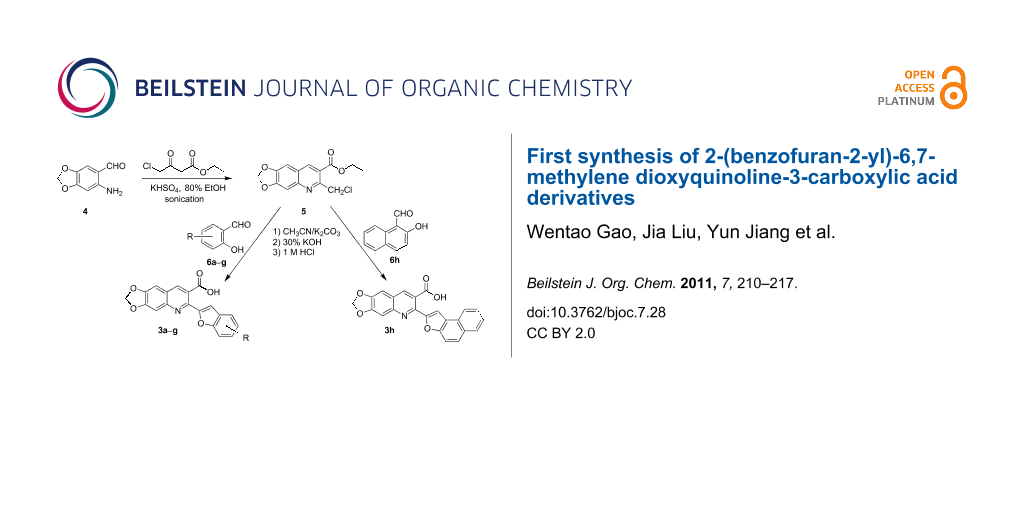
Abstract
A facile and inexpensive synthesis of a series of novel methylenedioxy-bearing 2-(benzofuran-2-yl)-quinoline-3-carboxylic acid derivatives 3a–h via the one-pot reaction of ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5) with various substituted salicylaldehydes 6a–g as well as 2-hydroxy-1-naphthaldehyde (6h) is described. Substrate 5 was synthesized by the Friedländer condensation reaction of 2-amino-4,5-methylenedioxybenzaldehyde (4) with ethyl 4-chloro-3-oxobutanoate using KHSO4 as catalyst under ultrasound irradiation conditions. The targeted compounds 3a–h were obtained in good yields of 52–82% and their structures were established based on spectral data and elemental analyses.

Graphical Abstract
Introduction
Heterocyclic systems containing a quinoline nucleus are an important group of compounds in medicinal chemistry, and are ubiquitous sub-structures associated with biologically active natural products [1-4]. Some quinoline compounds especially those containing heterocyclic systems at 2-position have been shown to display a wide spectrum of biological activities such as cytotoxic, anti-inflammatory and antifungal behavior [5,6]. For example, 2-(furan-2-yl)quinoline-4-carboxylic acid (and analogues) (1, Figure 1) was reported to inhibit C. albicans prolyl-tRNA synthetase and displayed potent in vitro antifungal activity against dermatophytes [6]. Consequently, studies concerning novel 2-heteroarylquinoline derivatives appear frequently in the literature [7,8].
![[1860-5397-7-28-1]](/bjoc/content/figures/1860-5397-7-28-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of compounds 1, 2 and 3.
Figure 1: Structures of compounds 1, 2 and 3.
In a nature’s collection of biologically active heterocycles, 2-heteroarylbenzofuran ring systems are widely distributed in nature and have been reported to have antiviral, antioxidant and antifungal activities [9-13]. For example, cicerfuran (2, Figure 1), obtained from the roots of a wild species of chickpea, Cier bijugum, was reported to be a major factor in the defense system against Fusarium wilt [12,13]. On the other hand, the methylenedioxy unit, present in many natural products such as safrole [14], Leucettamine B [15], and Steganacin [16], exhibits a wide variety of biological activities [17-20]. The methylenedioxy unit can be identified in the clinical antitumor agents etoposide, teniposide [21] and lignan lactone podophyllotoxin [22], and structure–activity relationships have shown that the methylenedioxy moiety is fundamental for cytotoxic activity since it can be metabolized by CYP to form metallo–carbene intermediates which may be responsible for the observed antitumor activity of lignans [23,24]. The presence of the methylenedioxy moiety in some other bioactive molecules drastically alters their pharmacological properties. For example, among the tested 2-phenylquinolin-4-ones (2-PQs) with potent cytotoxicity against human cancer cell lines, the methylenedioxy-bearing 2-PQ was identified as the most active compound in vivo [25].
In light of these findings and in view of structural diversity which plays a prominent role in medicinal and combinatorial chemistry and which leads to a faster and more efficient lead generation in new drug discovery [26], we felt that it would be of interest to construct new prototypes combining the methylenedioxy moiety, quinoline ring system and benzo[b]furan framework in the same molecule. Such compounds might be important for pharmacological studies or in the development of new medicinal products with interesting properties. Therefore, in continuation of our studies concerning the preparation of potential biologically active heterocyclic compounds [27-30], we now report herein a facile and inexpensive procedure for the preparation of novel hybrid molecules, i.e., 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid derivatives (3, Figure 1) under mild conditions.
Results and Discussion
The targeted 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid derivatives 3a–g were synthesized via a two-step procedure, starting from ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5), which was obtained by the Friedländer condensation reaction of 2-amino-4,5-methylenedioxybenzaldehyde (4) with ethyl 4-chloro-3-oxobutanoate as outlined in Scheme 1. In this reaction, we found that the best results could be achieved when the reaction was carried out under ultrasound irradiation conditions at 80 °C using KHSO4 as catalyst in 80% EtOH as solvent. The resulting product 5 was obtained in good yield (74%) after purification by a flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5:1).
![[1860-5397-7-28-i1]](/bjoc/content/inline/1860-5397-7-28-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5).
Scheme 1: Synthesis of ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5).
The next step involves the construction of the benzofuran moiety from intermediate 5. For the construction of 2-substituted benzofurans, the most widely used approach involves the palladium-catalyzed heteroannulation of 2-halophenols with a terminal alkyne via a tandem Sonogashira coupling-5-endo-dig-cyclization, largely based on the methods of Larock and his co-workers [31-34]. Recently, others less popular approaches for the synthesis of 2-substituted benzofurans have included p-toluenesulfonic acid-mediated cyclization of o-(1-alkynyl)anisoles to obtain 2-arylsubstituted benzofurans [35], rearrangement and cyclization reactions of 2-hydroxybenzophenones with Corey–Chaykovsky reagent [36], cyclization of 2-acyloxy-1-bromomethylarenes with Cr(II)Cl2/BF3·OEt2 catalyst [37], and boron tribromide-promoted tandem deprotection–cyclization of 2-methoxyphenylacetones [38], 2-methoxyphenylmethanols [39] and 2-hydroxy-3-arylpropenoic acids to yield 2-methyl, 2-carboxy, and 2-arylbenzo[b]furans, respectively [40]. However, these methods often require expensive catalysts and/or multi-step syntheses. Herein, we demonstrate an attractive one-pot procedure to afford the desired 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid derivatives (3a–g) by the reaction of 5 with substituted salicylaldehydes 6a–g. The reaction proceeds via in situ Williamson ether formation followed by ester hydrolysis and intramolecular cyclization (Scheme 2). The procedure uses an inexpensive inorganic base as catalyst under mild reaction conditions and gives straightforward and easy access for the incorporation of benzofuran core onto the quinoline nucleus at 2-position, to give the desired compounds in with good yields via a simple workup.
![[1860-5397-7-28-i2]](/bjoc/content/inline/1860-5397-7-28-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: One-pot synthesis of the targeted compounds 3a–g.
Scheme 2: One-pot synthesis of the targeted compounds 3a–g.
In this one-pot method, the 2-chloromethylquinoline 5 was first subjected to the Williamson reaction with various salicylaldehydes 6a–g in the presence of K2CO3 as catalyst in refluxing CH3CN. Acetonitrile was employed in our method because of its low boiling point and leads to a much more convenient workup procedure. Thus, once the Williamson reaction was complete as observed on TLC, we simply evaporated CH3CN to dryness, added 30% ethanolic potassium hydroxide solution (10 mL) to the residue and heated the mixture under reflux for 4 hours. Thus, the targeted products 3a–g were obtained in 52–82% overall yields. Encouraged by these results, we also attempted the reaction of 5 with 2-hydroxy-1-naphthaldehyde (6h) under similar reaction conditions with the aim of constructing a novel naphthofuran derivative. Interestingly, 2-hydroxy-1-napthaldehyde was equally amenable to the conditions and gave the corresponding 2-(naphtho[2,1-b]furan-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3h) in good yield (69%). The yields and melting points of all the synthesized products 3a–h are listed in Table 1. It is noteworthy that in the one-pot reaction the use of 30% ethanolic potassium hydroxide solution is sufficient to promote the reaction and there were no improvement in the reaction rates and yields by increasing the amount of potassium hydroxide or by using other bases. Compounds 3a–h are novel and their structures were established based on spectral data and elemental analyses. For example, the IR spectrum of 3a exhibited the presence of hydroxyl and carbonyl groups of carboxyl moiety at 3449 and 1697 cm−1, respectively. Its structure was unequivocal proven by the 1H NMR spectrum. Its 1H NMR spectrum showed no signals attributable to chloromethyl and ester groups but contained a broad singlet at 13.42 ppm for carboxylic proton. Particularly characteristic was the presence of the furan proton singlet at 7.52 ppm along with signals for seven aromatic ring protons between 7.37–8.57 ppm, which is consistent with the attachment of the nascent benzofuran ring moiety to the quinoline substrate. In addition, the structure of 3a was further confirmed by its 13C NMR spectrum, which revealed the presence of carboxyl carbon and methylenedioxy carbon at 168.12 and 102.68 ppm, respectively, along with the signals due to the aromatic carbons. The mass spectrum of 3a contained a quasi-molecular ion peak at m/z 334.1 ([M + H]+) which was also indicative of the proposed structure. The other synthesized compounds exhibited similar spectral characteristics. Elemental analysis was in agreement with the theoretical values.
Table 1: Synthesis of 2-benzofuranyl-6,7-methylenedioxyquinoline-3-carboxylic acids 3a–h.
| Entry | Aldehyde 6 | Product 3 | Yield (%)a | Mp (°C) | ||
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-28-i4.svg?max-width=637&scale=1.0)
|
6a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-28-i5.svg?max-width=637&scale=1.0)
|
3a | 74 | 201–202 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-28-i6.svg?max-width=637&scale=1.0)
|
6b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-28-i7.svg?max-width=637&scale=1.0)
|
3b | 76 | 241–242 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-28-i8.svg?max-width=637&scale=1.0)
|
6c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-28-i9.svg?max-width=637&scale=1.0)
|
3c | 82 | 260–261 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-28-i10.svg?max-width=637&scale=1.0)
|
6d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-28-i11.svg?max-width=637&scale=1.0)
|
3d | 80 | 233–234 |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-28-i12.svg?max-width=637&scale=1.0)
|
6e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-28-i13.svg?max-width=637&scale=1.0)
|
3e | 73 | 192–193 |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-28-i14.svg?max-width=637&scale=1.0)
|
6f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-28-i15.svg?max-width=637&scale=1.0)
|
3f | 64 | 239–240 |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-28-i16.svg?max-width=637&scale=1.0)
|
6g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-28-i17.svg?max-width=637&scale=1.0)
|
3g | 52 | 291–292 |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-28-i18.svg?max-width=637&scale=1.0)
|
6h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-28-i19.svg?max-width=637&scale=1.0)
|
3h | 69 | 232–233 |
aIsolated yield.
As shown in Table 1, with the exception of 3g (entry 7), a series of 2-(benzofuran-2-yl)quinoline-3-carboxylic acid derivatives 3 were readily prepared under mild reaction conditions in good to high yield from the one-pot reaction of 2-chloromethylquinoline 5 with a variety of salicylaldehydes. In the case of entry 7, the reaction of 3,5-di-tert-butylsalicylaldedyde (6g) with 5 gave the corresponding product 3g in a moderate yield of 52%, which might be attributed to the sterically hindered nature of the bulky tert-butyl group at 3-position of 6g. The ease of isolation of all the products is notable; after acidification with 1 M HCl the products were isolated as the major reaction products. The method failed when strong electron-withdrawing groups such as nitro or cyano groups were present on the salicylaldehyde. These reactions were found to be very complex and we could not separate and obtain any of the desired products in appreciable yields in these cases. Instead, intractable complex mixtures were observed on TLC.
To illustrate the reaction mechanism, the formation of 3a is outlined in Scheme 3. The in situ base-mediated ester hydrolysis of the initially formed Williamson product A resulted in the abstraction of acidic proton from the active methylene group in C and subsequent attack of the carbanion at the aldehyde carbonyl carbon with the formation of the five-membered cyclic system E. This is then followed by dehydration to yield 3a. The proposed mechanism is very similar to the formation of naphthofurans via intramolecular condensation in the presence of triethylamine reported by Srivastava et al [41].
![[1860-5397-7-28-i3]](/bjoc/content/inline/1860-5397-7-28-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible mechanistic pathway of formation of 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3a).
Scheme 3: Possible mechanistic pathway of formation of 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carb...
Conclusion
In summary, the present method offers a straightforward and facile synthetic route for the preparation of a variety of 2-benzofuranyl-6,7-methylenedioxyquinoline-3-carboxylic acids 3a–h. Ready availability of starting materials, mild reaction conditions, experimental simplicity and satisfactory yields contribute to the usefulness of this method. Biological activity of the synthesized compounds remains to be studied.
Experimental
Melting points (uncorrected) were determined with a WRS-1B melting point apparatus and are uncorrected. Ultrasonication was performed in a KQ-250B medical ultrasound cleaner with a frequency of 40 KHz and output power of 250W (built in heating 30–80 °C thermostatically adjustable). 1H NMR and 13C NMR spectra were recorded on a Bruker Avance NMR spectrometer using CDCl3 or DMSO-d6 as the solvent. The reported chemical shifts (δ values) are given in parts per million downfield from tetramethylsilane (TMS) as the internal standard. The mass spectra were determined using a MSD VL ESI1 spectrometer. Elemental analyses was performed for C, H, and N using an Elementar Vario EL-III element analyzer and were within ± 0.4% of the calculated values. The progress of reactions was monitored by thin layer chromatography (TLC) on silica gel GF254 using ethyl acetate as mobile phase.
Preparation of ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5)
To a stirred solution of 2-amino-4,5-methylenedioxybenzaldehyde (4) (4.73 g, 29 mmol) and ethyl 4-chloro-3-oxobutanoate (8.11 g, 49 mmol) in 80% ethanol (160 mL), was added a solution of potassium hydrogen sulfate (2.27 g, 17 mmol) in water (20 mL). The resulting mixture was sonicated at 80 °C for 3 h. After the reaction was complete (TLC), the mixture was cooled to room temperature, poured into water and filtered to give the crude product, which was then purified by silica gel column chromatography with ethyl acetate/petroleum ether (5:1) as eluent. The title compound was obtained as a yellow solid in 74% yield, mp 199–203 °C, IR (KBr) ν/cm−1: 1710, 1251, 1231, 948; 1H NMR (CDCl3, 300 MHz): δ (ppm) 1.48 (t, J = 7.1 Hz, 3H, CH3), 4.48 (q, J = 7.1 Hz, 2H, CH2), 5.22 (s, 2H, ArCH2Cl), 6.17 (s, 2H, OCH2O), 7.13 (s, 1H, quinoline-H), 7.40 (s, 1H, quinoline-H), 8.62 (s, 1H, quinoline-H); MS (ESI, m/z): 294.0 [M + H]+; Anal. Calcd for C14H12ClNO4: C, 57.25; H, 4.12; N, 4.77. Found: C, 57.07; H, 4.29; N, 4.70.
General procedure for the synthesis of 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acids 3a–h
A mixture of ethyl 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5) (0.5 mmol, 0.147 g), the corresponding salicylaldehyde or 2-hydroxy-1-naphthaldehyde (0.5 mmol) and anhydrous K2CO3 (2.5 mmol, 0.400 g) was stirred in refluxing CH3CN (12 mL). The conversion was monitored by TLC. After completion, CH3CN was evaporated to dryness. Then 30% ethanolic potassium hydroxide solution (15 mL) was added to the residue and the mixture heated under reflux for 4 h, cooled, and acidified with 1 M hydrochloric acid solution. The resulting crude product was recrystalized from ethyl acetate to afford 3a–h. The melting points and yields of all the compounds are summarized in Table 1 and the spectral and analytical data are given below.
2-(Benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3a)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3449, 1697, 1258, 1110; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 6.32 (s, 2H, OCH2O), 7.37 (t, J = 7.2 Hz, 1H, benzofuran-H), 7.44 (t, J = 7.2 Hz, 1H, benzofuran-H), 7.50 (s, 1H, quinoline-H), 7.52 (s, 1H, furan-H), 7.56 (s, 1H, quinoline-H), 7.68 (d, J = 8.4 Hz, 1H, benzofuran-H), 7.82 (d, J = 7.8 Hz, 1H, benzofuran-H), 8.57 (s, 1H, quinoline-H), 13.42 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 102.68, 103.11, 104.82, 106.49, 111.38, 121.90, 123.35, 123.65, 125.32, 128.32, 135.97, 143.69, 146.06, 148.80, 152.42, 154.53, 154.63, 168.12; MS (ESI, m/z): 334.1 [M + H]+; Anal. Calcd for C19H11NO5: C, 68.47; H, 3.33; N, 4.20. Found: C, 68.33; H, 3.57; N, 4.12.
2-(5-Methoxybenzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3b)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3439, 1618, 1236, 1037; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 3.82 (s, 3H, OMe), 6.29 (s, 2H, OCH2O), 6.97 (dd, J = 8.4, 2.4 Hz, 1H, benzofuran-H), 7.26 (d, J = 2.4 Hz, 1H, benzofuran-H), 7.36 (s, 1H, quinoline-H), 7.46 (s, 1H, quinoline-H), 7.50–7.52 (m, 2H, furan-H and benzofuran-H), 8.53 (s, 1H, quinoline-H), 13.36 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 55.68, 102.70, 103.13, 103.86, 104.80, 106.64, 111.93, 114.26, 123.55, 124.38, 128.91, 136.10, 143.69, 146.15, 148.80, 149.66, 152.51, 155.17, 155.93, 168.76; MS (ESI, m/z): 364.1 [M + H]+; Anal. Calcd for C20H13NO6: C, 66.12; H, 3.61; N, 3.86. Found: C, 66.06; H, 3.80; N, 3.69.
2-(5-tert-Butylbenzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3c)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3470, 1745, 1253, 1116; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 1.37 (s, 9H, tert-butyl), 6.29 (s, 2H, OCH2O), 7.38 (s, 1H, quinoline-H), 7.44 (dd, J = 9.0, 1.2 Hz, 1H, benzofuran-H), 7.46 (s, 1H, quinoline-H), 7.51 (s, 1H, furan-H), 7.53 (d, J = 9.0 Hz, 1H, benzofuran-H), 7.73 (d, J = 1.2 Hz, 1H, benzofuran-H), 8.53 (s, 1H, quinoline-H), 13.36 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 31.69, 34.59, 102.69, 103.13, 104.81, 106.66, 110.69, 117.84, 123.26, 123.54, 128.01, 136.10, 143.83, 145.93, 146.15, 148.77, 152.49, 152.95, 154.59, 168.80; MS (ESI, m/z): 390.1 [M + H]+; Anal. Calcd for C23H19NO5: C, 70.94; H, 4.92; N, 3.60. Found: C, 70.85; H, 5.07; N, 3.57.
2-(5-Chlorobenzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3d)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3445, 1696, 1247, 1119; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 6.30 (s, 2H, OCH2O), 7.40 (s, 1H, quinoline-H), 7.41 (dd, J = 9.0, 1.8 Hz, 1H, benzofuran-H), 7.47 (s, 1H, quinoline-H), 7.53 (s, 1H, furan-H), 7.66 (d, J = 9.0 Hz, 1H, benzofuran-H), 7.84 (d, J = 1.8 Hz, 1H, benzofuran-H), 8.60 (s, 1H, quinoline-H), 13.42 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 102.79, 103.16, 104.84, 106.07, 112.98, 121.28, 123.83, 124.38, 125.21, 127.73, 129.92, 136.42, 143.41, 146.23, 149.02, 152.67, 153.11, 156.10, 168.46; MS (ESI, m/z): 368.0 [M + H]+; Anal. Calcd for C19H10ClNO5: C, 62.06; H, 2.74; N, 3.81. Found: C, 61.98; H, 2.83; N, 3.77.
2-(5-Bromobenzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3e)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3442, 1693, 1247, 1121; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 6.35 (s, 2H, OCH2O), 7.46 (s, 1H, quinoline-H), 7.53 (s, 1H, furan-H), 7.57 (d, J = 9.0 Hz, 1H, benzofuran-H), 7.58 (s, 1H, quinoline-H), 7.67 (d, J = 8.4 Hz, 1H, benzofuran-H), 8.04 (s, 1H, benzofuran-H), 8.65 (s, 1H, quinoline-H), 13.49 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 102.78, 103.16, 104.84, 105.92, 113.44, 115.62, 123.83, 124.30, 127.86, 130.56, 136.43, 143.38, 146.24, 149.02, 152.66, 153.44, 155.90, 168.46; MS (ESI, m/z): 412.0 [M + H]+; Anal. Calcd for C19H10BrNO5: C, 55.36, H, 2.45, N, 3.40. Found: C, 55.27, H, 2.69, N, 3.33.
2-(7-Bromo-5-tert-butylbenzofuran-2-yl)-6,7-methylenedioxyquinoline-7-carboxylic acid (3f)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3445, 1741, 1242, 1112; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 1.36 (s, 9H, tert-butyl), 6.30 (s, 2H, OCH2O), 7.46 (s, 1H, quinoline-H), 7.48 (s, 1H, benzofuran-H), 7.53 (s, 1H, furan-H), 7.61 (s, 1H, quinoline-H), 7.74 (s, 1H, benzofuran-H), 8.60 (s, 1H, quinoline-H), 13.38 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 31.51, 34.79, 102.70, 102.77, 103.19, 104.85, 107.16, 117.59, 123.85, 124.56, 125.72, 129.42, 136.54, 143.64, 146.22, 148.11, 148.97, 149.92, 152.65, 155.48, 168.30; MS (ESI, m/z): 468.0 [M + H]+; Anal. Calcd for C23H18BrNO5: C, 58.99; H, 3.87; N, 2.99. Found: C 58.75; H, 3.96; N 2.69.
2-(5,7-Di-tert-butylbenzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3g)
The compound was obtained as a yellow solid. IR (KBr) ν/cm−1: 3457, 1710, 1251, 1113; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 1.37 (s, 9H, tert-butyl), 1.43 (s, 9H, tert-butyl), 6.29 (s, 2H, OCH2O), 7.26 (d, J = 1.2 Hz, 1H, benzofuran-H), 7.41 (s, 1H, quinoline-H), 7.44 (s, 1H, quinoline-H), 7.51 (s, 1H, furan-H), 7.56 (d, J = 1.2 Hz, 1H, benzofuran-H), 8.54 (s, 1H, quinoline-H), 13.27 (s, 1H, OH); 13C NMR (150 MHz, DMSO-d6): δ (ppm) 29.97, 31.75, 34.08, 34.67, 102.67, 103.13, 104.75, 106.25, 115.73, 119.39, 123.53, 128.57, 133.71, 136.14, 144.05, 145.55, 146.16, 148.69, 151.37, 152.46, 154.34, 168.53; MS (ESI, m/z): 446.2 [M + H]+; Anal. Calcd for C27H27NO5: C, 72.79; H, 6.11; N, 3.14. Found: C, 72.71; H, 6.26; N, 3.11.
2-(Naphtho[2,1-b]furan-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acid (3h)
The compound was obtained as an orange solid. IR (KBr) ν/cm−1: 3433, 1601, 1258, 1034; 1H NMR (DMSO-d6, 600 MHz): δ (ppm) 6.30 (s, 2H, OCH2O), 7.47 (s, 1H, quinoline-H), 7.53 (s, 1H, furan-H), 7.57 (t, J = 7.8 Hz, 1H, naphth-H), 7.68 (t, J = 7.8 Hz, 1H, naphth-H), 7.81 (d, J = 9.0 Hz, 1H, naphth-H), 7.92 (d, J = 9.0 Hz, 1H, naphth-H), 8.08 (d, J = 8.4 Hz, 1H, naphth-H), 8.11 (s, 1H, quinoline-H), 8.44 (d, J = 8.4 Hz, 1H, naphth-H), 8.58 (s, 1H, quinoline-H), 13.39 (s, 1H, OH); 13C NMR (DMSO-d6, 150 MHz): δ (ppm) 90.29, 102.72, 103.21, 104.72, 105.93, 112.42, 123.51, 123.73, 123.92, 125.11, 126.36, 126.90, 127.49, 128.82, 130.13, 136.29, 143.87, 146.26, 148.76, 150.23, 152.57, 154.12, 155.35, 168.82; MS (ESI, m/z): 384.1 [M + H]+; Anal. Calcd for C23H13NO5: C, 72.06; H, 3.42; N, 3.65. Found: C, 71.96; H, 3.69; N, 3.51.
Supporting Information
Supporting Information features 1H NMR and 13C NMR spectra of the substrate 2-chloromethyl-6,7-methylenedioxyquinoline-3-carboxylate (5) and 2-(benzofuran-2-yl)-6,7-methylenedioxyquinoline-3-carboxylic acids 3a–h.
| Supporting Information File 1: 1H NMR and 13C NMR spectra of the title compounds 5 and 3a–h. | ||
| Format: PDF | Size: 768.3 KB | Download |
References
-
Cimanga, K.; De Bruyne, T.; Pieters, L.; Vlietinck, A. J. J. Nat. Prod. 1997, 60, 688–691. doi:10.1021/np9605246
Return to citation in text: [1] -
Jonckers, T. H. M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M.-C.; van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E. L.; Rozenski, J.; Quirijnen, L.; Maes, L.; Dommisse, R.; Lemière, G. L. F.; Vlietinck, A.; Pieters, L. J. Med. Chem. 2002, 45, 3497–3508. doi:10.1021/jm011102i
Return to citation in text: [1] -
Witherup, K. M.; Ransom, R. W.; Graham, A. C.; Bernard, A. M.; Salvatore, M. J.; Lumma, W. C.; Anderson, P. S.; Pitzenberger, S. M.; Varga, S. L. J. Am. Chem. Soc. 1995, 117, 6682–6685. doi:10.1021/ja00130a005
Return to citation in text: [1] -
Balamurugan, K.; Jeyachandran, V.; Perumal, S.; Manjashetty, T. H.; Yogeeswari, P.; Sriram, D. Eur. J. Med. Chem. 2010, 45, 682–688. doi:10.1016/j.ejmech.2009.11.011
Return to citation in text: [1] -
Chen, Y.-L.; Zhao, Y.-L.; Lu, C.-M.; Tzeng, C.-C.; Wang, J.-P. Bioorg. Med. Chem. 2006, 14, 4373–4378. doi:10.1016/j.bmc.2006.02.039
Return to citation in text: [1] -
Meléndez Gómez, C. M.; Kouznetsov, V. V.; Sortino, M. A.; Álvarez, S. L.; Zacchino, S. A. Bioorg. Med. Chem. 2008, 16, 7908–7920. doi:10.1016/j.bmc.2008.07.079
Return to citation in text: [1] [2] -
Yadav, J. S.; Reddy, B. V. S.; Vishnumurthy, P.; Premalatha, K. Synthesis 2008, 719–724. doi:10.1055/s-2008-1032175
Return to citation in text: [1] -
Verniest, G.; Wang, X.; De Kimpe, N.; Padwa, A. J. Org. Chem. 2010, 75, 424–433. doi:10.1021/jo902287t
Return to citation in text: [1] -
Fuganti, C.; Serra, S. Tetrahedron Lett. 1998, 39, 5609–5610. doi:10.1016/S0040-4039(98)01053-3
Return to citation in text: [1] -
Abdel-Wahab, B. F.; Abdel-Aziz, H. A.; Ahmed, E. M. Eur. J. Med. Chem. 2009, 44, 2632–2635. doi:10.1016/j.ejmech.2008.09.029
Return to citation in text: [1] -
Akgul, Y. Y.; Anil, H. Phytochemistry 2003, 63, 939–943. doi:10.1016/S0031-9422(03)00357-1
Return to citation in text: [1] -
Aslam, S. N.; Stevenson, P. C.; Phythian, S. J.; Veitch, N. C.; Hall, D. R. Tetrahedron 2006, 62, 4214–4226. doi:10.1016/j.tet.2006.02.015
Return to citation in text: [1] [2] -
Stevenson, P. C.; Veitch, N. C. Phytochemistry 1998, 48, 947–951. doi:10.1016/S0031-9422(97)00920-5
Return to citation in text: [1] [2] -
Lages, A. S.; Silva, K. C. M.; Miranda, A. L. P.; Fraga, C. A. M.; Barreiro, E. J. Bioorg. Med. Chem. Lett. 1998, 8, 183–188. doi:10.1016/S0960-894X(97)10216-5
Return to citation in text: [1] -
Chan, G. W.; Mong, S.; Hemling, M. E.; Freyer, A. J.; Offen, P. M.; DeBrosse, C. W.; Sarau, H. M.; Westley, J. W. J. Nat. Prod. 1993, 56, 116–121. doi:10.1021/np50091a016
Return to citation in text: [1] -
Kupchan, S. M.; Britton, R. W.; Ziegler, M. F.; Gilmore, C. J.; Restivo, R. J.; Bryan, R. F. J. Am. Chem. Soc. 1973, 95, 1335–1336. doi:10.1021/ja00785a054
Return to citation in text: [1] -
Micale, N.; Zappalà, M.; Grasso, S. Farmaco 2002, 57, 853–859. doi:10.1016/S0014-827X(02)01276-4
Return to citation in text: [1] -
Leite, A. C. L.; da Silva, K. P.; de Souza, I. A.; de Araújo, J. M.; Brondani, D. J. Eur. J. Med. Chem. 2004, 39, 1059–1065. doi:10.1016/j.ejmech.2004.09.007
Return to citation in text: [1] -
Bezerra-Netto, H. J. C.; Lacerda, D. I.; Miranda, A. L. P.; Alves, H. M.; Barreiro, E. J.; Fraga, C. A. M. Bioorg. Med. Chem. 2006, 14, 7924–7935. doi:10.1016/j.bmc.2006.07.046
Return to citation in text: [1] -
Öztürk, S. E.; Akgül, Y.; Anil, H. Bioorg. Med. Chem. 2008, 16, 4431–4437. doi:10.1016/j.bmc.2008.02.057
Return to citation in text: [1] -
Chen, G. L.; Yang, L.; Rowe, T. C.; Halligan, B. D.; Tewey, K. M.; Liu, L. F. J. Biol. Chem. 1984, 259, 13560–13566.
Return to citation in text: [1] -
Capilla, A. S.; Sánchez, I.; Caignard, D. H.; Renard, P.; Pujol, M. D. Eur. J. Med. Chem. 2001, 36, 389–393. doi:10.1016/S0223-5234(01)01231-4
Return to citation in text: [1] -
Castro, A.; del Corral, J. M. M.; Gordaliza, M.; Grande, C.; Gómez-Zurita, A.; García-Grávalos, D.; San Feliciano, A. Eur. J. Med. Chem. 2003, 38, 65–74. doi:10.1016/S0223-5234(02)00007-7
Return to citation in text: [1] -
Imperio, D.; Pirali, T.; Galli, U.; Pagliai, F.; Cafici, L.; Canonico, P. L.; Sorba, G.; Genazzani, A. A.; Tron, G. C. Bioorg. Med. Chem. 2007, 15, 6748–6757. doi:10.1016/j.bmc.2007.08.020
Return to citation in text: [1] -
Chou, L.-C.; Chen, C.-T.; Lee, J.-C.; Way, T.-D.; Huang, C.-H.; Huang, S.-M.; Teng, C.-M.; Yamori, T.; Wu, T.-S.; Sun, C.-M.; Chien, D.-S.; Qian, K.; Morris-Natschke, S. L.; Lee, K.-H.; Huang, L.-J.; Kuo, S.-C. J. Med. Chem. 2010, 53, 1616–1626. doi:10.1021/jm901292j
Return to citation in text: [1] -
Dolle, R. E.; Nelson, K. H., Jr. J. Comb. Chem. 1999, 1, 235–282. doi:10.1021/cc9900192
Return to citation in text: [1] -
Li, Y.; Zhang, C.-H.; Sun, M.-C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203
Return to citation in text: [1] -
Gao, W. T.; Zhang, X.-F.; Li, Y.; Liu, H.-Y.; Imafuku, K. Heterocycles 2010, 81, 1689–1696. doi:10.3987/COM-10-11960
Return to citation in text: [1] -
Gao, W. T.; Cheng, X. P.; Li, Y. Heterocycles 2010, 81, 1923–1930. doi:10.3987/COM-10-11980
Return to citation in text: [1] -
Li, Y.; Gao, W. T. Beilstein J. Org. Chem. 2010, 6, 966–972. doi:10.3762/bjoc.6.108
Return to citation in text: [1] -
Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 10292–10296. doi:10.1021/jo051299c
Return to citation in text: [1] -
Larock, R. C.; Yum, E. K.; Doty, M. J.; Sham, K. K. C. J. Org. Chem. 1995, 60, 3270–3271. doi:10.1021/jo00116a003
Return to citation in text: [1] -
Yue, D.; Larock, R. C. J. Org. Chem. 2002, 67, 1905–1909. doi:10.1021/jo011016q
Return to citation in text: [1] -
Larock, R. C.; Harrison, L. W. J. Am. Chem. Soc. 1984, 106, 4218–4227. doi:10.1021/ja00327a026
Return to citation in text: [1] -
Jacubert, M.; Hamze, A.; Provot, O.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Tetrahedron Lett. 2009, 50, 3588–3592. doi:10.1016/j.tetlet.2009.03.087
Return to citation in text: [1] -
Chittimalla, S. K.; Chang, T.-C.; Liu, T.-C.; Hsieh, H.-P.; Liao, C.-C. Tetrahedron 2008, 64, 2586–2595. doi:10.1016/j.tet.2008.01.024
Return to citation in text: [1] -
Ledoussal, B.; Gorgues, A.; Le Coq, A. Tetrahedron 1987, 43, 5841–5852. doi:10.1016/S0040-4020(01)87790-5
Return to citation in text: [1] -
Dupont, R.; Cotelle, P. Synthesis 1999, 1651–1655. doi:10.1055/s-1999-3572
Return to citation in text: [1] -
Dupont, R.; Cotelle, P. Tetrahedron 2001, 57, 5585–5589. doi:10.1016/S0040-4020(01)00462-8
Return to citation in text: [1] -
Dupont, R.; Cotelle, P. Tetrahedron Lett. 2001, 42, 597–600. doi:10.1016/S0040-4039(00)02011-6
Return to citation in text: [1] -
Srivastava, V.; Negi, A. S.; Kumar, J. K.; Faridi, U.; Sisodia, B. S.; Darokar, M. P.; Luqman, S.; Khanuja, S. P. S. Bioorg. Med. Chem. Lett. 2006, 16, 911–914. doi:10.1016/j.bmcl.2005.10.105
Return to citation in text: [1]
| 36. | Chittimalla, S. K.; Chang, T.-C.; Liu, T.-C.; Hsieh, H.-P.; Liao, C.-C. Tetrahedron 2008, 64, 2586–2595. doi:10.1016/j.tet.2008.01.024 |
| 31. | Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 10292–10296. doi:10.1021/jo051299c |
| 32. | Larock, R. C.; Yum, E. K.; Doty, M. J.; Sham, K. K. C. J. Org. Chem. 1995, 60, 3270–3271. doi:10.1021/jo00116a003 |
| 33. | Yue, D.; Larock, R. C. J. Org. Chem. 2002, 67, 1905–1909. doi:10.1021/jo011016q |
| 34. | Larock, R. C.; Harrison, L. W. J. Am. Chem. Soc. 1984, 106, 4218–4227. doi:10.1021/ja00327a026 |
| 35. | Jacubert, M.; Hamze, A.; Provot, O.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Tetrahedron Lett. 2009, 50, 3588–3592. doi:10.1016/j.tetlet.2009.03.087 |
| 1. | Cimanga, K.; De Bruyne, T.; Pieters, L.; Vlietinck, A. J. J. Nat. Prod. 1997, 60, 688–691. doi:10.1021/np9605246 |
| 2. | Jonckers, T. H. M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M.-C.; van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E. L.; Rozenski, J.; Quirijnen, L.; Maes, L.; Dommisse, R.; Lemière, G. L. F.; Vlietinck, A.; Pieters, L. J. Med. Chem. 2002, 45, 3497–3508. doi:10.1021/jm011102i |
| 3. | Witherup, K. M.; Ransom, R. W.; Graham, A. C.; Bernard, A. M.; Salvatore, M. J.; Lumma, W. C.; Anderson, P. S.; Pitzenberger, S. M.; Varga, S. L. J. Am. Chem. Soc. 1995, 117, 6682–6685. doi:10.1021/ja00130a005 |
| 4. | Balamurugan, K.; Jeyachandran, V.; Perumal, S.; Manjashetty, T. H.; Yogeeswari, P.; Sriram, D. Eur. J. Med. Chem. 2010, 45, 682–688. doi:10.1016/j.ejmech.2009.11.011 |
| 9. | Fuganti, C.; Serra, S. Tetrahedron Lett. 1998, 39, 5609–5610. doi:10.1016/S0040-4039(98)01053-3 |
| 10. | Abdel-Wahab, B. F.; Abdel-Aziz, H. A.; Ahmed, E. M. Eur. J. Med. Chem. 2009, 44, 2632–2635. doi:10.1016/j.ejmech.2008.09.029 |
| 11. | Akgul, Y. Y.; Anil, H. Phytochemistry 2003, 63, 939–943. doi:10.1016/S0031-9422(03)00357-1 |
| 12. | Aslam, S. N.; Stevenson, P. C.; Phythian, S. J.; Veitch, N. C.; Hall, D. R. Tetrahedron 2006, 62, 4214–4226. doi:10.1016/j.tet.2006.02.015 |
| 13. | Stevenson, P. C.; Veitch, N. C. Phytochemistry 1998, 48, 947–951. doi:10.1016/S0031-9422(97)00920-5 |
| 26. | Dolle, R. E.; Nelson, K. H., Jr. J. Comb. Chem. 1999, 1, 235–282. doi:10.1021/cc9900192 |
| 7. | Yadav, J. S.; Reddy, B. V. S.; Vishnumurthy, P.; Premalatha, K. Synthesis 2008, 719–724. doi:10.1055/s-2008-1032175 |
| 8. | Verniest, G.; Wang, X.; De Kimpe, N.; Padwa, A. J. Org. Chem. 2010, 75, 424–433. doi:10.1021/jo902287t |
| 27. | Li, Y.; Zhang, C.-H.; Sun, M.-C.; Gao, W. T. J. Heterocycl. Chem. 2009, 46, 1190–1194. doi:10.1002/jhet.203 |
| 28. | Gao, W. T.; Zhang, X.-F.; Li, Y.; Liu, H.-Y.; Imafuku, K. Heterocycles 2010, 81, 1689–1696. doi:10.3987/COM-10-11960 |
| 29. | Gao, W. T.; Cheng, X. P.; Li, Y. Heterocycles 2010, 81, 1923–1930. doi:10.3987/COM-10-11980 |
| 30. | Li, Y.; Gao, W. T. Beilstein J. Org. Chem. 2010, 6, 966–972. doi:10.3762/bjoc.6.108 |
| 6. | Meléndez Gómez, C. M.; Kouznetsov, V. V.; Sortino, M. A.; Álvarez, S. L.; Zacchino, S. A. Bioorg. Med. Chem. 2008, 16, 7908–7920. doi:10.1016/j.bmc.2008.07.079 |
| 23. | Castro, A.; del Corral, J. M. M.; Gordaliza, M.; Grande, C.; Gómez-Zurita, A.; García-Grávalos, D.; San Feliciano, A. Eur. J. Med. Chem. 2003, 38, 65–74. doi:10.1016/S0223-5234(02)00007-7 |
| 24. | Imperio, D.; Pirali, T.; Galli, U.; Pagliai, F.; Cafici, L.; Canonico, P. L.; Sorba, G.; Genazzani, A. A.; Tron, G. C. Bioorg. Med. Chem. 2007, 15, 6748–6757. doi:10.1016/j.bmc.2007.08.020 |
| 41. | Srivastava, V.; Negi, A. S.; Kumar, J. K.; Faridi, U.; Sisodia, B. S.; Darokar, M. P.; Luqman, S.; Khanuja, S. P. S. Bioorg. Med. Chem. Lett. 2006, 16, 911–914. doi:10.1016/j.bmcl.2005.10.105 |
| 5. | Chen, Y.-L.; Zhao, Y.-L.; Lu, C.-M.; Tzeng, C.-C.; Wang, J.-P. Bioorg. Med. Chem. 2006, 14, 4373–4378. doi:10.1016/j.bmc.2006.02.039 |
| 6. | Meléndez Gómez, C. M.; Kouznetsov, V. V.; Sortino, M. A.; Álvarez, S. L.; Zacchino, S. A. Bioorg. Med. Chem. 2008, 16, 7908–7920. doi:10.1016/j.bmc.2008.07.079 |
| 25. | Chou, L.-C.; Chen, C.-T.; Lee, J.-C.; Way, T.-D.; Huang, C.-H.; Huang, S.-M.; Teng, C.-M.; Yamori, T.; Wu, T.-S.; Sun, C.-M.; Chien, D.-S.; Qian, K.; Morris-Natschke, S. L.; Lee, K.-H.; Huang, L.-J.; Kuo, S.-C. J. Med. Chem. 2010, 53, 1616–1626. doi:10.1021/jm901292j |
| 16. | Kupchan, S. M.; Britton, R. W.; Ziegler, M. F.; Gilmore, C. J.; Restivo, R. J.; Bryan, R. F. J. Am. Chem. Soc. 1973, 95, 1335–1336. doi:10.1021/ja00785a054 |
| 21. | Chen, G. L.; Yang, L.; Rowe, T. C.; Halligan, B. D.; Tewey, K. M.; Liu, L. F. J. Biol. Chem. 1984, 259, 13560–13566. |
| 39. | Dupont, R.; Cotelle, P. Tetrahedron 2001, 57, 5585–5589. doi:10.1016/S0040-4020(01)00462-8 |
| 15. | Chan, G. W.; Mong, S.; Hemling, M. E.; Freyer, A. J.; Offen, P. M.; DeBrosse, C. W.; Sarau, H. M.; Westley, J. W. J. Nat. Prod. 1993, 56, 116–121. doi:10.1021/np50091a016 |
| 22. | Capilla, A. S.; Sánchez, I.; Caignard, D. H.; Renard, P.; Pujol, M. D. Eur. J. Med. Chem. 2001, 36, 389–393. doi:10.1016/S0223-5234(01)01231-4 |
| 40. | Dupont, R.; Cotelle, P. Tetrahedron Lett. 2001, 42, 597–600. doi:10.1016/S0040-4039(00)02011-6 |
| 14. | Lages, A. S.; Silva, K. C. M.; Miranda, A. L. P.; Fraga, C. A. M.; Barreiro, E. J. Bioorg. Med. Chem. Lett. 1998, 8, 183–188. doi:10.1016/S0960-894X(97)10216-5 |
| 37. | Ledoussal, B.; Gorgues, A.; Le Coq, A. Tetrahedron 1987, 43, 5841–5852. doi:10.1016/S0040-4020(01)87790-5 |
| 12. | Aslam, S. N.; Stevenson, P. C.; Phythian, S. J.; Veitch, N. C.; Hall, D. R. Tetrahedron 2006, 62, 4214–4226. doi:10.1016/j.tet.2006.02.015 |
| 13. | Stevenson, P. C.; Veitch, N. C. Phytochemistry 1998, 48, 947–951. doi:10.1016/S0031-9422(97)00920-5 |
| 17. | Micale, N.; Zappalà, M.; Grasso, S. Farmaco 2002, 57, 853–859. doi:10.1016/S0014-827X(02)01276-4 |
| 18. | Leite, A. C. L.; da Silva, K. P.; de Souza, I. A.; de Araújo, J. M.; Brondani, D. J. Eur. J. Med. Chem. 2004, 39, 1059–1065. doi:10.1016/j.ejmech.2004.09.007 |
| 19. | Bezerra-Netto, H. J. C.; Lacerda, D. I.; Miranda, A. L. P.; Alves, H. M.; Barreiro, E. J.; Fraga, C. A. M. Bioorg. Med. Chem. 2006, 14, 7924–7935. doi:10.1016/j.bmc.2006.07.046 |
| 20. | Öztürk, S. E.; Akgül, Y.; Anil, H. Bioorg. Med. Chem. 2008, 16, 4431–4437. doi:10.1016/j.bmc.2008.02.057 |
© 2011 Gao et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)