Abstract
A variety of 2-amino-3-arylpropan-1-ols, anti-2-amino-3-aryl-3-methoxypropan-1-ols and anti-2-amino-1-arylpropan-1,3-diols were prepared selectively through elaboration of trans-4-aryl-3-chloro-β-lactams. In addition, a number of 2-(azidomethyl)aziridines was converted into novel 2-[(1,2,3-triazol-1-yl)methyl]aziridines by Cu(I)-catalyzed azide-alkyne cycloaddition, followed by microwave-assisted, regioselective ring opening by dialkylamine towards 1-(2,3-diaminopropyl)-1,2,3-triazoles. Although most of these compounds exhibited weak antiplasmodial activity, six representatives showed moderate antiplasmodial activity against both a chloroquine-sensitive and a chloroquine-resistant strain of Plasmodium falciparum with IC50-values of ≤25 μM.

Graphical Abstract
Introduction
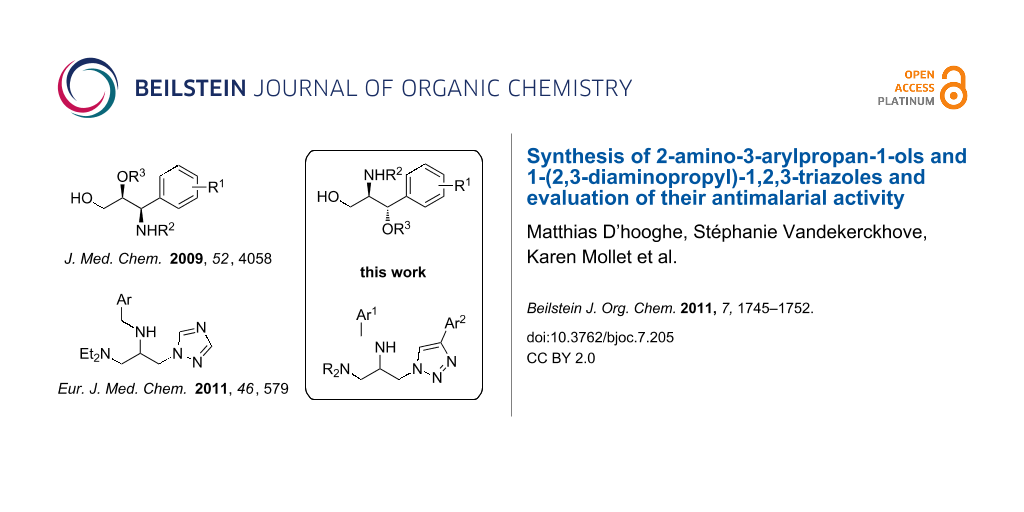
Malaria remains a major issue in health control, especially in developing countries. This disease affects 40% of the global population, causing an annual mortality of one million people [1]. Despite recent advances in the development of a vaccine against malaria, chemotherapy remains the most viable alternative towards treatment of the disease [2]. In light of the rapid emergence of multiple drug resistance to clinically established antimalarial drugs, however, there is a compelling need to introduce new chemicals that can overcome this resistance. In 2007, nitrogen-analogues of glycerol, which have a long-standing tradition in medicine as β-blockers, were introduced as a novel class of antimalarials [3]. Prior to this, the well known β-blocker propranolol was shown to inhibit infection of erythrocytes by P. falciparum, as well as to reduce the parasitaemia of P. berghei infections in vivo [4,5]. In light of the biological potential of these compounds, continuous efforts have been devoted to the preparation of structurally diverse analogues bearing a functionalized propane skeleton [6-8]. In that respect, we have been engaged in the stereoselective synthesis of syn-2-alkoxy-3-amino-3-arylpropan-1-ols 1 by reductive ring opening of the corresponding β-lactams, which were shown to be of great importance as novel antiplasmodial agents (Figure 1) [6]. More recently, we reported 1,2,3-triaminopropanes 2 as a new class of antimalarial compounds (Figure 1), prepared through microwave-assisted, regioselective ring opening of the corresponding 2-(aminomethyl)aziridines by diethylamine [8]. Nonetheless, a number of challenges with regard to structure–activity relationship studies of functionalized aminopropanes remain unaddressed, especially concerning the screening of structural analogues of aminopropanes 1 and 2.
![[1860-5397-7-205-1]](/bjoc/content/figures/1860-5397-7-205-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Functionalized aminopropanes as potential antimalarial agents.
Figure 1: Functionalized aminopropanes as potential antimalarial agents.
In the present paper, the synthesis of racemic anti-2-aminopropan-1-ols 3 is described as a variant of the syn-3-aminopropan-1-ols 1 synthesis (regio- and stereoisomerism with respect to the relative position of the amino group NHR2 and the oxygen substituent OR3, Figure 1), by applying a different synthetic route. Furthermore, a new synthetic approach is disclosed towards racemic aminopropanes 4 bearing a 1,2,3-triazole moiety, as structural analogues of the previously reported 1,2,4-triazoles 2 (Figure 1). Both classes of functionalized aminopropanes 3 and 4 were tested for their antiplasmodial activity.
Results and Discussion
Synthesis
Within azaheterocyclic chemistry, aziridines [9-17] and β-lactams [18-27] are extraordinary classes of strained compounds with diverse synthetic and biological applications. In previous works, we have elaborated the synthetic potential of 3-chloroazetidin-2-ones with a focus on stereoselectivity, thus providing convenient entries into, e.g., aziridines, azetidines and β-aminoalcohols [28-31]. In continuation of our interest in the use of functionalized β-lactams as synthons for further elaboration, racemic trans-4-aryl-3-chloro-β-lactams 5 were prepared by treatment of N-(arylmethylidene)alkylamines (synthesized in high yields through condensation of the corresponding benzaldehydes with the appropriate primary amines in CH2Cl2 in the presence of anhydrous MgSO4) with 1.5 equiv of chloroacetyl chloride and 3 equiv of 2,6-lutidine in benzene according to a literature protocol [30]. Subsequently, β-lactams 5 were subjected to LiAlH4-mediated reductive ring opening, furnishing either 2-aminopropan-1-ols 6a–c, by using two molar equiv of LiAlH4 in Et2O under reflux for 20–80 h, or trans-2-aryl-3-(hydroxymethyl)aziridines 7a–h by applying milder reactions conditions (i.e., one molar equiv of LiAlH4 in Et2O at room temperature for 5–8 h) (Scheme 1) [30].
![[1860-5397-7-205-i1]](/bjoc/content/inline/1860-5397-7-205-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of aminopropanols 6, 8 and 9.
Scheme 1: Synthesis of aminopropanols 6, 8 and 9.
As aziridines are known to be versatile synthetic intermediates for the preparation of a variety of ring-opened and ring-expanded amines, the aziridines 7 were deployed as substrates for the stereoselective synthesis of functionalized aminopropanols. In accordance with a literature approach [30], the nonactivated trans-2-aryl-3-(hydroxymethyl)aziridines 7 were regio- and stereoselectively converted into anti-2-amino-3-aryl-3-methoxypropan-1-ols 8a–e through heating in methanol under reflux (Scheme 1). Furthermore, in order to provide access to the class of 2-aminopropan-1,3-diols, aziridines 7 were evaluated for the first time as substrates for a water-induced aziridine ring opening in an acidic medium. Thus, treatment of trans-2-aryl-3-(hydroxymethyl)aziridines 7 with one equiv of para-toluenesulfonic acid in a H2O/THF (1/1) solvent system [32] furnished novel anti-2-amino-1-arylpropan-1,3-diols 9a–d in good yields after 3 h at 40 °C, again in a regio- and stereospecific way (Scheme 1). The observed regio- and stereoselectivity in aminopropanols 8 and 9 can be rationalized by considering the ring opening of the aziridine moiety at C2 due to benzylic stabilization of the developing carbenium ion in an SN2 fashion [30].
As the use of imines bearing a N-tert-butyl group in combination with a substituent in the ortho-position of the aromatic ring is known to afford the corresponding cis-4-aryl-3-chloroazetidin-2-ones as the major stereoisomers after condensation with chloroketene in benzene [30], racemic cis-3-chloro-β-lactams 10a,b were prepared and converted into cis-2-aryl-3-(hydroxymethyl)aziridines 11a,b upon treatment with two molar equiv of LiAlH4 in Et2O under reflux for 15 h (Scheme 2). Next, the aziridines 11, which have previously been shown to be unreactive towards LiAlH4 and methanol and thus unable to undergo ring opening [30], were used as substrates for a water-induced ring opening through initial protonation of the aziridine ring with p-TsOH. Although more drastic reaction conditions were required compared to the ring opening of trans-2-aryl-3-(hydroxymethyl)aziridines 7 (3 equiv p-TsOH, Δ, 30 h instead of 1 equiv p-TsOH, 40 °C, 3 h), novel syn-aminopropanols 12a,b were obtained in a selective and convenient way (Scheme 2, yields after purification), providing the first example of the ring opening of this type of aziridines. Also in this case, the observed regio- and stereoselectivity in the formation of aminopropanols 12 can be rationalized by considering the ring opening of the aziridine moiety at C2 due to benzylic stabilization of the developing carbenium ion in an SN2 fashion [30]. The formation of the other regio- and stereoisomers was excluded based on detailed spectroscopic analysis.
![[1860-5397-7-205-i2]](/bjoc/content/inline/1860-5397-7-205-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of syn-aminopropan-1,3-diols 12.
Scheme 2: Synthesis of syn-aminopropan-1,3-diols 12.
It should be noted that both diastereomeric antipodes of the class of 1-aryl-2-aminopropan-1,3-diols, i.e., anti- and syn-aminopropanols 9 and 12, can now be prepared selectively through choice of the appropriate imine for the Staudinger synthesis of the starting β-lactams.
Given the recently disclosed antiplasmodial activities of a number of 2,3-diamino-1-(1,2,4-triazol-1-yl)propanes [8], the second objective of this work was the preparation of new analogues bearing a 1,2,3-triazole moiety instead. A powerful methodology towards the synthesis of functionalized 1,2,3-triazoles involves the Cu(I)-catalyzed azide-alkyne Huisgen cycloaddition (CuAAC) [33], which has gained major interest from the synthetic community due to its high efficiency and selectivity. Eligible substrates to perform “click chemistry” [34] incorporate an azide group and an aziridine ring in their structure, for example in 2-(azidomethyl)aziridines, thus providing a direct access to 2-[(1,2,3-triazol-1-yl)methyl]aziridines through Cu-catalyzed reaction with alkynes [35].
In this work, nonactivated N-(arylmethyl)aziridines were selected as substrates, as previous research had revealed the importance of N-benzyl, N-chlorobenzyl and N-methoxybenzyl groups in functionalized aminopropanes with regard to their antiplasmodial activity [6,8]. Thus, a number of racemic 2-(azidomethyl)aziridines 14 was prepared by reaction of sodium azide with 2-(bromomethyl)aziridines 13 [36-38], employing the electrophilicity of the latter as a convenient handle for their connection to other moieties. In this way, novel 2-(azidomethyl)aziridines 14a–e were prepared in good yields by treatment of bromides 13 with two equiv of sodium azide in DMSO at 80 °C for 16 h (CAUTION) (Scheme 3). Subsequently, a CuI-catalyzed 1,3-cycloaddition of N-(arylmethyl)aziridine azides 14 was evaluated for the first time by utilizing one equiv of an arylacetylene in CH3CN under reflux for 16 h, furnishing a direct entry towards new 2-[(1,2,3-triazol-1-yl)methyl]aziridines 15a–f in a highly efficient and selective way (Scheme 3).
![[1860-5397-7-205-i3]](/bjoc/content/inline/1860-5397-7-205-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2-[(1,2,3-triazol-1-yl)methyl]aziridines 15 and their transformation into 1,2,3-triaminopropanes 16.
Scheme 3: Synthesis of 2-[(1,2,3-triazol-1-yl)methyl]aziridines 15 and their transformation into 1,2,3-triami...
The final step comprised ring opening of the aziridine moiety in compounds 15 by diethylamine to afford functionalized aminopropanes as potential antimalarial agents. With the intention to introduce a diethylamino group, a microwave-promoted ring opening of analogous aziridines by diethylamine with the aid of the Et2NH·HCl/Et2NH system was employed [8]. In this way, protonation of the aziridine ring provides a highly electrophilic aziridinium intermediate, which is prone to undergo nucleophilic ring opening. In order to drive the reaction to completion, and to avoid competition between chloride- and diethylamine-induced ring opening, a large excess of Et2NH·HCl (20 equiv) and an additional amount of diethylamine (10 equiv) was used. Thus, heating of the aziridines 15 at 140 °C in CH3CN under microwave irradiation resulted in full and selective conversion to the desired new triaminopropanes 16a–e after 2 h (Scheme 3), which were purified by column chromatography (SiO2) in order to obtain analytically pure samples. Furthermore, in addition to the use of diethylamine, the introduction of a dimethylamino group was considered in order to compare the contribution of this moiety to the potential antiplasmodial activity with that of a diethylamino group. This objective was achieved by treatment of aziridines 15 with 20 equiv of Me2NH·HCl in CH3CN at 140 °C for 2 h under microwave irradiation, resulting in dimethylaminopropanes 16f,g in good yields (Scheme 3). This result showed that this microwave-assisted methodology for the ring opening of nonactivated aziridines can be further extended towards the use of other secondary amines. In order to introduce structural diversity within these molecules, different substituent patterns at the aromatic rings (R1, R2) in aminopropanes 16a–g were realized as well.
In view of the biological potential of aminopropanes in general, compounds 6, 8, 9, 12 and 16 were subsequently screened for their antiplasmodial activity.
In addition, aziridines 14 and 15 were tested against the malaria parasite Plasmodium falciparum, too.
Biological evaluation
At first, compounds 6a–c, 8a–e, 9a–d, 12a,b, 14a–e, 15a–f and 16a–g were screened for in vitro antiplasmodial activity. All samples were tested in triplicate on one occasion against a chloroquine-sensitive (CQS) strain of P. falciparum (D10). Those samples showing antiplasmodial activity were then tested in triplicate on one occasion against a chloroquine-resistant (CQR) strain of P. falciparum (Dd2) and screened for in vitro cytotoxicity against a Chinese hamster ovary (CHO) cell-line, in triplicate on one occasion. The antiplasmodial and cytotoxicity assays were performed as described previously [8,39,40].
The results from the biological study are summarized in Table 1. Although most of these compounds were shown to possess weak or no antiplasmodial activity, eight of them (i.e., compounds 6a, 9a,b, 15c,d, 16a,b,f) were identified as potentially interesting for further study with IC50-values of ≤25 μM. Moreover, these compounds, with the exception of triaminopropanes 16a,b, also proved to be active against a chloroquine-resistant strain of P. falciparum (Dd2). In addition, the in vitro cytotoxicity results showed that only compounds 6a and 16f have lower selectivity with SI’s of 11 and 16, respectively, whereas the other compounds did not show cytotoxicity at the concentrations tested.
Table 1: IC50-values of compounds 6, 8, 9, 12, 14, 15 and 16 tested for in vitro antiplasmodial activity and cytotoxicity.
| Compound | R1 | R2 | R3 |
D10:
IC50 (μM) |
Dd2:
IC50 (μM) |
CHO:
IC50 (μM) |
RIa | SIb |
|---|---|---|---|---|---|---|---|---|
| 6a | 4-Cl | Bn | – | 12.58 | 10.88 | 137.90 | 0.9 | 11 |
| 6b | 4-OMe | iBu | – | 281.58 | ND | ND | ND | ND |
| 6c | 2-F | n-Pr | – | 217.34 | ND | ND | ND | ND |
| 8a | H | iPr | – | 369.70 | ND | ND | ND | ND |
| 8b | H | Bn | – | 143.79 | ND | ND | ND | ND |
| 8c | 4-Cl | Bn | – | 198.56 | ND | ND | ND | ND |
| 8d | 4-OMe | iBu | – | 38.56 | 25.69 | >530 | 0.7 | ND |
| 8e | 2-F | n-Pr | – | 38.54 | 21.92 | >530 | 0.6 | ND |
| 9a | 4-Cl | Bn | – | 25.22 | 8.47 | >530 | 0.3 | ND |
| 9b | 2-Cl | Bn | – | 21.18 | 13.57 | >530 | 0.6 | ND |
| 9c | 4-Me | Bn | – | 129.86 | ND | ND | ND | ND |
| 9d | H | 4-ClBn | – | 80.68 | ND | ND | ND | ND |
| 12a | OMe | – | – | 98.68 | ND | ND | ND | ND |
| 12b | F | – | – | 60.46 | ND | ND | ND | ND |
| 14a | H | – | – | 230.40 | 466.81 | >530 | 2 | ND |
| 14b | 4-Cl | – | – | 100.82 | ND | ND | ND | ND |
| 14c | 4-OMe | – | – | 160.50 | ND | ND | ND | ND |
| 14d | 2-Cl | – | – | 93.68 | ND | ND | ND | ND |
| 14e | 3-Cl | – | – | 179.86 | ND | ND | ND | ND |
| 15a | H | H | – | 34.44 | 106.97 | >530 | 3.1 | ND |
| 15b | 4-Cl | H | – | 41.32 | ND | ND | ND | ND |
| 15c | 4-OMe | H | – | 25.69 | 55.65 | >530 | 2.2 | ND |
| 15d | H | Me | – | 20.43 | 20.99 | >530 | 1 | ND |
| 15e | H | OMe | – | 40.54 | ND | ND | ND | ND |
| 15f | 2-Cl | H | – | 32.33 | 25.00 | >530 | 0.8 | ND |
| 16a | H | H | Et | 22.09 | 231.47 | >530 | 10.5 | ND |
| 16b | 4-Cl | H | Et | 25.86 | 176.40 | >530 | 6.8 | ND |
| 16c | 4-OMe | H | Et | 139.61 | ND | ND | ND | ND |
| 16d | H | Me | Et | 32.55 | ND | ND | ND | ND |
| 16e | H | OMe | Et | 171.37 | ND | ND | ND | ND |
| 16f | 2-Cl | H | Me | 11.33 | 13.03 | 181.83 | 1.2 | 16.1 |
| 16g | H | H | Me | 69.58 | ND | ND | ND | ND |
| CQ |
19.14 ng/mL
(n = 6) |
75.56 ng/mL
(n = 5) |
3.9 | |||||
| Emetine |
0.27
(n = 6) |
|||||||
aRI (Resistance Index) = IC50 Dd2/IC50 D10; bSI (Selectivity Index) = IC50 CHO/IC50 D10; ND = not determined; n = number of data sets averaged. The more hydrophobic samples were added to the parasites as a suspension, meaning that for these samples the reported IC50-value might be an underestimation of the activity.
Although the aziridine moiety was initially only considered as a synthetically useful entity, two 2-[(1,2,3-triazol-1-yl)methyl]aziridines (15c and 15d) were also found to exhibit weak antiplasmodial activity. On the other hand, the aminopropane unit has again proven its value as a template for the preparation of novel antimalarial agents, as a variety of structurally different aminopropanes were demonstrated to exhibit weak to moderate antiplasmodial activity. In particular, antiplasmodial assays against a chloroquine-sensitive strain of P. falciparum (D10) showed activity for 2-aminopropan-1-ol 6a, 2-aminopropan-1,3-diols 9a,b and triaminopropanes 16a,b,f with IC50-values between 11.3 and 25.9 μM. Moreover, screening against a chloroquine-resistant strain of P. falciparum (Dd2) revealed antiplasmodial activity for 2-aminopropan-1-ol 6a, 2-aminopropan-1,3-diols 9a,b and triaminopropane 16f with IC50-values between 8.5 and 13.6 μM.
From a structure–activity relationship viewpoint, the presence of a chlorinated aromatic ring seems to contribute to the antiplasmodial activity of these functionalized aminopropanes, and the introduction of a dimethylamino group at the expense of a diethylamino moiety might provide better activities in some cases. It is noteworthy that these compounds were synthesized in racemic form, and it is conceivable that enantiomerically pure variants could deliver superior activities. It should also be noted that, in general, the bioactivities reported in this paper are less pronounced as compared to those described in literature precedents on the synthesis and evaluation of functionalized aminopropanes [6,8].
Conclusion
In summary, a variety of 2-amino-3-arylpropan-1-ols, anti-2-amino-3-aryl-3-methoxypropan-1-ols and anti-2-amino-1-arylpropan-1,3-diols were prepared selectively through elaboration of trans-4-aryl-3-chloro-β-lactams. Furthermore, a number of 2-(azidomethyl)aziridines were converted into novel 2-[(1,2,3-triazol-1-yl)methyl]aziridines by Cu(I)-catalyzed azide-alkyne cycloaddition, followed by microwave-assisted, regioselective ring opening by diethyl- or dimethylamine towards the corresponding 1-(2,3-diaminopropyl)-1,2,3-triazoles. From a synthetic viewpoint, new insights were provided concerning the water-induced ring opening of nonactivated cis- and trans-2-aryl-3-(hydroxymethyl)aziridines and with respect to the synthesis and use of 1-arylmethyl-2-(azidomethyl)aziridines for azide-alkyne cycloaddition reactions. From a biological viewpoint, most of these compounds exhibited weak antiplasmodial activity, although six representatives showed moderate antiplasmodial activity against both a chloroquine-sensitive and a chloroquine-resistant strain of P. falciparum with IC50-values of ≤25 μM.
Experimental
General information regarding NMR, IR, MS and elemental analyses, melting point measurements, and microwave reaction conditions can be found in the literature [8].
anti-2-(N-Benzylamino)-1-(4-chlorophenyl)propan-1,3-diol (9a)
Recrystallization from hexane/EtOAc (1:25), white crystals, 95%. Mp 192.3 °C; 1H NMR (300 MHz, CDCl3) δ 2.39 (s, 2H), 3.12–3.18 (m, 1H), 3.54 (dd, J = 13.0, 3.6 Hz, 1H), 3.82 (dd, J = 13.0, 5.8 Hz, 1H), 4.27 and 4.32 (2 d, J = 13.2 Hz, 2 × 1H), 5.24 (d, J = 2.2 Hz, 1H), 7.04–7.07, 7.19–7.36, 7.46–7.49 and 7.69–7.71 (4 m, 2H, 3H, 2H, 2H); 13C NMR (75 MHz, CDCl3) δ 51.0, 59.4, 62.6, 72.0, 127.2, 128.1, 128.7, 128.8, 128.9, 133.4, 137.0, 139.1; IR (cm−1) νmax: 3437 (OH), 3342 (NH), 2926, 1448, 1162, 1008, 681; MS (70 eV) m/z (%): 292/4 (M+ + 1, 100); HRMS (ESI): [M + H]+ calcd for C16H19ClNO2, 292.1104; found, 292.1112.
syn-2-(N-tert-Butylamino)-1-(2-methoxyphenyl)propan-1,3-diol (12a)
Rf 0.07 (EtOAc), white crystals, 30%. Mp 112.8 °C; 1H NMR (300 MHz, CDCl3) δ 1.03 (s, 9H), 2.77 (ddd, J = 7.1, 3.0, 3.0 Hz, 1H), 3.50–3.51 (m, 2H), 3.86 (s, 3H), 4.80 (d, J = 7.1 Hz, 1H), 6.87–6.89, 6.98–7.03, 7.22–7.28 and 7.47–7.50 (4 m, 4 × 1H); 13C NMR (75 MHz, CDCl3) δ 30.0, 50.8, 55.6, 58.1, 63.7, 68.5, 110.4, 121.3, 127.7, 128.4, 130.5, 156.5; IR (cm−1) νmax: 3391 (NH), 3313 (OH), 3058, 3004, 2987, 2957, 2927, 2873, 2838, 1492, 1467, 1438, 1368, 1242, 1066, 1050, 1027, 755; MS (70 eV) m/z (%): 254 (M+ + 1, 100); HRMS (ESI): [M + H]+calcd for C14H24NO3, 254.1756; found, 254.1766.
2-Azidomethyl-1-(phenylmethyl)aziridine (14a)
Rf 0.20 (hexane/EtOAc 4:1), yellow oil, 70%. 1H NMR (300 MHz, CDCl3) δ 1.51 (d, J = 6.1 Hz, 1H), 1.79 (d, J = 3.3 Hz, 1H), 1.79–1.86 (m, 1H), 3.18 and 3.27 (2 dd, J = 12.9, 6.6, 4.4 Hz, 2H) 3.38 and 3.58 (2 d, J = 13.2 Hz, 2H), 7.24–7.36 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 32.2, 37.9, 53.8, 64.3, 127.4, 128.3, 128.6, 138.8; IR (cm−1) νmax: 2088 (N3), 1453, 1357, 1322, 1255, 1159, 1062, 1028, 907, 732, 697; MS (70 eV) m/z (%): 189 (M+ + 1, 100); HRMS (ESI): [M + H]+ calcd for C10H13N4, 189.1140; found, 189.1141.
1-Phenylmethyl-2-[(4-phenyl-1,2,3-triazol-1-yl)methyl]aziridine (15a)
Rf 0.23 (CHCl3/MeOH 98:2), viscous light-brown oil, 61%. 1H NMR (300 MHz, CDCl3) δ 1.65 (d, J = 6.6 Hz, 1H), 1.88 (d, J = 3.3 Hz, 1H), 2.01–2.08 (m, 1H), 3.09 and 3.67 (2 d, J = 12.9 Hz, 2H), 3.93 and 4.70 (2 dd, J = 14.3, 8.2, 3.3 Hz, 2H), 7.06–7.43 (m, 8H), 7.49 (s, 1H), 7.66–7.69 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 32.4, 38.3, 53.5, 64.5, 119.9, 125.9, 127.7, 128.1, 128.4, 128.6, 128.7, 130.8, 138.2, 147.9; IR (cm−1) νmax: 2919, 1453, 1358, 1225, 1075, 1046, 1027, 763, 731, 694; MS (70 eV) m/z (%): 291 (M+ + 1, 100); HRMS (ESI): [M + H]+ calcd for C18H19N4, 291.1610; found, 291.1613.
3-Diethylamino-2-(phenylmethyl)amino-1-(4-phenyl-1,2,3-triazol-1-yl)propane (16a)
Rf 0.19 (CHCl3/MeOH 97:3), light-brown oil, 56%. 1H NMR (300 MHz, CDCl3) δ 0.92 (t, J = 7.2 Hz, 6H), 2.26–2.52 (m, 6H), 3.05–3.13 (m, 1H), 3.74 and 3.81 (2 d, J = 13.5 Hz, 2H), 4.36 and 4.44 (2 dd, J = 14.2, 4.9, 4.7 Hz, 2H), 7.21–7.44 and 7.82–7.84 (2 m, 10H), 7.86 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 11.8, 47.1, 52.0, 52.3, 55.0, 55.4, 121.2, 125.8, 127.2, 128.1, 128.2, 128.6, 128.9, 130.9, 140.2, 147.5; IR (cm−1) νmax: 2967, 1462, 1454, 1073, 762, 734, 694; MS (70 eV) m/z (%): 364 (M+ + 1, 100); HRMS (ESI): [M + H]+ calcd for C22H30N5, 364.2501; found, 364.2503.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 127.5 KB | Download |
References
-
WHO Fact sheet on malaria N°94, January 2009.
Return to citation in text: [1] -
Gemma, S.; Travagli, V.; Savini, L.; Novellino, E.; Campiani, G.; Butini, S. Recent Pat. Antiinfect. Drug. Discov. 2010, 5, 195.
Return to citation in text: [1] -
Robin, A.; Brown, F.; Bahamontes-Rosa, N.; Wu, B.; Beitz, E.; Kun, J. F. J.; Flitsch, S. L. J. Med. Chem. 2007, 50, 4243. doi:10.1021/jm070553l
Return to citation in text: [1] -
Murphy, S. C.; Harrison, T.; Hamm, H. E.; Lomasney, J. W.; Mohandas, N.; Haldar, K. PLoS Med. 2006, 3, 2403. doi:10.1371/journal.pmed.0030528
Return to citation in text: [1] -
Harrison, T.; Samuel, B. U.; Akompong, T.; Hamm, H.; Mohandas, N.; Lomasney, J. W.; Haldar, K. Science 2003, 301, 1734. doi:10.1126/science.1089324
Return to citation in text: [1] -
D’hooghe, M.; Dekeukeleire, S.; Mollet, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. J. Med. Chem. 2009, 52, 4058. doi:10.1021/jm9002632
Return to citation in text: [1] [2] [3] [4] -
Pérez-Silanes, S.; Berrade, L.; García-Sánchez, R. N.; Mendoza, A.; Galiano, S.; Pérez-Solórzano, B. M.; Nogal-Ruiz, J. J.; Martínez-Fernández, A. R.; Aldana, I.; Monge, A. Molecules 2009, 14, 4120. doi:10.3390/molecules14104120
Return to citation in text: [1] -
D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Zwanenburg, B.; ten Holte, P. Top. Curr. Chem. 2001, 216, 93. doi:10.1007/3-540-44726-1_3
Return to citation in text: [1] -
Sweeney, J. B. Chem. Soc. Rev. 2002, 31, 247. doi:10.1039/b006015l
Return to citation in text: [1] -
Hu, X. E. Tetrahedron 2004, 60, 2701. doi:10.1016/j.tet.2004.01.042
Return to citation in text: [1] -
Tanner, D. Angew. Chem., Int. Ed. Engl. 1994, 33, 599. doi:10.1002/anie.199405991
Return to citation in text: [1] -
Osborn, H. M. I.; Sweeney, J. Tetrahedron: Asymmetry 1997, 8, 1693. doi:10.1016/S0957-4166(97)00177-8
Return to citation in text: [1] -
McCoull, W. M.; Davis, F. A. Synthesis 2000, 1347. doi:10.1055/s-2000-7097
Return to citation in text: [1] -
Watson, I. D. G.; Yu, L.; Yudin, A. K. Acc. Chem. Res. 2006, 39, 194. doi:10.1021/ar050038m
Return to citation in text: [1] -
Padwa, A.; Murphree, S. S. ARKIVOC 2006, No. iii, 6.
Return to citation in text: [1] -
Singh, G. S.; D’hooghe, M.; De Kimpe, N. Chem. Rev. 2007, 107, 2080. doi:10.1021/cr0680033
Return to citation in text: [1] -
Ojima, I.; Delaloge, F. Chem. Soc. Rev. 1997, 26, 377. doi:10.1039/cs9972600377
Return to citation in text: [1] -
Alcaide, B.; Almendros, P. Synlett 2002, 381. doi:10.1055/s-2002-20448
Return to citation in text: [1] -
Fisher, J. F.; Meroueh, S. O.; Mobashery, S. Chem. Rev. 2005, 105, 395. doi:10.1021/cr030102i
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Rev. 2007, 107, 4437. doi:10.1021/cr0307300
Return to citation in text: [1] -
Ojima, I. Acc. Chem. Res. 1995, 28, 383. doi:10.1021/ar00057a004
Return to citation in text: [1] -
Singh, G. S. Tetrahedron 2003, 59, 7631. doi:10.1016/S0040-4020(03)01099-8
Return to citation in text: [1] -
Singh, G. S.; D'hooghe, M.; De Kimpe, N. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.; Ramsden, C.; Scriven, E.; Taylor, R., Eds.; Elsevier: Oxford, 2008; Vol. 2, p 1. doi:10.1016/B978-008044992-0.00201-7
Return to citation in text: [1] -
Xing, B.; Rao, J.; Liu, R. Mini-Rev. Med. Chem. 2008, 8, 455. doi:10.2174/138955708784223558
Return to citation in text: [1] -
Aranda, M. T.; Perez-Faginas, P.; Gonzalez-Muniz, R. Curr. Org. Synth. 2009, 6, 325. doi:10.2174/157017909788921899
Return to citation in text: [1] -
D’hooghe, M.; Dekeukeleire, S.; Leemans, E.; De Kimpe, N. Pure Appl. Chem. 2010, 82, 1749. doi:10.1351/PAC-CON-09-09-39
Return to citation in text: [1] -
Van Driessche, B.; Van Brabandt, W.; D’hooghe, M.; Dejaegher, Y.; De Kimpe, N. Tetrahedron 2006, 62, 6882. doi:10.1016/j.tet.2006.04.104
Return to citation in text: [1] -
D’hooghe, M.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2008, 6, 1190. doi:10.1039/b719686e
Return to citation in text: [1] -
D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Mollet, K.; D’hooghe, M.; De Kimpe, N. J. Org. Chem. 2011, 76, 264. doi:10.1021/jo1020932
Return to citation in text: [1] -
Manaka, T.; Nagayama, S.-I.; Desadee, W.; Yajima, N.; Kumamoto, T.; Watanabe, T.; Ishikawa, T.; Kawahata, M.; Yamaguchi, K. Helv. Chim. Acta 2007, 90, 128. doi:10.1002/hlca.200790006
Return to citation in text: [1] -
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302. doi:10.1039/b904091a
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
Return to citation in text: [1] -
Jamookeeah, C. E.; Beadle, C. D.; Harrity, J. P. A. Synthesis 2009, 133. doi:10.1055/s-0028-1083270
Return to citation in text: [1] -
De Kimpe, N.; Jolie, R.; De Smaele, D. J. Chem. Soc., Chem. Commun. 1994, 1221. doi:10.1039/C39940001221
Return to citation in text: [1] -
D’hooghe, M.; Waterinckx, A.; Kimpe, N. J. Org. Chem. 2005, 70, 227. doi:10.1021/jo048486f
Return to citation in text: [1] -
D’hooghe, M.; Rottiers, M.; Jolie, R.; De Kimpe, N. Synlett 2005, 931. doi:10.1055/s-2005-864801
Return to citation in text: [1] -
Kamdem Waffo, A. F.; Coombes, P. H.; Crouch, N. R.; Mulholland, D. A.; El Amin, S. M. M.; Smith, P. J. Phytochemistry 2007, 68, 663. doi:10.1016/j.phytochem.2006.10.011
Return to citation in text: [1] -
Khanye, S. D.; Smith, G. S.; Lategan, C.; Smith, P. J.; Gut, J.; Rosenthal, P. J.; Chibale, K. J. Inorg. Biochem. 2010, 104, 1079. doi:10.1016/j.jinorgbio.2010.06.005
Return to citation in text: [1]
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 6. | D’hooghe, M.; Dekeukeleire, S.; Mollet, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. J. Med. Chem. 2009, 52, 4058. doi:10.1021/jm9002632 |
| 7. | Pérez-Silanes, S.; Berrade, L.; García-Sánchez, R. N.; Mendoza, A.; Galiano, S.; Pérez-Solórzano, B. M.; Nogal-Ruiz, J. J.; Martínez-Fernández, A. R.; Aldana, I.; Monge, A. Molecules 2009, 14, 4120. doi:10.3390/molecules14104120 |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 4. | Murphy, S. C.; Harrison, T.; Hamm, H. E.; Lomasney, J. W.; Mohandas, N.; Haldar, K. PLoS Med. 2006, 3, 2403. doi:10.1371/journal.pmed.0030528 |
| 5. | Harrison, T.; Samuel, B. U.; Akompong, T.; Hamm, H.; Mohandas, N.; Lomasney, J. W.; Haldar, K. Science 2003, 301, 1734. doi:10.1126/science.1089324 |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 3. | Robin, A.; Brown, F.; Bahamontes-Rosa, N.; Wu, B.; Beitz, E.; Kun, J. F. J.; Flitsch, S. L. J. Med. Chem. 2007, 50, 4243. doi:10.1021/jm070553l |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 2. | Gemma, S.; Travagli, V.; Savini, L.; Novellino, E.; Campiani, G.; Butini, S. Recent Pat. Antiinfect. Drug. Discov. 2010, 5, 195. |
| 32. | Manaka, T.; Nagayama, S.-I.; Desadee, W.; Yajima, N.; Kumamoto, T.; Watanabe, T.; Ishikawa, T.; Kawahata, M.; Yamaguchi, K. Helv. Chim. Acta 2007, 90, 128. doi:10.1002/hlca.200790006 |
| 18. | Ojima, I.; Delaloge, F. Chem. Soc. Rev. 1997, 26, 377. doi:10.1039/cs9972600377 |
| 19. | Alcaide, B.; Almendros, P. Synlett 2002, 381. doi:10.1055/s-2002-20448 |
| 20. | Fisher, J. F.; Meroueh, S. O.; Mobashery, S. Chem. Rev. 2005, 105, 395. doi:10.1021/cr030102i |
| 21. | Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Rev. 2007, 107, 4437. doi:10.1021/cr0307300 |
| 22. | Ojima, I. Acc. Chem. Res. 1995, 28, 383. doi:10.1021/ar00057a004 |
| 23. | Singh, G. S. Tetrahedron 2003, 59, 7631. doi:10.1016/S0040-4020(03)01099-8 |
| 24. | Singh, G. S.; D'hooghe, M.; De Kimpe, N. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.; Ramsden, C.; Scriven, E.; Taylor, R., Eds.; Elsevier: Oxford, 2008; Vol. 2, p 1. doi:10.1016/B978-008044992-0.00201-7 |
| 25. | Xing, B.; Rao, J.; Liu, R. Mini-Rev. Med. Chem. 2008, 8, 455. doi:10.2174/138955708784223558 |
| 26. | Aranda, M. T.; Perez-Faginas, P.; Gonzalez-Muniz, R. Curr. Org. Synth. 2009, 6, 325. doi:10.2174/157017909788921899 |
| 27. | D’hooghe, M.; Dekeukeleire, S.; Leemans, E.; De Kimpe, N. Pure Appl. Chem. 2010, 82, 1749. doi:10.1351/PAC-CON-09-09-39 |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 9. | Zwanenburg, B.; ten Holte, P. Top. Curr. Chem. 2001, 216, 93. doi:10.1007/3-540-44726-1_3 |
| 10. | Sweeney, J. B. Chem. Soc. Rev. 2002, 31, 247. doi:10.1039/b006015l |
| 11. | Hu, X. E. Tetrahedron 2004, 60, 2701. doi:10.1016/j.tet.2004.01.042 |
| 12. | Tanner, D. Angew. Chem., Int. Ed. Engl. 1994, 33, 599. doi:10.1002/anie.199405991 |
| 13. | Osborn, H. M. I.; Sweeney, J. Tetrahedron: Asymmetry 1997, 8, 1693. doi:10.1016/S0957-4166(97)00177-8 |
| 14. | McCoull, W. M.; Davis, F. A. Synthesis 2000, 1347. doi:10.1055/s-2000-7097 |
| 15. | Watson, I. D. G.; Yu, L.; Yudin, A. K. Acc. Chem. Res. 2006, 39, 194. doi:10.1021/ar050038m |
| 16. | Padwa, A.; Murphree, S. S. ARKIVOC 2006, No. iii, 6. |
| 17. | Singh, G. S.; D’hooghe, M.; De Kimpe, N. Chem. Rev. 2007, 107, 2080. doi:10.1021/cr0680033 |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 6. | D’hooghe, M.; Dekeukeleire, S.; Mollet, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. J. Med. Chem. 2009, 52, 4058. doi:10.1021/jm9002632 |
| 28. | Van Driessche, B.; Van Brabandt, W.; D’hooghe, M.; Dejaegher, Y.; De Kimpe, N. Tetrahedron 2006, 62, 6882. doi:10.1016/j.tet.2006.04.104 |
| 29. | D’hooghe, M.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2008, 6, 1190. doi:10.1039/b719686e |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 31. | Mollet, K.; D’hooghe, M.; De Kimpe, N. J. Org. Chem. 2011, 76, 264. doi:10.1021/jo1020932 |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 30. | D’hooghe, M.; Mollet, K.; Dekeukeleire, S.; De Kimpe, N. Org. Biomol. Chem. 2010, 8, 607. doi:10.1039/b919864d |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 39. | Kamdem Waffo, A. F.; Coombes, P. H.; Crouch, N. R.; Mulholland, D. A.; El Amin, S. M. M.; Smith, P. J. Phytochemistry 2007, 68, 663. doi:10.1016/j.phytochem.2006.10.011 |
| 40. | Khanye, S. D.; Smith, G. S.; Lategan, C.; Smith, P. J.; Gut, J.; Rosenthal, P. J.; Chibale, K. J. Inorg. Biochem. 2010, 104, 1079. doi:10.1016/j.jinorgbio.2010.06.005 |
| 6. | D’hooghe, M.; Dekeukeleire, S.; Mollet, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. J. Med. Chem. 2009, 52, 4058. doi:10.1021/jm9002632 |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 36. | De Kimpe, N.; Jolie, R.; De Smaele, D. J. Chem. Soc., Chem. Commun. 1994, 1221. doi:10.1039/C39940001221 |
| 37. | D’hooghe, M.; Waterinckx, A.; Kimpe, N. J. Org. Chem. 2005, 70, 227. doi:10.1021/jo048486f |
| 38. | D’hooghe, M.; Rottiers, M.; Jolie, R.; De Kimpe, N. Synlett 2005, 931. doi:10.1055/s-2005-864801 |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 35. | Jamookeeah, C. E.; Beadle, C. D.; Harrity, J. P. A. Synthesis 2009, 133. doi:10.1055/s-0028-1083270 |
| 6. | D’hooghe, M.; Dekeukeleire, S.; Mollet, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. J. Med. Chem. 2009, 52, 4058. doi:10.1021/jm9002632 |
| 8. | D’hooghe, M.; Kenis, S.; Vervisch, K.; Lategan, C.; Smith, P. J.; Chibale, K.; De Kimpe, N. Eur. J. Med. Chem. 2011, 46, 579. doi:10.1016/j.ejmech.2010.11.037 |
| 33. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302. doi:10.1039/b904091a |
| 34. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5 |
© 2011 D’hooghe et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)