'Brien, P.; Pickett, N. L. Chem. Mater. 2001, 13, 3843–3858. doi:10.1021/cm000843p |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 19. | Holder, E.; Tessler, N.; Rogach, A. L. J. Mater. Chem. 2008, 18, 1064–1078. doi:10.1039/b712176h |
| 20. | Gaponik, N. P.; Talapin, D. V.; Rogach, A. L.; Eychmüller, A. J. Mater. Chem. 2000, 10, 2163–2166. doi:10.1039/b001958p |
| 21. | Colvin, V. L.; Schlamp, M. C.; Alivisatos, A. P. Nature 1994, 370, 354–357. doi:10.1038/370354a0 |
| 22. | Liu, J.; Tanaka, T.; Sivula, K.; Alivisatos, A. P.; Fréchet, J. M. J. J. Am. Chem. Soc. 2004, 126, 6550–6551. doi:10.1021/ja0489184 |
| 23. | Reiss, P.; Couderc, E.; De Girolamo, J.; Pron, A. Nanoscale 2011, 3, 446–489. doi:10.1039/c0nr00403k |
| 12. | Facchetti, A.; Deng, Y.; Wang, A.; Koide, Y.; Sirringhaus, H.; Marks, T. J.; Friend, R. H. Angew. Chem., Int. Ed. 2000, 39, 4547–4551. doi:10.1002/1521-3773(20001215)39:24<4547::AID-ANIE4547>3.0.CO;2-J |
| 13. | Facchetti, A.; Yoon, M.-H.; Stern, C. L.; Hutchinson, G. R.; Ratner, M. A.; Marks, T. J. J. Am. Chem. Soc. 2004, 126, 13480–13501. doi:10.1021/ja048988a |
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 11. | Garnier, F.; Yassar, A.; Hajlaoui, R.; Horowitz, G.; Deloffre, F.; Servet, B.; Ries, S.; Alnot, P. J. Am. Chem. Soc. 1993, 115, 8716–8721. doi:10.1021/ja00072a026 |
| 17. | Greenham, N. C.; Peng, X.; Alivisatos, A. P. Phys. Rev. B 1996, 54, 17628–17637. doi:10.1103/PhysRevB.54.17628 |
| 18. | Ginger, D. S.; Greenham, N. C. Phys. Rev. B 1999, 59, 10622–10629. doi:10.1103/PhysRevB.59.10622 |
| 26. | Malik, M. A.; O'Brien, P.; Revaprasadu, N. Chem. Mater. 2002, 14, 2004–2010. doi:10.1021/cm011154w |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 25. | Cumberland, S. L.; Hanif, K. M.; Javier, A.; Khitrov, G. A.; Strouse, G. F.; Woessner, S. M.; Yun, C. S. Chem. Mater. 2002, 14, 1576–1584. doi:10.1021/cm010709k |
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 30. | Pozo-Gonzalo, C.; Khan, T.; McDouall, J. J. W.; Skabara, P. J.; Roberts, D. M.; Light, M. E.; Coles, S. J.; Hursthouse, M. B.; Neugebauer, H.; Cravino, A.; Sariciftci, N. S. J. Mater. Chem. 2002, 12, 500–510. doi:10.1039/b109017h |
| 29. | Skabara, P. J.; Serebryakov, I. M.; Roberts, D. M.; Perepichka, I. F.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 1999, 64, 6418–6424. doi:10.1021/jo990198+ |
| 29. | Skabara, P. J.; Serebryakov, I. M.; Roberts, D. M.; Perepichka, I. F.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 1999, 64, 6418–6424. doi:10.1021/jo990198+ |
| 14. | Mason, C. R.; Skabara, P. J.; Cupertino, D.; Schofield, J.; Meghdadi, F.; Ebner, B.; Serdar Saricifti, N. J. Mater. Chem. 2005, 15, 1446–1453. doi:10.1039/b415610b |
| 29. | Skabara, P. J.; Serebryakov, I. M.; Roberts, D. M.; Perepichka, I. F.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 1999, 64, 6418–6424. doi:10.1021/jo990198+ |
| 27. | Hikmet, R. A. M.; Talapin, D. V.; Weller, H. J. Appl. Phys. 2003, 93, 3509–3514. doi:10.1063/1.1542940 |
| 28. | Milliron, D. J.; Alivisatos, A. P.; Pitios, C.; Edder, C.; Fréchet, J. M. J. Adv. Mater. 2003, 15, 58–61. doi:10.1002/adma.200390011 |
© 2011 Mason et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)