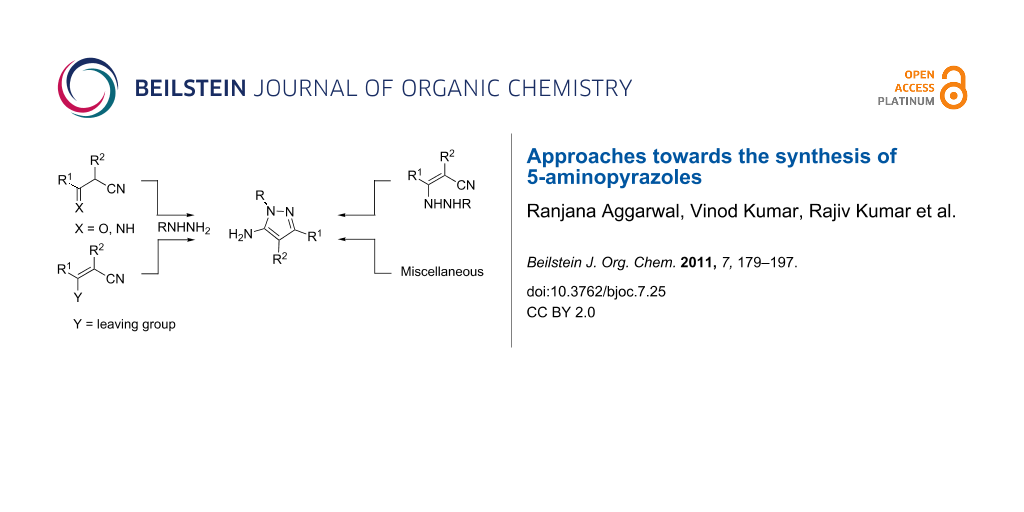
Abstract
The biological and medicinal properties of 5-aminopyrazoles have prompted enormous research aimed at developing synthetic routes to these heterocyles. This review focuses on the biological properties associated with this system. Various synthetic methods developed up to 2010 for these compounds are described, particularly those that involve the reactions of β-ketonitriles, malononitrile, alkylidenemalononitriles and their derivatives with hydrazines, as well as some novel miscellaneous methods.

Graphical Abstract
Review
The 5-aminopyrazole system represents an important heterocyclic template that has attracted considerable interest because of its long history of application in the pharmaceutical and agrochemical industries [1-4]. These compounds have been extensively investigated over the past one hundred years and their chemistry has been reviewed in two books published in 1964 [5] and in 1967 [6].
Structurally simple 5-amino-1-tert-butylpyrazole-4-carboxamide I was found to inhibit p56 Lck [7] (Figure 1). 5-Amino-1-(4-methylphenyl) pyrazole II has been tested as an NPY5 antagonist [8]. 5-Amino-4-benzoyl-3-methylthio-1-(2,4,6-trichlorophenyl)pyrazole III has been reported as a potent corticotrophin-releasing factor-1 (CRF-1) receptor antagonist [9]. 5-Amino-1-(2,6-dichloro-4-(trifluoromethyl)phenyl)-4-(3-methoxyphenyl)-3-methylthiopyrazole IV has been described as a potent GABA inhibitor with selectivity towards insect versus mammalian receptors [10]. The simple N-phenyl amide of 5-amino -1,3-dimethylpyrazole-4-carboxylic acid V has been shown to exhibit antifungal activity [11] (Figure 1). The 5-amino-1-pyrazinyl-3-carboxamidopyrazole derivative VI has been recently reported as a potent antibacterial agent with a very broad spectrum [12]. Recently, components of the mitotic machinery have been targeted in an attempt to develop novel anticancer agents. These include critical signaling kinases such as the Aurora, PLK, and the cyclin-dependent kinases (CDK). Compound VII (AZD1152) is the first Aurora-B selective inhibitor to enter clinical trials [13] (Figure 1).
![[1860-5397-7-25-1]](/bjoc/content/figures/1860-5397-7-25-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Pharmacologically active 5-aminopyrazoles.
Figure 1: Pharmacologically active 5-aminopyrazoles.
Besides the importance of 5-aminopyrazoles as biologically active agents, they are also useful synthons and building blocks for many heterocyclic products and can act as a binucleophile [14-18]. Cyclocondensation of 5-aminopyrazoles with 1,3-dielectrophiles has been extensively used for the preparation of bicyclic nitrogen heterocycles, especially in the preparation of condensed heterocycles such as pyrazolo[3,4-d]pyrimidines, pyrazolo[3,4-b]pyridines, imidazopyrazoles etc.
In view of significant interest in the synthesis of these heterocyclics, we herein report a detailed account of the synthetic methods available for 5-aminopyrazoles.
As pyrazole derivatives do not exist in nature, probably, due to the difficulty in the construction of N–N bond by living organisms, their availability depends on the synthetic methods. A large number of synthetic methods have recently appeared. Some of the important methods are outlined below.
1. Reaction of β-ketonitriles with hydrazines
The most versatile method available for the synthesis of 5-aminopyrazoles involves the condensation of β-ketonitriles with hydrazines. β-Ketonitriles 1 react smoothly with hydrazines to yield 5-aminopyrazoles 3 [19-28]. The reaction apparently involves the nucleophilic attack of the terminal nitrogen of the hydrazine on the carbonyl carbon with the formation of hydrazones 2, which subsequently undergo cyclization by the attack of the other nitrogen on the nitrile carbon to produce 5-aminopyrazoles 3 (Scheme 1). Utilizing this reaction, a large number of 5-amino-1-heteroarylpyrazoles have been synthesized in our laboratory by the reaction of several heteroarylhydrazines with α-cyanoacetophenones [29,30]. The intermediate hydrazones 2 are rarely isolated, though their formation has been reported in the reaction of 2-nitro/2,4-dinitrophenylhydrazines and aryl-α-cyanoacetaldehydes 1 (R2 = aryl, R1 = H) [31].
![[1860-5397-7-25-i1]](/bjoc/content/inline/1860-5397-7-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General equation for the condensation of β-ketonitriles with hydrazines.
Scheme 1: General equation for the condensation of β-ketonitriles with hydrazines.
Recently, the synthesis of biologically active 5-amino-1-heteroaryl-3-trifluoromethylpyrazoles 6 has been achieved by us by the reaction of trifluoroacetylbenzyl cyanide 4 with heteroarylhydrazines [32]. The reaction of 2-hydrazino-4-methylquinoline with α-trifluoroacetylbenzyl cyanide (R = CF3) (4) at room temperature afforded the intermediate hydrazone 5. The hydrazone 5 was characterized by IR and NMR spectroscopy. The IR spectrum of 5 showed a fundamental stretching band due to C≡N at 2179 cm−1. The 19F NMR spectrum of compound 5 showed fluorine signal at δ −65 ppm due to CF3 group confirming the formation of hydrazone 5, which exists as the Z-isomer. From the literature [33], the signal for the CF3 group in trifluoromethylhydrazones appears at δ −64 to −66 ppm for Z-isomers and at δ −67 to −71 ppm for E-isomers. As expected, 5 underwent cyclization in refluxing ethanol to give the corresponding 5-aminopyrazole 6 [32]. α-Acetyl/formylbenzyl cyanide (R = H/CH3) 4 on reaction with heteroarylhydrazines in refluxing ethanol yielded the corresponding 5-amino-4-phenylpyrazoles 6. These compounds were found to be good antibacterial agents (Scheme 2) [34].
![[1860-5397-7-25-i2]](/bjoc/content/inline/1860-5397-7-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of hydrazinoheterocycles with α-phenyl-β-cyanoketones (4).
Scheme 2: Reaction of hydrazinoheterocycles with α-phenyl-β-cyanoketones (4).
The isolation of hydrazones 8 has also been reported during the condensation of cyanoacetaldehyde (7) with hydrazines [35]. These hydrazones 8 were cyclized to the corresponding 5-aminopyrazoles 9 under basic conditions (Scheme 3).
![[1860-5397-7-25-i3]](/bjoc/content/inline/1860-5397-7-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Condensation of cyanoacetaldehyde (7) with hydrazines.
Scheme 3: Condensation of cyanoacetaldehyde (7) with hydrazines.
Recently, Kordik et al [36] treated α-cyano-4-nitroacetophenone (10) with aryl hydrazines in the presence of triethylamine and obtained the corresponding 5-aminopyrazoles 11 in excellent yields. The latter were further converted into their sulfonamide derivatives 12 by reducing the nitro group to an amino group by catalytic hydrogenation followed by treatment with an arylsulfonyl chloride (Scheme 4).
![[1860-5397-7-25-i4]](/bjoc/content/inline/1860-5397-7-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 5-aminopyrazoles and their sulfonamide derivatives.
Scheme 4: Synthesis of 5-aminopyrazoles and their sulfonamide derivatives.
Alternatively, 5-aminopyrazoles 17 containing a cyclohexylmethyl- or phenylmethyl- sulfonamido group at position-3 were prepared by treating β-ketonitriles 16 with a substituted hydrazine in the presence of Et3N in ethanol under reflux conditions. The intermediate 16 was obtained from β-ketoester 15 on treatment with TFA, which in turn was synthesized by condensing 4-(phenylsulfonamidomethyl)cyclohexane carboxylic acid or benzoic acid 13, respectively, with tert-butyl cyanoacetate (14), as illustrated in Scheme 5 [36].
![[1860-5397-7-25-i5]](/bjoc/content/inline/1860-5397-7-25-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 5-aminopyrazoles, containing a cyclohexylmethyl- or phenylmethyl- sulfonamido group at position-3.
Scheme 5: Synthesis of 5-aminopyrazoles, containing a cyclohexylmethyl- or phenylmethyl- sulfonamido group at...
Baraldi et al. [37] utilized this method for the regioselective synthesis of 2-alkyl- or 2-aryl-3-aminothieno[3,4-c]pyrazoles 19. Several alkyl- or arylhydrazine hydrochlorides on condensation with 4-cyano-3-oxotetrahydrothiophene (18) in refluxing ethanol gave the thienopyrazoles in excellent yields. The regioselectivity of this process has been confirmed by the treatment of 18 with phenylhydrazine, which generated a mixture of intermediate hydrazone 20 and 2-phenyl-3-aminothieno[3,4-c]pyrazole (21) (Scheme 6). Hydrazones 20 on treatment with 5% HCl in ethanol underwent cyclization to afford 21.
![[1860-5397-7-25-i6]](/bjoc/content/inline/1860-5397-7-25-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Regioselective synthesis of 3-amino-2-alkyl (or aryl) thieno[3,4-c]pyrazoles 19.
Scheme 6: Regioselective synthesis of 3-amino-2-alkyl (or aryl) thieno[3,4-c]pyrazoles 19.
A novel solid phase synthesis of some 5-aminopyrazoles 24 and their N-acyl and N-sulfonyl derivatives has recently been reported by Watson et al. [38] via the resin supported β-ketonitriles 22 (Scheme 7). The resin supported aminopyrazoles 23 were hydrolysed to yield 24 in excellent yields. The synthesis is versatile and affords compoundswith a known pharmacophoric template ideally suited for combinatorial library generation.
![[1860-5397-7-25-i7]](/bjoc/content/inline/1860-5397-7-25-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Solid supported synthesis of 5-aminopyrazoles.
Scheme 7: Solid supported synthesis of 5-aminopyrazoles.
Another solid phase synthesis of 5-aminopyrazoles has been reported [39] by utilizing enamine nitrile 25 as the starting material (Scheme 8). In this reaction, compound 25 was readily hydrolyzed to afford the β-ketonitrile derivative, i.e., 4-(1-cyano-2-oxoethyl)benzamide 26 which reacted efficiently with hydrazines to give the corresponding 5-aminopyrazoles 27. Subsequent cleavage from the resin afforded 5-aminopyrazoles 28. This new 5-aminopyrazole synthesis is more versatile and efficient than its predecessor as it avoids the use of troublesome β-ketonitrile functionality. This new route is also ideally suited for the synthesis of combinatorial libraries for drug target screening.
![[1860-5397-7-25-i8]](/bjoc/content/inline/1860-5397-7-25-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of 5-aminopyrazoles from resin supported enamine nitrile 25 as the starting material.
Scheme 8: Synthesis of 5-aminopyrazoles from resin supported enamine nitrile 25 as the starting material.
In 2009, an efficient three-component, two-step “catch and release” solid-phase synthesis of 3,4,5-trisubstituted pyrazoles was reported which involved a base-promoted condensation of a 2-sulfonyl- or a 2-carbonyl-acetonitrile derivative (29 or 33) with an isothiocyanate and in situ immobilization of the resulting thiolate anion (30 or 34) on Merrifield resin in the first step. Reaction of the resin-bound sulfonyl intermediate 31 with hydrazine, followed by release from the resin and intramolecular cyclization, afforded 4-arylsulfonyl-3,5-diamino-1H-pyrazoles 32. Reaction of the resin-bound carbonyl intermediate 35 with hydrazine, on the other hand, led to 5-aryl-3-arylamino-1H-pyrazole-4-carbonitriles 36, instead of the 5-aminopyrazole 37, which can be rationalized in terms of the higher reactivity of the carbonyl group of 35 toward hydrazine compared to the cyano group (Scheme 9) [40].
![[1860-5397-7-25-i9]](/bjoc/content/inline/1860-5397-7-25-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Two-step “catch and release” solid-phase synthesis of 3,4,5-trisubstituted pyrazoles.
Scheme 9: Two-step “catch and release” solid-phase synthesis of 3,4,5-trisubstituted pyrazoles.
Gao and Lam recently reported a solid-phase synthesis of 5-aminopyrazoles 42 which were used as precursors for the preparation of pyrazolo[5,1-d][1,2,3,5]tetrazine-4(3H)-ones 43. Resin 39, obtained from Wang resin 38 and a 5-10 fold excess of 1,1′-carbonyldiimidazole (CDI), was treated with hydrazine hydrate in THF at room temperature to give hydrazide resin 40, which on further treatment with 2-(1-ethoxyethylidene)malononitrile in ethanol-CH2Cl2 (v/v 1:1) mixture at room temperature for 5 h provided resin bound 5-aminopyrazole 41. Resin 41 was easily cleaved with isopropylamine to give crude 42, which was diazotized with 4 M HCl and sodium nitrite in water at 0–5 °C to provide an intermediate diazonium salt. The latter underwent cycloaddition with an isocyanate in a one-pot reaction to give compound 43 (Scheme 10) [41].
![[1860-5397-7-25-i10]](/bjoc/content/inline/1860-5397-7-25-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of pyrazolo[5,1-d][1,2,3,5]tetrazine-4(3H)-ones.
Scheme 10: Synthesis of pyrazolo[5,1-d][1,2,3,5]tetrazine-4(3H)-ones.
5-Aminopyrazoles 45 have recently been prepared by Boc deprotection of the α-hydrazino acids 44 with TFA in methylene chloride followed by condensation with β-ketonitriles 1 (Scheme 11) [42]. 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide hydrochloride (EDCI) mediated intramolecular cyclodehydration resulted in the formation of the 5,5-ring system, imidazo[1,2-b]pyrazol-2-one 46.
![[1860-5397-7-25-i11]](/bjoc/content/inline/1860-5397-7-25-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of the 5,5-ring system, imidazo[1,2-b]pyrazol-2-ones.
Scheme 11: Synthesis of the 5,5-ring system, imidazo[1,2-b]pyrazol-2-ones.
3-Oxopropanenitriles 16 on coupling with aromatic diazonium salts gave the corresponding 2-arylhydrazones 47, which on treatment with hydrazine hydrate formed the 5-amino-4-arylazopyrazoles 48. 3-Oxo-3-(pyrrol-2-yl)propanenitrile (16) reacted with trichloroacetonitrile to yield enamine 49, which on further treatment with hydrazine hydrate afforded 5-amino-3-(pyrrol-2-yl)pyrazole-4-carbonitrile (50) (Scheme 12) [43].
![[1860-5397-7-25-i12]](/bjoc/content/inline/1860-5397-7-25-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of 5-amino-3-(pyrrol-2-yl)pyrazole-4-carbonitrile.
Scheme 12: Synthesis of 5-amino-3-(pyrrol-2-yl)pyrazole-4-carbonitrile.
Synthesis of 5-amino-3-aryl-1H-pyrazoles 53 has been reported using benzoylacetonitrile 51 as starting material. Substituted phenylhydrazines on reaction with substituted 1-aminocinnamonitriles 52, obtained from base catalyzed reaction of benzoylacetonitrile 51 and acetonitrile, yielded 5-amino-3-aryl-1H-pyrazoles 53. Corresponding amide derivatives, i.e., N-(1,3-diaryl-1H-pyrazol-5-yl)benzamides 54 were prepared by further treating aminopyrazoles 53 with substituted benzoyl chlorides in DCM (Scheme 13) [44].
![[1860-5397-7-25-i13]](/bjoc/content/inline/1860-5397-7-25-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of N-(1,3-diaryl-1H-pyrazol-5-yl)benzamide.
Scheme 13: Synthesis of N-(1,3-diaryl-1H-pyrazol-5-yl)benzamide.
3-Iminobutyronitrile (55) couples with aromatic diazonium salts in a similar manner to yield 2-arylhydrazono-3-iminobutyronitriles 56. Treatment of hydrazones 56 with hydrazine hydrate in refluxing ethanol afforded the corresponding 5-amino-4-arylazo-3-methylpyrazoles 57 in good yields [45]. Pyrazoles 57 further reacted with N-aryl-2-oxo-2-phenylethanehydrazonoyl bromides 58 to yield 3,7-bis(arylazo)-6-methyl-2-phenyl-1H-imidazo[1,2-b]pyrazoles 59 (Scheme 14).
![[1860-5397-7-25-i14]](/bjoc/content/inline/1860-5397-7-25-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of 3,7-bis(arylazo)-6-methyl-2-phenyl-1H-imidazo[1,2-b]pyrazoles.
Scheme 14: Synthesis of 3,7-bis(arylazo)-6-methyl-2-phenyl-1H-imidazo[1,2-b]pyrazoles.
2. Reaction of malononitrile and its derivatives with hydrazines
Malononitrile (60) and its derivatives have been shown to react smoothly with hydrazines to yield 3,5-diaminopyrazoles that possess a wide spectrum of biological activity. As early as in 1884, Rothenburg [46] reported the simplest reaction, i.e., the condensation of malononitrile with hydrazine to give 3,5-diaminopyrazole (61) (Scheme 15).
![[1860-5397-7-25-i15]](/bjoc/content/inline/1860-5397-7-25-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of 3,5-diaminopyrazole.
Scheme 15: Synthesis of 3,5-diaminopyrazole.
The work was subsequently reinvestigated by Sato [47] who found that instead of 3,5-diaminopyrazole, two other products were produced. These compounds were characterized as 5-amino-4-cyanopyrazole 64 and 5-amino-3-hydrazinopyrazole (65). It was suggested that the formation of 64 resulted when two moles of malononitrile condensed with one mole of hydrazine. In this reaction dimerization of malonitrile 62 occurs before the reaction with hydrazine to give 63. However, when one mole of malononitrile condenses with two moles of hydrazine, the formation of 65 takes place via the mechanistic pathway outlined in Scheme 16.
![[1860-5397-7-25-i16]](/bjoc/content/inline/1860-5397-7-25-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of 5-amino-4-cyanopyrazole and 5-amino-3-hydrazinopyrazole.
Scheme 16: Synthesis of 5-amino-4-cyanopyrazole and 5-amino-3-hydrazinopyrazole.
The reaction of substituted hydrazines with malononitrile follows a similar course to yield 67, [48,49] which is the 1-substituted analog of 64 (Scheme 17). However, with substituted malononitriles 66 no such dimerization is possible and the condensation with hydrazine hydrate results in the smooth formation of 3,5-diaminopyrazoles 68 (Scheme 17) [50-53].
![[1860-5397-7-25-i17]](/bjoc/content/inline/1860-5397-7-25-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of 3,5-diaminopyrazoles with substituted malononitriles.
Scheme 17: Synthesis of 3,5-diaminopyrazoles with substituted malononitriles.
Arulsamy and Bohle [54] have reported that the reaction of oximinomalononitrile (69) with hydrazine gives 3,5-diamino-4-oximinopyrazole (70) as the sole product (Scheme 18).
![[1860-5397-7-25-i18]](/bjoc/content/inline/1860-5397-7-25-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of 3,5-diamino-4-oximinopyrazole.
Scheme 18: Synthesis of 3,5-diamino-4-oximinopyrazole.
Shvekhgeimer and Ushakova [55] have reported the synthesis of 4-arylazo-3,5-diaminopyrazoles 73 starting from substituted sulfonamides 71. Sulfonamides 71 after diazotization undergo a coupling reaction with malononitrile to generate the hydrazones 72, which on cycloaddition with hydrazine hydrate give the corresponding pyrazoles (Scheme 19).
![[1860-5397-7-25-i19]](/bjoc/content/inline/1860-5397-7-25-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of 4-arylazo-3,5-diaminopyrazoles.
Scheme 19: Synthesis of 4-arylazo-3,5-diaminopyrazoles.
Reaction of ketenes, particularly those with a cyano group at one end and a leaving group such as alkoxy, alkylthio or halogen at the other, with hydrazine and its derivatives has assumed great importance in the synthesis of 5-aminopyrazoles [56,57]. The advantage of this procedure resides in the frequent possibility of forecasting the structure of the reaction product.
Cheng and Robins [58] have reported the synthesis of 5-amino-4-cyanopyrazoles 76 by the reaction of hydrazines with alkoxymethylenemalononitriles 74a (Y = OR', Scheme 20). Similar results were obtained when aminomethylenemalononitriles 74b (Y = NHR') were treated with hydrazine indicating that reaction is initiated on the vinyl ether (vinylamine) group of 74a/b to give 5-aminopyrazole-4-carbonitrile 76 through the intermediacy of 75 [59]. However, Elnagdi et al. [60] have reported that when ethyl hydrazinoacetate condenses with 74a or b, a change in regiochemistry occurs to yield 3-amino-4-cyanopyrazoles 77 (Scheme 20).
![[1860-5397-7-25-i20]](/bjoc/content/inline/1860-5397-7-25-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of 3- or 5-amino-4-cyanopyrazoles.
Scheme 20: Synthesis of 3- or 5-amino-4-cyanopyrazoles.
Ethoxymethylenemalononitrile (R = OC2H5, R1 = H) 74c and bis(methylthio)- methylenemalononitrile (R = R1 = SCH3) 74d on condensation with hydrazine hydrate yield 5-aminopyrazole-4-carbonitrile 76 (R1 = H) and 5-amino-3-methylthiopyrazole-4-carbonitrile 76 (R1 = SCH3), respectively. These compounds were further treated with nitrous acid and coupled with different secondary amines to yield the triazenopyrazoles 78. Compounds 78 were tested for biological activity against HIV-1 and herpes simplex viruses, and showed moderate activity against HIV-1 virus (Scheme 21) [61,62].
![[1860-5397-7-25-i21]](/bjoc/content/inline/1860-5397-7-25-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of triazenopyrazoles.
Scheme 21: Synthesis of triazenopyrazoles.
An interesting synthesis of 5(3)-aminopyrazoles 81 and 82 [63] has been developed using thioacetals 79 and 80 of malononitrile, which are conveniently obtained by the reaction of aniline and diethyl phosphite with bis(methylthio)methylenemalononitrile 74d, respectively. Reaction with hydrazine monohydrate was thought to occur with loss of the methylthio group by nucleophilic attack of hydrazine and subsequent cyclization by attack on the cyano group (Scheme 22).
![[1860-5397-7-25-i22]](/bjoc/content/inline/1860-5397-7-25-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of 5(3)-aminopyrazoles.
Scheme 22: Synthesis of 5(3)-aminopyrazoles.
The synthesis of a few 3-substituted 5-amino-4-cyanopyrazoles 84 has recently been reported by the treatment of 1,1-dicyano-2-methoxy-3-substituted propenes 83 with hydrazine hydrate in ethanolic TEA (Scheme 23) [64,65].
![[1860-5397-7-25-i23]](/bjoc/content/inline/1860-5397-7-25-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of 3-substituted 5-amino-4-cyanopyrazoles.
Scheme 23: Synthesis of 3-substituted 5-amino-4-cyanopyrazoles.
Acylated hydrazine, as expected, reacts with ethoxymethylenemalononitrile 74a in a similar manner. However, the reaction proceeds only in refluxing phosphorus oxychloride to produce compound 85 with a vinylated amino group (Scheme 24) [66].
![[1860-5397-7-25-i24]](/bjoc/content/inline/1860-5397-7-25-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of 2-{[(1-acetyl-4-cyano-1H-pyrazol-5-yl)amino]methylene}malononitrile.
Scheme 24: Synthesis of 2-{[(1-acetyl-4-cyano-1H-pyrazol-5-yl)amino]methylene}malononitrile.
Ketene dithioacetals 86 were utilized for the synthesis of corresponding pyrazole carbodithioates 88 by cyclization with methyl- or benzylhydrazine carbodithioate 87 in ethanolic TEA at room temperature. As before, the reaction proceeds via the nucleophilic substitution of the alkylthio group by the unsubstituted nitrogen of the hydrazine. The reaction of bis(methylthio)methylenecyanoacetamide 86 (R = CH3, X = CONH2) with aromatic amines gave the corresponding 3-N-substituted aminoacrylamides 89, which on further treatment with phenylhydrazine furnished the corresponding 5-amino-3-arylamino-1-phenylpyrazole-4-carboxamides 90 (Scheme 25) [67].
![[1860-5397-7-25-i25]](/bjoc/content/inline/1860-5397-7-25-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis of 5-aminopyrazole carbodithioates and 5-amino-3-arylamino-1-phenylpyrazole-4-carboxamides.
Scheme 25: Synthesis of 5-aminopyrazole carbodithioates and 5-amino-3-arylamino-1-phenylpyrazole-4-carboxamide...
Ketene S,S- and S,N-acetals or tetracyanoethylene 91 on reaction with 3-hydrazino-6-(p-tolyl)pyridazine afforded the 5-amino-4-cyanopyrazoles 92 (Scheme 26) [68].
![[1860-5397-7-25-i26]](/bjoc/content/inline/1860-5397-7-25-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Synthesis of 5-amino-4-cyanopyrazoles.
Scheme 26: Synthesis of 5-amino-4-cyanopyrazoles.
Several thiazolylpyrazoles 97–100 bearing a variety of substituents at positions 3 and 4 were prepared by the condensation of 2-hydrazino-4-phenylthiazole (93) in presence of TEA with arylidenenitriles 94, cyclohexylidene malononitrile (95), ethyl dimethylthiomethylene cyanoacetate (96) and ethoxymethylenemalononitrile (74), respectively, (Scheme 27) [69].
![[1860-5397-7-25-i27]](/bjoc/content/inline/1860-5397-7-25-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of thiazolylpyrazoles.
Scheme 27: Synthesis of thiazolylpyrazoles.
Synthesis of 5-amino-1-heteroaryl-3-methyl/aryl-4-cyanopyrazoles 102 has been carried out by us by treating various heteroarylhydrazines with alkylidenemalononitriles 101 in refluxing ethanol (Scheme 28) [70]. The starting material 101a (R = C2H5, R1 = CH3) was obtained by the reaction of malononitrile with triethyl orthoacetate in acetic anhydride whilst methoxyarylmethylidenemalonitriles 101b,c were obtained via a two step procedure involving the aroylation of the malonitrile with aroyl chlorides in the presence of NaH, followed by the treatment of the resulting intermediate with dimethyl sulfate.
![[1860-5397-7-25-i28]](/bjoc/content/inline/1860-5397-7-25-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of 5-amino-1-heteroaryl-3-methyl/aryl-4-cyanopyrazoles.
Scheme 28: Synthesis of 5-amino-1-heteroaryl-3-methyl/aryl-4-cyanopyrazoles.
Nilov et al. [71] have reported that the reaction of α-cyano-β-dimethylaminocrotonamide (103) with hydrazine hydrate yields 5-amino-3-methylpyrazole-4-carboxamide (104). The reaction proceeds by loss of dimethylamine in first step followed by cyclization via nucleophilic attack on cyano group (Scheme 29).
![[1860-5397-7-25-i29]](/bjoc/content/inline/1860-5397-7-25-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Synthesis of 5-amino-3-methylpyrazole-4-carboxamide.
Scheme 29: Synthesis of 5-amino-3-methylpyrazole-4-carboxamide.
3. Miscellaneous
In addition to methods involving the reaction of hydrazine with β-ketonitriles, malononitrile and its derivatives, a number of other procedures have also been developed for the synthesis of 5-aminopyrazoles. These methods are summarized below.
Synthesis of 4-acylamino-3(5)-amino-5(3)-arylsulfanylpyrazoles 107 by the reaction of 2-acylamino-3-arylsulfanyl-3-chloroacrylonitriles 106 with hydrazine hydrate has been described. Compounds 106 were readily obtained from 105, the addition products of carboxylic acid amides and trichloroacetaldehyde, by the reaction sequence shown in the Scheme 30 [72].
![[1860-5397-7-25-i30]](/bjoc/content/inline/1860-5397-7-25-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Synthesis of 4-acylamino-3(5)-amino-5(3)-arylsulfanylpyrazoles.
Scheme 30: Synthesis of 4-acylamino-3(5)-amino-5(3)-arylsulfanylpyrazoles.
The reaction of 2-chloro-2-chlorodifluoro/trifluoromethyl-1-cyano-1-diethoxy phosphorylethylene 108 with arylhydrazines in refluxing carbon tetrachloride results in the rapid replacement of the chlorine atom with the terminal NH2 group of arylhydrazines to give intermediates 109, which is slowly transformed into 5-amino-1-aryl-4-diethoxyphosphoryl-3-halomethylpyrazoles 110. 2,6-Dichloro-4-trifluoromethylphenylhydrazine undergoes this reaction under more drastic conditions, i.e., prolonged refluxing (16–20 h) in carbon tetrachloride (Scheme 31) [73].
![[1860-5397-7-25-i31]](/bjoc/content/inline/1860-5397-7-25-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Synthesis of 5-amino-1-aryl-4-diethoxyphosphoryl-3-halomethylpyrazoles.
Scheme 31: Synthesis of 5-amino-1-aryl-4-diethoxyphosphoryl-3-halomethylpyrazoles.
Heterocyclization reactions of trifluoromethylcyanovinyl phosphonates (TFMCPs) 111 with arylhydrazines have been studied: TFMCPs 111 can be used as precursors of 2,3-dihydro-1H-pyrazoles 114 modified by both trifluoromethyl and diethoxyphosphoryl groups. Arylhydrazines add rapidly to the alkene double bond of 111 (X = CF3) at room temperature to produce an adduct which slowly cyclizes to afford 2,3-dihydro-1H-pyrazoles 113 in good yields. 4-Trifluoromethylphenylhydrazine also adds to ethylene 111 (X = CO2Et), however, the resulting adduct 112 is formed primarily as a single diastereomer and does not undergo intramolecular cyclization to pyrazoline 113 even in refluxing benzene. Further, the reaction of isomeric alkene 115 with an arylhydrazine initially forms the unstable pyrazoline 116 that transforms into pyrazole 118. Firstly, the C–P bond apparently undergoes hydrolysis and the resulting 117 is slowly oxidized by atmospheric oxygen to yield pyrazole 118 (Scheme 32) [74].
![[1860-5397-7-25-i32]](/bjoc/content/inline/1860-5397-7-25-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of substituted 5-amino-3-trifluoromethylpyrazoles 114 and 118.
Scheme 32: Synthesis of substituted 5-amino-3-trifluoromethylpyrazoles 114 and 118.
Dodd et al. [75] have reported an efficient solid-support synthesis of 5-N-alkylamino and 5-N-arylaminopyrazoles 123. Heating the β-ketoesters 120 with resin-bound amines 119 in resin-compatible solvents, such as NMP or toluene, in the presence of DMAP gave the corresponding resin-immobilized β-ketoamides 121. The latter β-ketoamides 121, aryl- or alkylhydrazines and Lawesson’s reagent were suspended in a mixture of THF/Py and heated at 50–55 °C to afford resin-bound 5-aminopyrazoles 122. The free 5-aminopyrazoles 123 were liberated from the solid support by treatment with TFA (Scheme 33).
![[1860-5397-7-25-i33]](/bjoc/content/inline/1860-5397-7-25-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Solid-support synthesis of 5-N-alkylamino and 5-N-arylaminopyrazoles.
Scheme 33: Solid-support synthesis of 5-N-alkylamino and 5-N-arylaminopyrazoles.
The reaction of cyanoacetylhydrazine (125) with α-bromoacetophenone (124) gave the N-[2-bromo-1-phenylethylidene]-2-cyanoacetohydrazide (126). Compound 126 readily underwent cyclization when treated with potassium cyanide to give 5-amino-1-cyanoacetyl-3-phenyl-1H-pyrazole (128) through the intermediacy of the acyclic cyano derivative 127 (Scheme 34) [76].
![[1860-5397-7-25-i34]](/bjoc/content/inline/1860-5397-7-25-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Synthesis of 5-amino-1-cyanoacetyl-3-phenyl-1H-pyrazole.
Scheme 34: Synthesis of 5-amino-1-cyanoacetyl-3-phenyl-1H-pyrazole.
Hydrazonoyl chlorides 129 on treatment with benzothiazole-2-acetonitrile in ethanolic sodium ethoxide solution at room temperature afforded intermediate hydrazones 130 which on cyclization gave products identified as 3-substituted 5-amino-1-aryl-4-(benzothiazol-2-yl)pyrazoles 131 (Scheme 35) [77].
![[1860-5397-7-25-i35]](/bjoc/content/inline/1860-5397-7-25-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Synthesis of 3-substituted 5-amino-1-aryl-4-(benzothiazol-2-yl)pyrazoles.
Scheme 35: Synthesis of 3-substituted 5-amino-1-aryl-4-(benzothiazol-2-yl)pyrazoles.
Similarly, hydrazonyl chloride 132 on treatment with ethyl cyanoacetate in NaH/DMF at 0 °C gave intermediate 133 which underwent cyclization to afford 5-amino-4-carbethoxy-3-methyl-1-(4-sulfamoylphenyl)pyrazole 134 (Scheme 36) [78].
![[1860-5397-7-25-i36]](/bjoc/content/inline/1860-5397-7-25-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Synthesis of 5-amino-4-carbethoxy-3-methyl-1-(4-sulfamoylphenyl)pyrazole.
Scheme 36: Synthesis of 5-amino-4-carbethoxy-3-methyl-1-(4-sulfamoylphenyl)pyrazole.
The synthetic precursor 136 for preparation of 5-aminopyrazole 137 was obtained as the major product from the acidic cyclization of the hydrazine with enol 135 (R = H). By contrast, cyclization of the hydrazine with methyl ether 135 (R = Me) under basic conditions, completely reverts the regioselectivity of this reaction and the 3-aminopyrazole intermediate 136 was obtained in excellent yield (93%) as a single isomer. The new derivatives 137 were shown to inhibit intracellular phosphorylation of hsp27 as well as LPS-induced TNFa release in cells (Scheme 37) [79].
![[1860-5397-7-25-i37]](/bjoc/content/inline/1860-5397-7-25-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Synthesis of inhibitors of hsp27-phosphorylation and TNFa-release.
Scheme 37: Synthesis of inhibitors of hsp27-phosphorylation and TNFa-release.
The potassium salt of ethyl cyanopyruvate 138 on reaction with methyl carbazate 139 in a mixture of chloroform and ethyl acetate, saturated with hydrogen chloride resulted in situ protonation of the potassium salt followed by formation of intermediate hydrazone. Further treatment with TEA in acetonitrile resulted in cyclization and furnished the new 5-aminopyrazole acid ester 140. Reaction of 140 with the acid chloride of (9-fluoroenylmethyl)carbamate(Fmoc)-protected glycine led to peptide coupling and subsequent Fmoc deprotection with piperidine gave 141. A second coupling step can also be performed with Fmoc-protected glycine acid chloride, which affords, again after Fmoc removal, the diglycylpyrazole 142 (Scheme 38) [80].
![[1860-5397-7-25-i38]](/bjoc/content/inline/1860-5397-7-25-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Synthesis of the diglycylpyrazole 142.
Scheme 38: Synthesis of the diglycylpyrazole 142.
A new synthetic route [81] to 5-amino-1-aryl-4-benzoyl pyrazole derivatives 144 involves the reaction of β-ketonitriles with N,N’-diphenylformamidine to give initially the cyclocondensation precursors 143 which is then transformed to 144 by reaction with hydrazines (Scheme 39).
![[1860-5397-7-25-i39]](/bjoc/content/inline/1860-5397-7-25-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Synthesis of 5-amino-1-aryl-4-benzoylpyrazole derivatives.
Scheme 39: Synthesis of 5-amino-1-aryl-4-benzoylpyrazole derivatives.
The enamine nitrile 145 reacts readily with 3-hydrazinopropanenitrile to yield 146 via elimination of chloroform by the attack of less hindered nitrogen of reagent. Cyclization by treatment of the latter with 3% NaOH solution gave 4-benzoyl-3,5-diamino-1-(2-cyanoethyl)pyrazole 147 (Scheme 40) [82].
![[1860-5397-7-25-i40]](/bjoc/content/inline/1860-5397-7-25-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Synthesis of 4-benzoyl-3,5-diamino-1-(2-cyanoethyl)pyrazole.
Scheme 40: Synthesis of 4-benzoyl-3,5-diamino-1-(2-cyanoethyl)pyrazole.
2-Cyano-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)acetamide (148) was utilized for the synthesis of the 5-aminopyrazole 150. Treatment of 148 with phenyl isothiocyanate in DMF in the presence of potassium hydroxide at room temperature, followed by treatment with methyl iodide afforded the novel ketene N,S-acetal 149. Reaction of 149 with hydrazine in refluxing ethanol gave the corresponding 5-aminopyrazole derivative 150. The reaction proceeds in the usual manner, i.e., loss of methylthio group by nucleophilic attack of hydrazine in the first step followed by the cyclization (Scheme 41) [83].
![[1860-5397-7-25-i41]](/bjoc/content/inline/1860-5397-7-25-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Synthesis of the 5-aminopyrazole derivative 150.
Scheme 41: Synthesis of the 5-aminopyrazole derivative 150.
Hutaud et al. [84] have reported a unique method for the preparation of 3,5-diaminopyrazoles 153 in good yields by the treatment of the enamine nitrile 151 with trifluoroacetic acid. Boc deprotection by trifluoroacetic acid to 152 is followed by spontaneously nucleophilic attack on the cyano group by the N-terminal nitrogen of the hydrazine substitutent (Scheme 42).
![[1860-5397-7-25-i42]](/bjoc/content/inline/1860-5397-7-25-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Synthesis of 3,5-diaminopyrazoles 153.
Scheme 42: Synthesis of 3,5-diaminopyrazoles 153.
Beam et al. [85] have reported a novel synthesis of 5-aminopyrazoles 155 from polylithiated C(α), N-thiosemicarbazones (X = S) or C(α), N-semicarbazones (X = O). The polylithiated intermediates, prepared from C (α), N-thiosemicarbazones (X = S) or C (α), N-semicarbazones (X = O) 154 and an excess of lithium diisopropylamide (LDA), underwent cyclization and on subsequent hydrolysis gave the 5-aminopyrazole derivatives 155 (Scheme 43).
![[1860-5397-7-25-i43]](/bjoc/content/inline/1860-5397-7-25-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: Synthesis of 5-aminopyrazoles derivatives 155 via lithiated intermediates.
Scheme 43: Synthesis of 5-aminopyrazoles derivatives 155 via lithiated intermediates.
It has been reported that 1,2,4-oxadiazolylmethylenedioxolanes 156 undergo cyclization on treatment with 2-hydroxyethylhydrazine to give 5-amino-4-(1,2,4-oxadiazol-5-yl)-pyrazoles [86] 157 (Scheme 44).
![[1860-5397-7-25-i44]](/bjoc/content/inline/1860-5397-7-25-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: Synthesis of 5-amino-4-(1,2,4-oxadiazol-5-yl)-pyrazoles 157.
Scheme 44: Synthesis of 5-amino-4-(1,2,4-oxadiazol-5-yl)-pyrazoles 157.
The reaction of 3-aminothioacrylamide 158 with hydrazine hydrochloride has been reported to furnish the 5-aminopyrazole 159 in good yield. Various derivatives were tested for anticonvulsant activity in a variety of test models (Scheme 45) [87].
![[1860-5397-7-25-i45]](/bjoc/content/inline/1860-5397-7-25-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 45: Synthesis of a 5-aminopyrazole with anticonvulsant activity.
Scheme 45: Synthesis of a 5-aminopyrazole with anticonvulsant activity.
Another interesting synthesis that affords tetrasubstituted 5-aminopyrazole derivatives 162 involves the reaction of N,N-disubstituted hydrazines 160 with ketones [88]. The hydrazones 161 so formed undergo cyclization in the presence of base to yield the desired compounds 162 (Scheme 46).
![[1860-5397-7-25-i46]](/bjoc/content/inline/1860-5397-7-25-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 46: Synthesis of tetrasubstituted 5-aminopyrazole derivatives.
Scheme 46: Synthesis of tetrasubstituted 5-aminopyrazole derivatives.
Abdelhamid et al. [89,90] have reported the synthesis of substituted 5-aminopyrazoles 164 by the treatment of active methylene compounds such as malononitrile, ethyl cyanoacetate etc. with hydrazonoyl halides 163 in ethanolic sodium ethoxide (Scheme 47).
![[1860-5397-7-25-i47]](/bjoc/content/inline/1860-5397-7-25-i47.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 47: Synthesis of substituted 5-aminopyrazoles from hydrazonoyl halides.
Scheme 47: Synthesis of substituted 5-aminopyrazoles from hydrazonoyl halides.
Ioannidou and Koutentis [91] investigated the conversion of isothiazoles into pyrazoles on treatment with hydrazine. The influence of various C-3, C-4 and C-5 isothiazole substituents and some limitations of this ring transformation were investigated. When a good nucleofugal group (e.g., Cl, Br and I) is present at C-3 in the isothiazole 165, it is replaced by an amino group and 5-aminopyrazoles 166 are obtained. However, when the 3-substituent is not a good leaving group it is retained in the pyrazole product 167. A series of 3-chloro-5-substituted isothiazole-4-carbonitriles 168 bearing steric and/or electronic constraints at C-5 were also treated with anhydrous hydrazine and the corresponding 3-aminopyrazoles 169 were obtained in varying yields. However, when the substituent at C-5 in isothiazole was a better nucleofuge (e.g., PhO, PhS and Cl), the 5-hydrazinoisothiazole 170 was rapidly produced in good yield. Several isothiazoles 171 with a variety of C-4 substituents were also reacted with anhydrous hydrazine to yield the corresponding 3-amino-5-phenylpyrazoles 172. Reaction time and the yield of the reaction was dependent on the substituents present (Scheme 48).
![[1860-5397-7-25-i48]](/bjoc/content/inline/1860-5397-7-25-i48.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 48: Synthesis of 3-amino-5-phenylpyrazoles from isothiazoles.
Scheme 48: Synthesis of 3-amino-5-phenylpyrazoles from isothiazoles.
The reaction of hydroxylamine with 3-(4-phenyl-1,2,4-triazol-3-yl)chromones 173 has been reported to give the 2-aminochromones 174. The 2-aminochromones 174 undergo ring transformation to afford the 5-aminopyrazoles 175 but only upon prolonged heating with hydrazine hydrate in high boiling alcohols (2-propanol, butanol) or in DMF (Scheme 49) [92].
![[1860-5397-7-25-i49]](/bjoc/content/inline/1860-5397-7-25-i49.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 49: Synthesis of 5-aminopyrazoles via ring transformation.
Scheme 49: Synthesis of 5-aminopyrazoles via ring transformation.
Conclusion
5-Aminopyrazole is an important heterocyclic system which has great significance in pharmaceutical industry as well as being a useful synthon for the synthesis of many bridgehead heterocycles. This review describes new strategies and the development of novel concepts along with conventional methods to synthesize a wide variety of substituted 5-aminopyrazoles. Conventional methods such as condensation of β-ketonitriles, malononitrile and its derivatives with hydrazines in addition to modern methods of resin supported solid-phase synthesis, multi-component synthesis and ring transformations provide useful synthetic routes to 5-aminopyrazoles.
References
-
Elguero, J. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 5, pp 167–303. doi:10.1016/B978-008096519-2.00072-2
Return to citation in text: [1] -
Elguero, J. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Pergamon Press: Oxford, 1996; Vol. 3, pp 1–75. doi:10.1016/B978-008096518-5.00059-9
Return to citation in text: [1] -
Kost, A. N.; Grandberg, I. I. In Advances in Heterocyclic Chemistry; Katritzky, A. R.; Boulton, A. J., Eds.; Academic Press: New York, 1966; Vol. 6, pp 347 ff.
Return to citation in text: [1] -
Lee, K. Y.; Kim, J. M.; Kim, J. N. Tetrahedron Lett. 2003, 44, 6737–6740. doi:10.1016/S0040-4039(03)01648-4
Return to citation in text: [1] -
Wiley, R. H.; Wiley, P. Pyrazolones, Pyrazolidones and Derivatives; John Wiley and Sons: New York, 1964.
Return to citation in text: [1] -
Behr, L. C.; Fusco, R.; Jarboe, C. H. In The Chemistry of Heterocyclic Compounds, Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings; Weissberger, A., Ed.; Interscience Publishers: New York, 1967.
Return to citation in text: [1] -
David, D. P.; Martin, D. J.; Charles, M. D. F. 5-Aminopyrazoles useful as selective inhibitors of the protein tyrosine kinase P56ick. WO 9740019 (A1), Nov 30, 1997.
Return to citation in text: [1] -
Kordik, C. P.; Luo, C.; Zanoni, B. C.; Lovenberg, T. W.; Wilson, S. J.; Vaidya, A. H.; Crooke, J. J.; Rosenthal, D. I.; Reitz, A. B. Bioorg. Med. Chem. Lett. 2001, 11, 2287–2290. doi:10.1016/S0960-894X(01)00449-8
Return to citation in text: [1] -
Nakazato, A.; Okuyama, S. Drugs Future 1999, 24, 1089–1098. doi:10.1358/dof.1999.024.10.665576
Return to citation in text: [1] -
Meegalla, S. K.; Doller, D.; Sha, D.; Soll, R.; Wisnewski, N.; Silver, G. M.; Dhanoa, D. Bioorg. Med. Chem. Lett. 2004, 14, 4949–4953. doi:10.1016/j.bmcl.2004.07.033
Return to citation in text: [1] -
Huppatz, J. L. Aust. J. Chem. 1985, 38, 221–230. doi:10.1071/CH9850221
Return to citation in text: [1] -
Shamroukh, A. H.; Rashad, A. E.; Sayed, H. H. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 2347–2360. doi:10.1080/104265090921074
Return to citation in text: [1] -
Carter, T. A.; Wodicka, L. M.; Shah, N. P.; Velasco, A. M.; Fabian, M. A.; Treiber, D. K.; Milanov, Z. V.; Atteridge, C. E.; Biggs, W. H.; Edeen, P. T.; Floyd, M.; Ford, J. M.; Grotzfeld, R. M.; Herrgard, S.; Insko, D. E.; Mehta, S. A.; Patel, H. K.; Pao, W.; Sawyers, C. L.; Varmus, H.; Zarrinkar, P. P.; Lockhart, D. J. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11011–11016. doi:10.1073/pnas.0504952102
Return to citation in text: [1] -
An, H.; Eum, S.-J.; Koh, M.; Lee, S. K.; Park, S. B. J. Org. Chem. 2008, 73, 1752–1761. doi:10.1021/jo702196f
Return to citation in text: [1] -
Abu Elmaati, T. M.; El-Taweel, F. M. J. Heterocycl. Chem. 2004, 41, 109–134. doi:10.1002/jhet.5570410201
Return to citation in text: [1] -
Selleri, S.; Gratteri, P.; Costagli, C.; Bonaccini, C.; Costanzo, A.; Melani, F.; Guerrini, G.; Ciciani, G.; Costa, B.; Spinetti, F.; Martini, C.; Bruni, F. Bioorg. Med. Chem. 2005, 13, 4821–4834. doi:10.1016/j.bmc.2005.05.015
Return to citation in text: [1] -
Gopalsamy, A.; Yang, H.; Ellingboe, J. W.; Tsou, H.-R.; Zhang, N.; Honores, E.; Powell, D.; Miranda, M.; McGinnis, J. P.; Rabindran, S. K. Bioorg. Med. Chem. Lett. 2005, 15, 1591–1594. doi:10.1016/j.bmcl.2005.01.066
Return to citation in text: [1] -
Zhang, X.-Y.; Li, X.-Y.; Fan, X.-S.; Wang, X.; Qu, G.-R.; Wang, J.-J. Heterocycles 2009, 78, 923–936. doi:10.3987/COM-08-11561
Return to citation in text: [1] -
Elnagdi, M. H.; Elgemeie, G. E. H.; Abd-Elaal, F. A.-E. Heterocycles 1985, 23, 3121–3153. doi:10.3987/R-1985-12-3121
Return to citation in text: [1] -
Bagley, M. C.; Davis, T.; Dix, M. C.; Widdowson, C. S.; Kipling, D. Org. Biomol. Chem. 2006, 4, 4158–4164. doi:10.1039/b611493h
Return to citation in text: [1] -
Butler, D. E.; Alexander, S. M. J. Heterocycl. Chem. 1982, 19, 1173–1177. doi:10.1002/jhet.5570190537
Return to citation in text: [1] -
Elnagdi, M. H.; Elfahham, H. A.; Elgemeie, G. E. H. Heterocycles 1983, 20, 519–550. doi:10.3987/R-1983-03-0519
Return to citation in text: [1] -
Elnagdi, M. H.; Elmoghayar, M. R. H.; Elgemeie, G. E. H. Synthesis 1984, 1–26. doi:10.1055/s-1984-30717
Return to citation in text: [1] -
Sunder, S.; Peet, N. P. J. Heterocycl. Chem. 1980, 17, 1527–1529. doi:10.1002/jhet.5570170734
Return to citation in text: [1] -
Elderfield, R. C., Ed. Heterocyclic Compounds; John Wiley and Sons: New York, 1957; Vol. 5.
Return to citation in text: [1] -
Snyder, H. R., Jr. J. Heterocycl. Chem. 1975, 12, 1303–1304. doi:10.1002/jhet.5570120640
Return to citation in text: [1] -
Senga, K.; Robins, R. K.; Brien, D. E. J. Heterocycl. Chem. 1975, 12, 899–901. doi:10.1002/jhet.5570120517
Return to citation in text: [1] -
Joshi, K. C.; Pathak, V. N.; Garg, U. J. Heterocycl. Chem. 1979, 16, 1141–1145. doi:10.1002/jhet.5570160611
Return to citation in text: [1] -
Singh, S. P.; Prakash, O.; Tomar, R. K.; Sawhney, S. N. Indian J. Chem. 1978, 16B, 733–735.
Return to citation in text: [1] -
Singh, S. P.; Tomar, R. K.; Prakash, O.; Sawhney, S. N. Indian J. Chem. 1979, 17B, 372–374.
Return to citation in text: [1] -
Smith, P. A. S.; Ahmad, Y. J. Org. Chem. 1971, 36, 2972–2974. doi:10.1021/jo00819a014
Return to citation in text: [1] -
Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021
Return to citation in text: [1] [2] -
Sosnovskikh, V. Y.; Sizon, A. Y.; Usachev, B. I. Russ. Chem. Bull. 2002, 51, 1270–1279. doi:10.1023/A:1020908831426
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Tyagi, P.; Singh, S. P. Bioorg. Med. Chem. 2005, 14, 1785–1791. doi:10.1016/j.bmc.2005.10.026
Return to citation in text: [1] -
Jachak, M.; Krießmann, U.; Mittelbach, M.; Junek, H. Monatsh. Chem. 1993, 124, 199–207. doi:10.1007/BF00808679
Return to citation in text: [1] -
Kordik, C. P.; Luo, C.; Zanoni, B. C.; Dax, S. L.; McNally, J. J.; Lovenberg, T. W.; Wilson, S. J.; Reitz, A. B. Bioorg. Med. Chem. Lett. 2001, 11, 2283–2286. doi:10.1016/S0960-894X(01)00448-6
Return to citation in text: [1] [2] -
Baraldi, P. G.; El-Kashef, H.; Manfredini, S.; de las Infantas, M. J. P.; Romagnoli, R.; Spalluto, G. Synthesis 1998, 1331–1334. doi:10.1055/s-1998-6095
Return to citation in text: [1] -
Watson, S. P.; Wilson, R. D.; Judd, D. B.; Richards, S. A. Tetrahedron Lett. 1997, 38, 9065–9068. doi:10.1016/S0040-4039(97)10436-1
Return to citation in text: [1] -
Wilson, R. D.; Watson, S. P.; Richards, S. A. Tetrahedron Lett. 1998, 39, 2827–2830. doi:10.1016/S0040-4039(98)00257-3
Return to citation in text: [1] -
Ma, W.; Peterson, B.; Kelson, A.; Laborde, E. J. Comb. Chem. 2009, 11, 697–703. doi:10.1021/cc900045t
Return to citation in text: [1] -
Gao, Y.; Lam, Y. J. Comb. Chem. 2010, 12, 69–74. doi:10.1021/cc900063y
Return to citation in text: [1] -
Blass, B. E.; Srivastava, A.; Coburn, K. R.; Faulkner, A. L.; Janusz, J. J.; Ridgeway, J. M.; Seibel, W. L. Tetrahedron Lett. 2004, 45, 619–621. doi:10.1016/j.tetlet.2003.10.177
Return to citation in text: [1] -
Hassaneen, H. M. E. Synth. Commun. 2007, 37, 3579–3588. doi:10.1080/00397910701557564
Return to citation in text: [1] -
de Paulis, T.; Hemstapat, K.; Chen, Y.; Zhang, Y.; Saleh, S.; Alagille, D.; Baldwin, R. M.; Tamagnan, G. D.; Conn, P. J. J. Med. Chem. 2006, 49, 3332–3344. doi:10.1021/jm051252j
Return to citation in text: [1] -
Shawali, A. S.; Mosselhi, M. A.; Altablawy, F. M. A.; Farghaly, T. A.; Tawfik, N. M. Tetrahedron 2008, 64, 5524–5530. doi:10.1016/j.tet.2008.03.096
Return to citation in text: [1] -
Rothenburg, E. Chem. Ber. 1894, 27, 685–691. doi:10.1002/cber.189402701133
Return to citation in text: [1] -
Sato, T. J. Org. Chem. 1959, 24, 963–966. doi:10.1021/jo01089a019
Return to citation in text: [1] -
Grey, E. J.; Stevens, H. N. E.; Stevens, M. P. G. J. Chem. Soc., Perkin Trans. 1 1978, 885–888.
Return to citation in text: [1] -
Taylor, E. C.; Hartke, K. S. J. Am. Chem. Soc. 1959, 81, 2456–2464. doi:10.1021/ja01519a045
Return to citation in text: [1] -
Echevarría, A.; Martín, M.; Pérez, C.; Rozas, I. Arch. Pharm. 1994, 327, 303–305. doi:10.1002/ardp.19943270507
Return to citation in text: [1] -
Vaquero, J. J.; Fuentes, L.; Del Castillo, J. C.; Pérez, M. I.; García, J. L.; Soto, J. L. Synthesis 1987, 33–35. doi:10.1055/s-1987-27831
Return to citation in text: [1] -
Hassanien, A. A.; Amar, A. E.; Ghozlan, S. A. S. J. Chin. Chem. Soc. 2000, 47, 1273–1278.
Return to citation in text: [1] -
Elnagdi, M. H.; Abd Allah, S. O. J. Prakt. Chem. 1973, 315, 1009–1016. doi:10.1002/prac.19733150604
Return to citation in text: [1] -
Arulsamy, N.; Bohle, D. J. Org. Chem. 2000, 65, 1139–1143. doi:10.1021/jo991614t
Return to citation in text: [1] -
Shvekhgeimer, M.-G. A.; Ushakova, O. A. Chem. Heterocycl. Compd. 2001, 37, 370–371. doi:10.1023/A:1017527620923
Return to citation in text: [1] -
Cankar, P.; Wiedermannova, I.; Slouka, J. Acta Univ. Palacki. Olomuc., Fac. Rerum Nat., Chem. 2002, 41, 7–15.
Return to citation in text: [1] -
Dyachenko, V. D.; Tkachev, R. P. Russ. J. Org. Chem. 2006, 42, 149–171. doi:10.1134/S1070428002120011
Return to citation in text: [1] -
Cheng, C. C.; Robins, R. K. J. Org. Chem. 1956, 21, 1240–1256. doi:10.1021/jo01117a010
Return to citation in text: [1] -
Dooley, M. J.; Quinn, R. J.; Scammells, P. J. Aust. J. Chem. 1989, 42, 747–750. doi:10.1071/CH9890747
Return to citation in text: [1] -
Elnagdi, M. H.; Hafez, E. A. A.; El-Fahham, H. A.; Kandeel, E. M. J. Heterocycl. Chem. 1980, 17, 73–76. doi:10.1002/jhet.5570170115
Return to citation in text: [1] -
Larsen, J. S.; Zahran, M. A.; Pedersen, E. B.; Nielsen, C. Monatsh. Chem. 1999, 130, 1167–1173. doi:10.1007/PL00010295
Return to citation in text: [1] -
Howe, R. K.; Bolluyt, S. C. J. Org. Chem. 1969, 34, 1713–1716. doi:10.1021/jo01258a040
Return to citation in text: [1] -
Lu, R.-J.; Yang, H.-Z. Tetrahedron Lett. 1997, 38, 5201–5204. doi:10.1016/S0040-4039(97)01111-8
Return to citation in text: [1] -
Kraybill, B. C.; Elkin, L. L.; Blethrow, J. D.; Morgan, D. O.; Shokat, K. M. J. Am. Chem. Soc. 2002, 124, 12118–12128. doi:10.1021/ja0264798
Return to citation in text: [1] -
Kluge, R.; Schulz, M.; Pobišova, M.; Nüchter, M. Chem. Ber. 1994, 127, 1729–1733. doi:10.1002/cber.19941270924
Return to citation in text: [1] -
Quinn, R. J.; Scammells, P. J.; Kennard, C. H. L.; Smith, G. Aust. J. Chem. 1991, 44, 1795–1801. doi:10.1071/CH9911795
Return to citation in text: [1] -
Hassan, S. M.; Emam, H. A.; Abdelall, M. M. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 175, 109–127. doi:10.1080/10426500108040260
Return to citation in text: [1] -
Al-Afaleq, E. I.; Abubshait, S. A. Molecules 2001, 6, 621–638. doi:10.3390/60700621
Return to citation in text: [1] -
Amer, A. A. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 2330–2343. doi:10.1080/10426500801963608
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Singh, S. P. Indian J. Chem. 2006, 45B, 1426–1430.
Return to citation in text: [1] -
Nilov, D. B.; Solov’eva, N. P.; Nikolaeva, I. S.; Peters, V. V.; Krylova, L. Y.; Gus’kova, T. A.; Granik, V. G. Pharm. Chem. J. 1998, 32, 358–361. doi:10.1007/BF02645992
Return to citation in text: [1] -
Popil’nichenko, S. V.; Pil’o, S. G.; Brovarets, G. B. S.; Chernega, A. N.; Drach, B. S. Russ. J. Gen. Chem. 2005, 75, 1816–1820. doi:10.1007/s11176-005-0517-2
Return to citation in text: [1] -
Shidlovskii, A. F.; Peregudov, A. S.; Averkiev, B. B.; Antipin, M. Y.; Chkanikov, N. D. Russ. Chem. Bull. 2004, 53, 2060–2070. doi:10.1007/s11172-005-0073-2
Return to citation in text: [1] -
Shidlovskii, A. F.; Peregudov, A. S.; Bulychev, Y. N.; Chkanikov, N. D. Pharm. Chem. J. 2009, 43, 549–559. doi:10.1007/s11094-010-0349-1
Return to citation in text: [1] -
Dodd, D. S.; Martinez, R. L.; Kamau, M.; Ruan, Z.; Van Kirk, K.; Cooper, C. B.; Hermsmeier, M. A.; Traeger, S. C.; Poss, M. A. J. Comb. Chem. 2005, 7, 584–588. doi:10.1021/cc049814s
Return to citation in text: [1] -
Wardakhan, W. W.; Louca, N. A. J. Chil. Chem. Soc. 2007, 52, 1145–1149. doi:10.4067/S0717-97072007000200006
Return to citation in text: [1] -
Dawood, K. M. J. Chem. Res., Synop. 1998, 128–129. doi:10.1039/A706107B
Return to citation in text: [1] -
Organ, M. G.; Mayer, S. J. Comb. Chem. 2003, 5, 118–124. doi:10.1021/cc020045r
Return to citation in text: [1] -
Velcicky, J.; Feifel, R.; Hawtin, S.; Heng, R.; Huppertz, C.; Koch, G.; Kroemer, M.; Moebitz, H.; Revesz, L.; Scheufler, C.; Schlapbach, A. Bioorg. Med. Chem. Lett. 2010, 20, 1293–1297. doi:10.1016/j.bmcl.2009.10.138
Return to citation in text: [1] -
Rzepecki, P.; Gallmeier, H.; Geib, N.; Cernovska, K.; König, B.; Schrader, T. J. Org. Chem. 2004, 69, 5168–5178. doi:10.1021/jo0496603
Return to citation in text: [1] -
Bagley, M. C.; Davis, T.; Dix, M. C.; Murziani, P. G. S.; Rokicki, M. J.; Kipling, D. Bioorg. Med. Chem. Lett. 2008, 18, 3745–3748. doi:10.1016/j.bmcl.2008.05.037
Return to citation in text: [1] -
Elmoghayar, M. R. H.; Elghandour, A. H. H. Monatsh. Chem. 1986, 117, 201–204. doi:10.1007/BF00809440
Return to citation in text: [1] -
Bondock, S.; Rabie, R.; Etman, H. A.; Fadda, A. A. Eur. J. Med. Chem. 2008, 43, 2122–2129. doi:10.1016/j.ejmech.2007.12.009
Return to citation in text: [1] -
Hutaud, D. H.; Baudy-Floc’h, M.; Gougeon, P.; Gall, P.; Le Grel, P. Synthesis 2001, 2435–2440. doi:10.1055/s-2001-18723
Return to citation in text: [1] -
Beam, C. F.; Davis, S. E.; Cordray, T. L.; Chan, K. W.; Kassis, C. M.; Freeman-Davis, J. G.; Latham, G. M.; Guion, T. S.; Hilderbran, K. C.; Church, A. C.; Koller, M. U.; Metz, C. R.; Pennington, W. T.; Schey, K. L. J. Heterocycl. Chem. 1997, 34, 1549–1554. doi:10.1002/jhet.5570340527
Return to citation in text: [1] -
Neidlein, R.; Li, S. J. Heterocycl. Chem. 1996, 33, 1943–1949. doi:10.1002/jhet.5570330663
Return to citation in text: [1] -
Unverferth, K.; Engel, J.; Höfgen, N.; Rostock, A.; Günther, R.; Lankau, H.-J.; Menzer, M.; Rolfs, A.; Liebscher, J.; Müller, B.; Hofmann, H.-J. J. Med. Chem. 1998, 41, 63–73. doi:10.1021/jm970327j
Return to citation in text: [1] -
Fouqué, D.; About-Jaudet, E.; Collingnon, N. Synth. Commun. 1995, 25, 3443–3455. doi:10.1080/00397919508013868
Return to citation in text: [1] -
Abdelhamid, A. O.; Zohdi, H. F.; Sallam, M. M.; Ahmed, N. A. Molecules 2000, 5, 967–973. doi:10.3390/50700967
Return to citation in text: [1] -
Abdelhamid, A. O. J. Chem. Res., Synop. 1993, 208–209.
Return to citation in text: [1] -
Ioannidou, H. A.; Koutentis, P. A. Tetrahedron 2009, 65, 7023–7037. doi:10.1016/j.tet.2009.06.041
Return to citation in text: [1] -
Shokol, T. V.; Turov, V. A.; Semeniuchenko, V. V.; Krivokhizha, N. V.; Khilya, V. P. Chem. Heterocycl. Compd. 2006, 42, 500–505. doi:10.1007/s10593-006-0117-z
Return to citation in text: [1]
| 43. | Hassaneen, H. M. E. Synth. Commun. 2007, 37, 3579–3588. doi:10.1080/00397910701557564 |
| 44. | de Paulis, T.; Hemstapat, K.; Chen, Y.; Zhang, Y.; Saleh, S.; Alagille, D.; Baldwin, R. M.; Tamagnan, G. D.; Conn, P. J. J. Med. Chem. 2006, 49, 3332–3344. doi:10.1021/jm051252j |
| 45. | Shawali, A. S.; Mosselhi, M. A.; Altablawy, F. M. A.; Farghaly, T. A.; Tawfik, N. M. Tetrahedron 2008, 64, 5524–5530. doi:10.1016/j.tet.2008.03.096 |
| 56. | Cankar, P.; Wiedermannova, I.; Slouka, J. Acta Univ. Palacki. Olomuc., Fac. Rerum Nat., Chem. 2002, 41, 7–15. |
| 57. | Dyachenko, V. D.; Tkachev, R. P. Russ. J. Org. Chem. 2006, 42, 149–171. doi:10.1134/S1070428002120011 |
| 58. | Cheng, C. C.; Robins, R. K. J. Org. Chem. 1956, 21, 1240–1256. doi:10.1021/jo01117a010 |
| 54. | Arulsamy, N.; Bohle, D. J. Org. Chem. 2000, 65, 1139–1143. doi:10.1021/jo991614t |
| 55. | Shvekhgeimer, M.-G. A.; Ushakova, O. A. Chem. Heterocycl. Compd. 2001, 37, 370–371. doi:10.1023/A:1017527620923 |
| 48. | Grey, E. J.; Stevens, H. N. E.; Stevens, M. P. G. J. Chem. Soc., Perkin Trans. 1 1978, 885–888. |
| 49. | Taylor, E. C.; Hartke, K. S. J. Am. Chem. Soc. 1959, 81, 2456–2464. doi:10.1021/ja01519a045 |
| 50. | Echevarría, A.; Martín, M.; Pérez, C.; Rozas, I. Arch. Pharm. 1994, 327, 303–305. doi:10.1002/ardp.19943270507 |
| 51. | Vaquero, J. J.; Fuentes, L.; Del Castillo, J. C.; Pérez, M. I.; García, J. L.; Soto, J. L. Synthesis 1987, 33–35. doi:10.1055/s-1987-27831 |
| 52. | Hassanien, A. A.; Amar, A. E.; Ghozlan, S. A. S. J. Chin. Chem. Soc. 2000, 47, 1273–1278. |
| 53. | Elnagdi, M. H.; Abd Allah, S. O. J. Prakt. Chem. 1973, 315, 1009–1016. doi:10.1002/prac.19733150604 |
| 59. | Dooley, M. J.; Quinn, R. J.; Scammells, P. J. Aust. J. Chem. 1989, 42, 747–750. doi:10.1071/CH9890747 |
| 60. | Elnagdi, M. H.; Hafez, E. A. A.; El-Fahham, H. A.; Kandeel, E. M. J. Heterocycl. Chem. 1980, 17, 73–76. doi:10.1002/jhet.5570170115 |
| 61. | Larsen, J. S.; Zahran, M. A.; Pedersen, E. B.; Nielsen, C. Monatsh. Chem. 1999, 130, 1167–1173. doi:10.1007/PL00010295 |
| 62. | Howe, R. K.; Bolluyt, S. C. J. Org. Chem. 1969, 34, 1713–1716. doi:10.1021/jo01258a040 |
| 71. | Nilov, D. B.; Solov’eva, N. P.; Nikolaeva, I. S.; Peters, V. V.; Krylova, L. Y.; Gus’kova, T. A.; Granik, V. G. Pharm. Chem. J. 1998, 32, 358–361. doi:10.1007/BF02645992 |
| 68. | Al-Afaleq, E. I.; Abubshait, S. A. Molecules 2001, 6, 621–638. doi:10.3390/60700621 |
| 69. | Amer, A. A. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 2330–2343. doi:10.1080/10426500801963608 |
| 66. | Quinn, R. J.; Scammells, P. J.; Kennard, C. H. L.; Smith, G. Aust. J. Chem. 1991, 44, 1795–1801. doi:10.1071/CH9911795 |
| 67. | Hassan, S. M.; Emam, H. A.; Abdelall, M. M. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 175, 109–127. doi:10.1080/10426500108040260 |
| 63. | Lu, R.-J.; Yang, H.-Z. Tetrahedron Lett. 1997, 38, 5201–5204. doi:10.1016/S0040-4039(97)01111-8 |
| 64. | Kraybill, B. C.; Elkin, L. L.; Blethrow, J. D.; Morgan, D. O.; Shokat, K. M. J. Am. Chem. Soc. 2002, 124, 12118–12128. doi:10.1021/ja0264798 |
| 65. | Kluge, R.; Schulz, M.; Pobišova, M.; Nüchter, M. Chem. Ber. 1994, 127, 1729–1733. doi:10.1002/cber.19941270924 |
| 73. | Shidlovskii, A. F.; Peregudov, A. S.; Averkiev, B. B.; Antipin, M. Y.; Chkanikov, N. D. Russ. Chem. Bull. 2004, 53, 2060–2070. doi:10.1007/s11172-005-0073-2 |
| 74. | Shidlovskii, A. F.; Peregudov, A. S.; Bulychev, Y. N.; Chkanikov, N. D. Pharm. Chem. J. 2009, 43, 549–559. doi:10.1007/s11094-010-0349-1 |
| 72. | Popil’nichenko, S. V.; Pil’o, S. G.; Brovarets, G. B. S.; Chernega, A. N.; Drach, B. S. Russ. J. Gen. Chem. 2005, 75, 1816–1820. doi:10.1007/s11176-005-0517-2 |
| 1. | Elguero, J. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 5, pp 167–303. doi:10.1016/B978-008096519-2.00072-2 |
| 2. | Elguero, J. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Pergamon Press: Oxford, 1996; Vol. 3, pp 1–75. doi:10.1016/B978-008096518-5.00059-9 |
| 3. | Kost, A. N.; Grandberg, I. I. In Advances in Heterocyclic Chemistry; Katritzky, A. R.; Boulton, A. J., Eds.; Academic Press: New York, 1966; Vol. 6, pp 347 ff. |
| 4. | Lee, K. Y.; Kim, J. M.; Kim, J. N. Tetrahedron Lett. 2003, 44, 6737–6740. doi:10.1016/S0040-4039(03)01648-4 |
| 8. | Kordik, C. P.; Luo, C.; Zanoni, B. C.; Lovenberg, T. W.; Wilson, S. J.; Vaidya, A. H.; Crooke, J. J.; Rosenthal, D. I.; Reitz, A. B. Bioorg. Med. Chem. Lett. 2001, 11, 2287–2290. doi:10.1016/S0960-894X(01)00449-8 |
| 32. | Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021 |
| 81. | Bagley, M. C.; Davis, T.; Dix, M. C.; Murziani, P. G. S.; Rokicki, M. J.; Kipling, D. Bioorg. Med. Chem. Lett. 2008, 18, 3745–3748. doi:10.1016/j.bmcl.2008.05.037 |
| 7. | David, D. P.; Martin, D. J.; Charles, M. D. F. 5-Aminopyrazoles useful as selective inhibitors of the protein tyrosine kinase P56ick. WO 9740019 (A1), Nov 30, 1997. |
| 33. | Sosnovskikh, V. Y.; Sizon, A. Y.; Usachev, B. I. Russ. Chem. Bull. 2002, 51, 1270–1279. doi:10.1023/A:1020908831426 |
| 6. | Behr, L. C.; Fusco, R.; Jarboe, C. H. In The Chemistry of Heterocyclic Compounds, Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings; Weissberger, A., Ed.; Interscience Publishers: New York, 1967. |
| 29. | Singh, S. P.; Prakash, O.; Tomar, R. K.; Sawhney, S. N. Indian J. Chem. 1978, 16B, 733–735. |
| 30. | Singh, S. P.; Tomar, R. K.; Prakash, O.; Sawhney, S. N. Indian J. Chem. 1979, 17B, 372–374. |
| 79. | Velcicky, J.; Feifel, R.; Hawtin, S.; Heng, R.; Huppertz, C.; Koch, G.; Kroemer, M.; Moebitz, H.; Revesz, L.; Scheufler, C.; Schlapbach, A. Bioorg. Med. Chem. Lett. 2010, 20, 1293–1297. doi:10.1016/j.bmcl.2009.10.138 |
| 5. | Wiley, R. H.; Wiley, P. Pyrazolones, Pyrazolidones and Derivatives; John Wiley and Sons: New York, 1964. |
| 31. | Smith, P. A. S.; Ahmad, Y. J. Org. Chem. 1971, 36, 2972–2974. doi:10.1021/jo00819a014 |
| 80. | Rzepecki, P.; Gallmeier, H.; Geib, N.; Cernovska, K.; König, B.; Schrader, T. J. Org. Chem. 2004, 69, 5168–5178. doi:10.1021/jo0496603 |
| 12. | Shamroukh, A. H.; Rashad, A. E.; Sayed, H. H. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 2347–2360. doi:10.1080/104265090921074 |
| 14. | An, H.; Eum, S.-J.; Koh, M.; Lee, S. K.; Park, S. B. J. Org. Chem. 2008, 73, 1752–1761. doi:10.1021/jo702196f |
| 15. | Abu Elmaati, T. M.; El-Taweel, F. M. J. Heterocycl. Chem. 2004, 41, 109–134. doi:10.1002/jhet.5570410201 |
| 16. | Selleri, S.; Gratteri, P.; Costagli, C.; Bonaccini, C.; Costanzo, A.; Melani, F.; Guerrini, G.; Ciciani, G.; Costa, B.; Spinetti, F.; Martini, C.; Bruni, F. Bioorg. Med. Chem. 2005, 13, 4821–4834. doi:10.1016/j.bmc.2005.05.015 |
| 17. | Gopalsamy, A.; Yang, H.; Ellingboe, J. W.; Tsou, H.-R.; Zhang, N.; Honores, E.; Powell, D.; Miranda, M.; McGinnis, J. P.; Rabindran, S. K. Bioorg. Med. Chem. Lett. 2005, 15, 1591–1594. doi:10.1016/j.bmcl.2005.01.066 |
| 18. | Zhang, X.-Y.; Li, X.-Y.; Fan, X.-S.; Wang, X.; Qu, G.-R.; Wang, J.-J. Heterocycles 2009, 78, 923–936. doi:10.3987/COM-08-11561 |
| 19. | Elnagdi, M. H.; Elgemeie, G. E. H.; Abd-Elaal, F. A.-E. Heterocycles 1985, 23, 3121–3153. doi:10.3987/R-1985-12-3121 |
| 20. | Bagley, M. C.; Davis, T.; Dix, M. C.; Widdowson, C. S.; Kipling, D. Org. Biomol. Chem. 2006, 4, 4158–4164. doi:10.1039/b611493h |
| 21. | Butler, D. E.; Alexander, S. M. J. Heterocycl. Chem. 1982, 19, 1173–1177. doi:10.1002/jhet.5570190537 |
| 22. | Elnagdi, M. H.; Elfahham, H. A.; Elgemeie, G. E. H. Heterocycles 1983, 20, 519–550. doi:10.3987/R-1983-03-0519 |
| 23. | Elnagdi, M. H.; Elmoghayar, M. R. H.; Elgemeie, G. E. H. Synthesis 1984, 1–26. doi:10.1055/s-1984-30717 |
| 24. | Sunder, S.; Peet, N. P. J. Heterocycl. Chem. 1980, 17, 1527–1529. doi:10.1002/jhet.5570170734 |
| 25. | Elderfield, R. C., Ed. Heterocyclic Compounds; John Wiley and Sons: New York, 1957; Vol. 5. |
| 26. | Snyder, H. R., Jr. J. Heterocycl. Chem. 1975, 12, 1303–1304. doi:10.1002/jhet.5570120640 |
| 27. | Senga, K.; Robins, R. K.; Brien, D. E. J. Heterocycl. Chem. 1975, 12, 899–901. doi:10.1002/jhet.5570120517 |
| 28. | Joshi, K. C.; Pathak, V. N.; Garg, U. J. Heterocycl. Chem. 1979, 16, 1141–1145. doi:10.1002/jhet.5570160611 |
| 78. | Organ, M. G.; Mayer, S. J. Comb. Chem. 2003, 5, 118–124. doi:10.1021/cc020045r |
| 10. | Meegalla, S. K.; Doller, D.; Sha, D.; Soll, R.; Wisnewski, N.; Silver, G. M.; Dhanoa, D. Bioorg. Med. Chem. Lett. 2004, 14, 4949–4953. doi:10.1016/j.bmcl.2004.07.033 |
| 75. | Dodd, D. S.; Martinez, R. L.; Kamau, M.; Ruan, Z.; Van Kirk, K.; Cooper, C. B.; Hermsmeier, M. A.; Traeger, S. C.; Poss, M. A. J. Comb. Chem. 2005, 7, 584–588. doi:10.1021/cc049814s |
| 9. | Nakazato, A.; Okuyama, S. Drugs Future 1999, 24, 1089–1098. doi:10.1358/dof.1999.024.10.665576 |
| 13. | Carter, T. A.; Wodicka, L. M.; Shah, N. P.; Velasco, A. M.; Fabian, M. A.; Treiber, D. K.; Milanov, Z. V.; Atteridge, C. E.; Biggs, W. H.; Edeen, P. T.; Floyd, M.; Ford, J. M.; Grotzfeld, R. M.; Herrgard, S.; Insko, D. E.; Mehta, S. A.; Patel, H. K.; Pao, W.; Sawyers, C. L.; Varmus, H.; Zarrinkar, P. P.; Lockhart, D. J. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11011–11016. doi:10.1073/pnas.0504952102 |
| 76. | Wardakhan, W. W.; Louca, N. A. J. Chil. Chem. Soc. 2007, 52, 1145–1149. doi:10.4067/S0717-97072007000200006 |
| 35. | Jachak, M.; Krießmann, U.; Mittelbach, M.; Junek, H. Monatsh. Chem. 1993, 124, 199–207. doi:10.1007/BF00808679 |
| 32. | Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021 |
| 34. | Aggarwal, R.; Kumar, V.; Tyagi, P.; Singh, S. P. Bioorg. Med. Chem. 2005, 14, 1785–1791. doi:10.1016/j.bmc.2005.10.026 |
| 84. | Hutaud, D. H.; Baudy-Floc’h, M.; Gougeon, P.; Gall, P.; Le Grel, P. Synthesis 2001, 2435–2440. doi:10.1055/s-2001-18723 |
| 85. | Beam, C. F.; Davis, S. E.; Cordray, T. L.; Chan, K. W.; Kassis, C. M.; Freeman-Davis, J. G.; Latham, G. M.; Guion, T. S.; Hilderbran, K. C.; Church, A. C.; Koller, M. U.; Metz, C. R.; Pennington, W. T.; Schey, K. L. J. Heterocycl. Chem. 1997, 34, 1549–1554. doi:10.1002/jhet.5570340527 |
| 82. | Elmoghayar, M. R. H.; Elghandour, A. H. H. Monatsh. Chem. 1986, 117, 201–204. doi:10.1007/BF00809440 |
| 83. | Bondock, S.; Rabie, R.; Etman, H. A.; Fadda, A. A. Eur. J. Med. Chem. 2008, 43, 2122–2129. doi:10.1016/j.ejmech.2007.12.009 |
| 42. | Blass, B. E.; Srivastava, A.; Coburn, K. R.; Faulkner, A. L.; Janusz, J. J.; Ridgeway, J. M.; Seibel, W. L. Tetrahedron Lett. 2004, 45, 619–621. doi:10.1016/j.tetlet.2003.10.177 |
| 39. | Wilson, R. D.; Watson, S. P.; Richards, S. A. Tetrahedron Lett. 1998, 39, 2827–2830. doi:10.1016/S0040-4039(98)00257-3 |
| 91. | Ioannidou, H. A.; Koutentis, P. A. Tetrahedron 2009, 65, 7023–7037. doi:10.1016/j.tet.2009.06.041 |
| 40. | Ma, W.; Peterson, B.; Kelson, A.; Laborde, E. J. Comb. Chem. 2009, 11, 697–703. doi:10.1021/cc900045t |
| 92. | Shokol, T. V.; Turov, V. A.; Semeniuchenko, V. V.; Krivokhizha, N. V.; Khilya, V. P. Chem. Heterocycl. Compd. 2006, 42, 500–505. doi:10.1007/s10593-006-0117-z |
| 37. | Baraldi, P. G.; El-Kashef, H.; Manfredini, S.; de las Infantas, M. J. P.; Romagnoli, R.; Spalluto, G. Synthesis 1998, 1331–1334. doi:10.1055/s-1998-6095 |
| 88. | Fouqué, D.; About-Jaudet, E.; Collingnon, N. Synth. Commun. 1995, 25, 3443–3455. doi:10.1080/00397919508013868 |
| 38. | Watson, S. P.; Wilson, R. D.; Judd, D. B.; Richards, S. A. Tetrahedron Lett. 1997, 38, 9065–9068. doi:10.1016/S0040-4039(97)10436-1 |
| 89. | Abdelhamid, A. O.; Zohdi, H. F.; Sallam, M. M.; Ahmed, N. A. Molecules 2000, 5, 967–973. doi:10.3390/50700967 |
| 90. | Abdelhamid, A. O. J. Chem. Res., Synop. 1993, 208–209. |
| 36. | Kordik, C. P.; Luo, C.; Zanoni, B. C.; Dax, S. L.; McNally, J. J.; Lovenberg, T. W.; Wilson, S. J.; Reitz, A. B. Bioorg. Med. Chem. Lett. 2001, 11, 2283–2286. doi:10.1016/S0960-894X(01)00448-6 |
| 86. | Neidlein, R.; Li, S. J. Heterocycl. Chem. 1996, 33, 1943–1949. doi:10.1002/jhet.5570330663 |
| 36. | Kordik, C. P.; Luo, C.; Zanoni, B. C.; Dax, S. L.; McNally, J. J.; Lovenberg, T. W.; Wilson, S. J.; Reitz, A. B. Bioorg. Med. Chem. Lett. 2001, 11, 2283–2286. doi:10.1016/S0960-894X(01)00448-6 |
| 87. | Unverferth, K.; Engel, J.; Höfgen, N.; Rostock, A.; Günther, R.; Lankau, H.-J.; Menzer, M.; Rolfs, A.; Liebscher, J.; Müller, B.; Hofmann, H.-J. J. Med. Chem. 1998, 41, 63–73. doi:10.1021/jm970327j |
© 2011 Aggarwal et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)