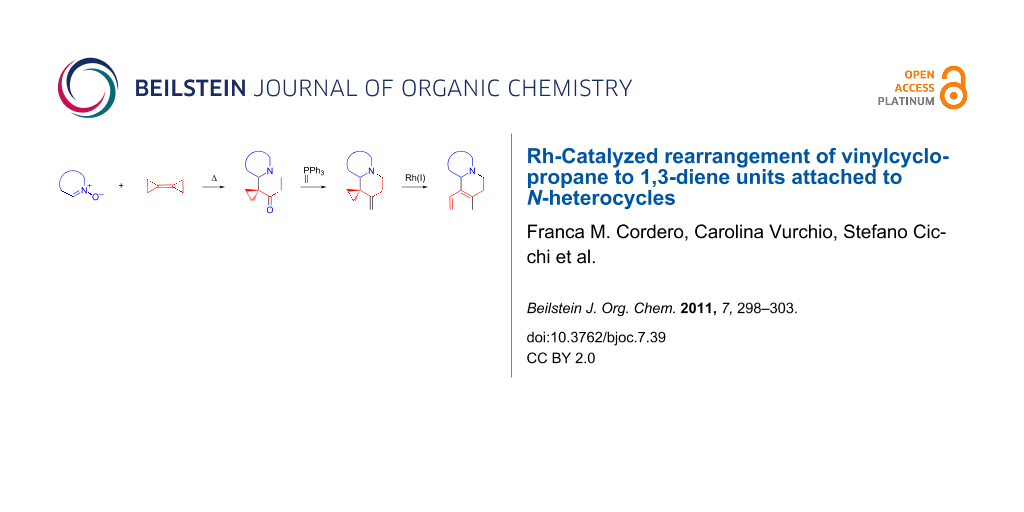
Abstract
Dienes embedded in quinolizidine and indolizidine structures can be prepared in four steps from cyclic nitrones and bicyclopropylidene. The key intermediates α-spirocyclopropanated N-heterocyclic ketones, generated via a domino 1,3-dipolar cycloaddition/thermal rearrangement sequence, were converted by Wittig methylenation to the corresponding vinylcyclopropanes (VCPs), which underwent rearrangement to 1,3-dienes in the presence of the Wilkinson Rh(I) complex under microwave heating. The previously unexplored Rh(I)-catalyzed opening of the VCP moiety embedded in an azapolycyclic system occurs at high temperature (110–130 °C) to afford the corresponding 1,3-dienes in moderate yield (34–53%).

Graphical Abstract
Introduction
The cyclopropyl group endows many natural and synthetic compounds with a broad spectrum of interesting properties, mainly related to its unusual bonding and inherent ring strain [1-3]. This characteristic confers on molecules containing this moiety high reactivity, especially towards ring expansion and ring-opening transformations. The smallest carbocycle can therefore be considered as a peculiar functional group that can promote unique reactivities and synthetic possibilities [4]. The main obstacle to full exploitation of this chemistry is the difficulty of selectively introducing a cyclopropyl group into a given substrate so that the various specific cyclopropane transformations can be used as a synthetic tool. In recent years we have shown that 1,3-dipolar cycloadditions of nitrones 1 to the highly strained alkene bicyclopropylidene (BCP, 2) [5-7] afford spirocyclopropanated isoxazolidines 3 [8,9] which, on heating, rearrange [10] to yield a large variety of spirocyclopropanated heterocyclic ketones 4 depending on the nature of the starting nitrone (Scheme 1) [11-17].
![[1860-5397-7-39-i1]](/bjoc/content/inline/1860-5397-7-39-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General approach to spirocyclopropanated tetrahydropyridones by 1,3-dipolar cycloaddition/thermal rearrangement.
Scheme 1: General approach to spirocyclopropanated tetrahydropyridones by 1,3-dipolar cycloaddition/thermal r...
This rather general and convenient access to spirocyclopropane-annelated heterocyclic ketones 4 makes it attractive for the construction of other heterocyclic compounds by selective elaboration of the α-oxocyclopropane functionality, for example, to vinylcyclopropane (VCP) by simple Wittig olefination. The rearrangement of VCPs to cyclopentenes and dienes are well known processes [18-25] that occur thermally or under catalysis by various transition metals including Rh, Ni, Pd, Cu, Cr, Mo, and Fe [26-34]. To date the metal-catalyzed rearrangement of azaheterocyclic VCP has not been reported. In the context of our interest in the VCP chemistry of spirocyclopropane-annelated heterocyclic compounds [35], we started to investigate some metal-catalyzed rearrangements. The first choice was the readily available so-called Wilkinson catalyst Rh(PPh3)3Cl, because of its documented efficiency in catalyzing the rearrangement [26] and of the possibility to extend its use to other interesting transformations, such as the [5 + 2] cycloadditions of vinylcyclopropanes to alkynes developed by Wender and co-workers [36,37]. It is known, that rhodium-catalyzed rearrangements of unactivated VCPs, without any functional substituent, usually afford dienes. In order to evaluate the influence of the N-heterocyclic system on the rearrangement, some model VCPs were generated by Wittig olefination of the α-oxocyclopropane group of functionalized oligocyclic spirocyclopropane-tetrahydropyridones and converted into the corresponding 1,3-dienes by treatment with Rh(PPh3)3Cl.
Results and Discussion
The tetrahydropyridones employed in this study were prepared according to published procedures with slight modifications. In particular, oxidation of the tetrahydroquinoline 5 with oxone [38] afforded the nitrone 6 [39-41] in 66% yield (Scheme 2, see Supporting Information File 1 for full experimental data). Treatment of 6 (1.2–1.7 equiv) with BCP (2) in xylenes at 125 °C for 64 h directly afforded the α-oxocyclopropane derivative 8 [13] (51–72% yield) along with a minor amount of the open-chain isomer 9 (20–23% yield).
![[1860-5397-7-39-i2]](/bjoc/content/inline/1860-5397-7-39-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tetrahydrospiro[cyclopropane-1,1’(2’H,6’H)-pyrido[2,1-a]isoquinolin]-2’-one 8.
Scheme 2: Synthesis of tetrahydrospiro[cyclopropane-1,1’(2’H,6’H)-pyrido[2,1-a]isoquinolin]-2’-one 8.
The open-chain isomer 9 is derived from a rarely observed 1,5-hydrogen shift in the cyclopropanated 1,6-diradical intermediate, which in this case is probably facilitated by the enhanced mobility of the benzylic hydrogen and by the formation of the conjugationally stabilized imine 9 [11].
The 1,3-dipolar cycloaddition/thermal rearrangement domino reaction of BCP (2) with the enantiopure nitrone 10 [42] derived from L-tartaric acid was complete within only 1.5 h at 120–125 °C under microwave (MW) heating and afforded the oxospirocyclopropanes anti-12 and syn-12 in 55% overall yield along with the 1,5-hydrogen shift product 13 (13%) (Scheme 3, see Supporting Information File 1 for full experimental data). The two diastereomeric indolizidinones anti-12 and syn-12 are formed by the thermal rearrangement of the cycloadducts anti-(3-t-BuO)-11 and syn-(3-t-BuO)-11, respectively.
![[1860-5397-7-39-i3]](/bjoc/content/inline/1860-5397-7-39-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 7’-oxohexahydro spiro[cyclopropane-1-8’(5’H)indolizines] 12.
Scheme 3: Synthesis of 7’-oxohexahydro spiro[cyclopropane-1-8’(5’H)indolizines] 12.
Wittig olefination of ketones 8, anti-12 and 16 [43] with MePPh3Br/t-BuOK in THF at room temperature gave the VCPs 14, 15, and 17 in good yields (53–96%) (Scheme 4, see Supporting Information File 1 for full experimental data). The configuration was retained under the reaction conditions in compounds 15 and 17, as ascertained by the unique set of 1H NMR signals in the crude reaction mixture.
![[1860-5397-7-39-i4]](/bjoc/content/inline/1860-5397-7-39-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Olefination of spirocyclopropanated heterocyclic ketones 8, 12 and 16.
Scheme 4: Olefination of spirocyclopropanated heterocyclic ketones 8, 12 and 16.
The tricyclic compound 14 was then treated with a catalytic amount of Rh(PPh3)3Cl (Table 1). In toluene at room temperature no reaction occurred, but on heating under reflux (110 °C) or at 130 °C in a microwave (MW) oven a rearrangement occurred leading to a main product assigned as the diene 18 and a mixture of the two diastereomers (E)- and (Z)-19. No trace of the cyclopentene-annelation product was observed in the reaction mixture. In refluxing toluene, the reaction was quite slow and after 15 h in the presence of 5 mol % of catalyst, a considerable amount of starting material still remained. Even after the addition of a second portion of the catalyst and further heating at 110 °C for 18 h, the conversion was incomplete, and the VCP 14 was recovered in 18% yield after chromatography (Table 1, entry 1). The dienes 18 and 19 were obtained in 35% overall yield (43% yield based on converted 14). The VCP was completely consumed after 58 h at 110 °C by adding three portions of the catalyst (5 mol % each at 0, 24 and 40 h, respectively) (Table 1, entry 2). In this case, the products were obtained in a lower yield (28%) probably because they partially decomposed upon prolonged heating. The reaction carried out at 130 °C in an MW reactor for shorter times actually gave better yields (Table 1, entries 3 and 4). The best result with a conversion of 78% and 59% yield was achieved by heating a mixture of the VCP 14 and 10 mol % of the catalyst in toluene at 130 °C for 140 min (Table 1, entry 4). When the reaction was allowed to continue until all the starting material was completely consumed led to complete decomposition of the products (Table 1, entry 5). Higher temperatures (160 °C in xylenes) or the addition of AgOTf [44] did neither improve the conversion rate nor the yield of the rearrangement products (Table 1, entries 6 and 7). A slight improvement was achieved by addition of trifluoroethanol (TFE, 5% of the total volume) as a co-solvent [45]. Under these conditions, the conversion of the VCP was complete after 5.5 h at 130 °C, and the dienes 18 and 19 were obtained in a 1:1 ratio in 46% overall yield after chromatography (Table 1, entry 8, see Supporting Information File 1 for full experimental data).
Table 1: Rearrangement of VCP 14 catalyzed by Rh(PPh3)3Cl.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-39-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Rh(I) (%) | Temp.a (°C) | Time | Conv.b (%) | 18/19 | Yieldc (%) |
|---|---|---|---|---|---|---|
| 1 | 5 + 5d | 110 | 15 + 18 h | 82 | 2.5:1 | 35 (43) |
| 2 | 5 + 5 + 5e | 110 | 24 + 16 + 18 h | 100 | 1.1:1 | 28 (28) |
| 3 | 5 | 130 (MW) | 1 h 30 min | 62 | 4.5:1 | 46 (74) |
| 4 | 10 | 130 (MW) | 2 h 20 min | 78 | 1.4:1 | 59 (75) |
| 5 | 5 + 5d | 130 (MW) | 3 + 4 h | 100 | decf | |
| 6g | 5 + 5d | 160 (MW) | 2 + 1 h | 72h | 3.3:1 | ndi |
| 7j | 5 | 130 (MW) | 2 h | 55h | 3:1 | ndi |
| 8k | 10 | 130 (MW) | 5 h 30 min | 100 | 1:1 | 46 (46) |
aMW: the reaction was carried out in a CEM Discover microwave reactor with IR temperature monitoring. bBased on recovered starting material after chromatography. cOverall yield after chromatography on SiO2. The yield based on converted VCP is given in parentheses. dThe catalyst was added in two batches (5 mol % each at 0 and 15 h in entry 1, at 0 and 3 h in entry 5, at 0 and 2 h in entry 6). eThe catalyst was added in three batches (5 mol % each at 0, 24 and 40 h). fDecomposition products. gThe reaction was run in xylenes. hDetermined by 1H NMR analysis of the crude reaction mixture. iNot determined. j5% AgOTf was added. k5% TFE was added.
The collected data show that longer reaction times significantly influence the product ratio in favour of the dienes 19 (Table 1). These results are in accord with an isomerization of 18 into 19 under the reaction conditions. However, in the absence of the catalyst, heating of diene 18 under otherwise identical conditions did not induce any isomerization of 18 to 19, which confirms that Rh also catalyzes the 1,5-hydrogen shift in 18.
The structure assignment was easily made on the basis of 1H NMR data. In particular, the diene 18 showed the typical signals of an exocyclic vinyl substituent (δ 7.00 (=CHv), 5.17 (=CHHcis), 5.17 (=CHHtrans) ppm; Jtrans = 17.5, Jcis = 11.3 Hz). Irradiation of 11b-H (δ 4.76 ppm) produced a positive NOE on the olefinic Htrans whereas irradiation of =CH resulted in enhancement of the 2-CH3 signal (δ 1.87 ppm) suggesting a preferred s-trans conformation for the diene moiety in solution (Figure 1).
![[1860-5397-7-39-1]](/bjoc/content/figures/1860-5397-7-39-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Key NOE interactions. 18: 11b-H/11-H, 11b-H/6-H, 11b-H/Ht, Hv/2-CH3; E-19: Hb/CH3, Hc/11b-H, Hc/11-H; Z-19: Hb’/Hc’, 11b-H/CH3.
Figure 1: Key NOE interactions. 18: 11b-H/11-H, 11b-H/6-H, 11b-H/Ht, Hv/2-CH3; E-19: Hb/CH3, Hc/11b-H, Hc/11-...
The 1H NMR spectra of the dienes 19 showed a quartet in the region of olefinic due to the resonance of the proton Hc coupled with the methyl group (δ 5.29 and 5.96 ppm, 3J = 6.9–7.0 Hz) and two signals due to the methylene protons Ha and Hb (δ 5.06 and 4.75; 4.89 and 4.65 ppm, 2J = 2.3–2.1 Hz).
The E- and Z-configuration of the dienes 19 was determined by NOESY 1D NMR spectroscopy. In the major compound (E)-19, irradiation of the olefinic Hb (δ 4.75 ppm) gave enhancement of the ethylidene methyl group (δ 1.76 ppm), and irradiation of Hc (δ 5.29 ppm) showed enhancement at 11-H and 11b-H (δ 6.56 and 4.41 ppm). For the isomer (Z)-19, irradiation of Hc’ (δ 5.96 ppm) gave enhancement of Hb’ (δ 4.89 ppm) and an NOE interaction was present between the methyl group and 11b-H (δ 5.02 ppm) in agreement with the assigned configuration (Figure 1).
The VCPs 15 and 17 were completely consumed on heating in toluene in a MW oven at 130 °C for 3 h and 110 °C for 3.5 h, respectively, in the presence of Rh(PPh3)3Cl (10%) and TFE (5%). In these cases, dienes 20 and 21 were obtained in 53 and 34% yield, respectively, as the sole reaction products (Scheme 5, see Supporting Information File 1 for full experimental data). Their structures were assigned analogously as before. Compounds 20 and 21 were found to be unstable upon standing for prolonged periods, even at low temperatures. This explains the low isolated yields in their syntheses.
![[1860-5397-7-39-i5]](/bjoc/content/inline/1860-5397-7-39-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Rearrangement of VCPs 15 and 17 catalyzed by Rh(PPh3)3Cl.
Scheme 5: Rearrangement of VCPs 15 and 17 catalyzed by Rh(PPh3)3Cl.
Analogously to other Rh(I)-catalyzed VCP rearrangements [46,47], the mechanism of the rearrangement likely involves insertion of the Rh(I) species into the cyclopropane ring of the VCP system, with or without incorporation of the double bond to form the intermediates C which can undergo metal hydride elimination or 1,3-hydride migration to the rhodium to give, respectively, the allyl- and alkylrhodium(III) hydride complexes D and F. Metal extrusion by reductive elimination leads to the observed dienes and regeneration of the catalyst (Scheme 6).
![[1860-5397-7-39-i6]](/bjoc/content/inline/1860-5397-7-39-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Mechanism of the rearrangement of heterocyclic VCPs catalyzed by Rh(PPh3)3Cl.
Scheme 6: Mechanism of the rearrangement of heterocyclic VCPs catalyzed by Rh(PPh3)3Cl.
Conclusion
Azaheterocycles 14, 15 and 17 containing a spiro-annelated VCP moiety have been synthesized starting from cyclic nitrones and BCP by a three-step two-pot sequence consisting of a 1,3-dipolar cycloaddition, thermal rearrangement and Wittig methylenation. These compounds in the presence of the Wilkinson Rh(I) complex at high temperatures (110–130 °C) under MW heating underwent a slow rearrangement to afford the corresponding azaheterocycles containing 1,3-diene units in moderate yields. The rearrangement produced mixtures of isomeric dienes from benzoquinolizidine 14 and was regioselective in the case of indolizidines 15 and 17, showing that the Wilkinson Rh(I) catalyst is also capable of inducing the VCP rearrangement in the presence of strongly nucleophilic azaheterocycles. Accordingly, new functionalised heterocyclic compounds can be produced by the straightforward methodology based on nitrones and bicyclopropylidene.
Supporting Information
Supporting information features experimental procedures and spectroscopic data.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 85.3 KB | Download |
Acknowledgements
Mr. Reent Michel, a Sokrates-exchange student from the Georg-August-Universität Göttingen (Germany), is acknowledged for his partial contribution to this work. Ente Cassa di Risparmio di Firenze (Florence, Italy) is acknowledged for partial financial support of a fellowship for C. V. Ministry of University and Research (MIUR Rome-Italy) is acknowledged for financial support (PRIN project).
References
-
de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/ange.19790911104
Return to citation in text: [1] -
Salaün, J. In The Chemistry of the Cyclopropyl Group; Patai, S.; Rappoport, Z., Eds.; Wiley: New York, 1987; pp 809–878.
Return to citation in text: [1] -
Salaün, J. Top. Curr. Chem. 2000, 207, 1–67. doi:10.1007/3-540-48255-5_1
Return to citation in text: [1] -
de Meijere, A., Ed. Houben−Weyl, Carbocyclic Three-Membered Ring Compounds; Thieme: Stuttgart, 1997; Vol. E17.
Return to citation in text: [1] -
Brandi, A.; Goti, A. Chem. Rev. 1998, 98, 589–636. doi:10.1021/cr940341t
Return to citation in text: [1] -
de Meijere, A.; Kozhushkov, S. I. Eur. J. Org. Chem. 2000, 3809–3822. doi:10.1002/1099-0690(200012)2000:23<3809::AID-EJOC3809>3.0.CO;2-X
Return to citation in text: [1] -
de Meijere, A.; Kozhushkov, S. I. Chem. Rev. 2000, 100, 93–142. doi:10.1021/cr960153y
Return to citation in text: [1] -
Brandi, A.; Cordero, F. M.; De Sarlo, F.; Goti, A.; Guarna, A. Synlett 1993, 1–8. doi:10.1055/s-1993-22329
Return to citation in text: [1] -
Goti, A.; Cordero, F. M.; Brandi, A. Top. Curr. Chem. 1996, 178, 1–97. doi:10.1007/3-540-60495-2_1
Return to citation in text: [1] -
Brandi, A.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. Rev. 2003, 103, 1213–1269. doi:10.1021/cr010005u
Return to citation in text: [1] -
Goti, A.; Anichini, B.; Brandi, A.; Kozhushkov, S. I.; Gratkowski, C.; de Meijere, A. J. Org. Chem. 1996, 61, 1665–1672. doi:10.1021/jo951838l
Return to citation in text: [1] [2] -
Goti, A.; Anichini, B.; Brandi, A.; de Meijere, A.; Citti, L.; Nevischi, S. Tetrahedron Lett. 1995, 36, 5811–5814. doi:10.1016/0040-4039(95)01103-O
Return to citation in text: [1] -
Zorn, C.; Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S. I.; de Meijere, A.; Citti, L. J. Org. Chem. 1999, 64, 7846–7855. doi:10.1021/jo990873f
Return to citation in text: [1] [2] -
Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S. I.; de Meijere, A. Synlett 1997, 25–26. doi:10.1055/s-1997-684
Return to citation in text: [1] -
Marradi, M.; Brandi, A.; Magull, J.; Schill, H.; de Meijere, A. Eur. J. Org. Chem. 2006, 5485–5494. doi:10.1002/ejoc.200600417
Return to citation in text: [1] -
Zorn, C.; Goti, A.; Brandi, A.; Johnsen, K.; Noltemeyer, M.; Kozhushkov, S. I.; de Meijere, A. J. Org. Chem. 1999, 64, 755–763. doi:10.1021/jo981366l
Return to citation in text: [1] -
Revuelta, J.; Cicchi, S.; Faggi, C.; Kozhushkov, S. I.; de Meijere, A.; Brandi, A. J. Org. Chem. 2006, 71, 2417–2423. doi:10.1021/jo052564x
Return to citation in text: [1] -
Neureiter, N. P. J. Org. Chem. 1959, 24, 2044–2046. doi:10.1021/jo01094a621
Return to citation in text: [1] -
Overberger, C. G.; Borchert, A. E. J. Am. Chem. Soc. 1960, 82, 1007–1008. doi:10.1021/ja01489a069
Return to citation in text: [1] -
Flowers, M. C.; Frey, H. M. J. Chem. Soc. 1961, 82, 3547–3548.
Return to citation in text: [1] -
Hudlicky, T.; Becker, D. A.; Fan, R. L.; Kozhushkov, S. I. In Houben−Weyl; de Meijere, A., Ed.; Thieme: Stuttgart, 1997; Vol. E17c, pp 2538–2565.
Return to citation in text: [1] -
Hudlicky, T.; Fan, R. L.; Reed, J. W.; Gadamasetti, K. G. Org. React. 1992, 41, 1–133. doi:10.1002/0471264180.or041.01
Return to citation in text: [1] -
Hudlicky, T.; Reed, J. W. In Comprehensive Organic Synthesis-Selectivity, Strategy & Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I.; Paquette, L. A., Eds.; Pergamon Press: Oxford, 1991; Vol. 5, pp 899–970.
Return to citation in text: [1] -
Baldwin, J. E. Chem. Rev. 2003, 103, 1197–1212. doi:10.1021/cr010020z
Return to citation in text: [1] -
Wang, S. C.; Tantillo, D. J. J. Organomet. Chem. 2006, 691, 4386–4392. doi:10.1016/j.jorganchem.2005.12.052
Return to citation in text: [1] -
Hayashi, M.; Ohmatsu, T.; Meng, Y.-P.; Saigo, K. Angew. Chem., Int. Ed. 1998, 110, 877–879. doi:10.1002/(SICI)1521-3773(19980403)37:6<837::AID-ANIE837>3.0.CO;2-R
Return to citation in text: [1] [2] -
Zuo, G.; Louie, J. Angew. Chem., Int. Ed. 2004, 43, 2277–2279. doi:10.1002/anie.200353469
Angew. Chem. 2004, 116, 2327–2329. doi:10.1002/ange.200353469
Return to citation in text: [1] -
Hiroi, K.; Arinaga, Y. Tetrahedron Lett. 1994, 35, 153–156. doi:10.1016/0040-4039(94)88188-X
Return to citation in text: [1] -
Morizawa, Y.; Oshima, K.; Nozaki, H. Tetrahedron Lett. 1982, 23, 2871–2874. doi:10.1016/S0040-4039(00)88436-1
Return to citation in text: [1] -
Davies, H. M. L.; Hu, B. J. Org. Chem. 1992, 57, 3186–3190. doi:10.1021/jo00037a041
Return to citation in text: [1] -
Hudlicky, T.; Koszyk, F. J.; Kutchan, T. M.; Sheth, J. P. J. Org. Chem. 1980, 45, 5020–5027. doi:10.1021/jo01313a003
Return to citation in text: [1] -
Buchert, M.; Reissig, H.-U. Liebigs Ann. 1996, 2007–2013. doi:10.1002/jlac.199619961210
Return to citation in text: [1] -
Davies, H. M. L.; Hu, B. Tetrahedron Lett. 1992, 33, 453–456.
Return to citation in text: [1] -
Doyle, M. P.; van Leusen, D. J. Org. Chem. 1982, 47, 5326–5339. doi:10.1021/jo00148a020
Return to citation in text: [1] -
Brandi, A.; Cicchi, S.; Brandl, M.; Kozhushkov, S. I.; de Meijere, A. Synlett 2001, 433–435. doi:10.1055/s-2001-11396
Example for a study of the thermal VCP to cyclopentene rearrangement in such compounds.
Return to citation in text: [1] -
Wender, P. A.; Takahashi, H.; Witulski, B. J. Am. Chem. Soc. 1995, 117, 4720–4721. doi:10.1021/ja00121a036
Return to citation in text: [1] -
Wegner, H. A.; de Meijere, A.; Wender, P. A. J. Am. Chem. Soc. 2005, 127, 6530–6531. doi:10.1021/ja043671w
Return to citation in text: [1] -
Sánchez-Izquierdo, F.; Blanco, P.; Busqué, F.; Alibés, R.; de March, P.; Figueredo, M.; Font, J.; Parella, T. Org. Lett. 2007, 9, 1769–1772. doi:10.1021/ol070486p
Return to citation in text: [1] -
Brandi, A.; Garro, S.; Guarna, A.; Goti, A.; Cordero, F.; De Sarlo, F. J. Org. Chem. 1988, 53, 2430–2434. doi:10.1021/jo00246a008
Return to citation in text: [1] -
Yamazaki, S. Bull. Chem. Soc. Jpn. 1997, 70, 877–883. doi:10.1246/bcsj.70.877
Return to citation in text: [1] -
Zhao, B.-X.; Yu, Y.; Eguchi, S. Org. Prep. Proced. Int. 1997, 29, 185–194. doi:10.1080/00304949709355182
Return to citation in text: [1] -
Cicchi, S.; Corsi, M.; Goti, A. J. Org. Chem. 1999, 64, 7243–7245. doi:10.1021/jo990417r
Return to citation in text: [1] -
Revuelta, J.; Cicchi, S.; de Meijere, A.; Brandi, A. Eur. J. Org. Chem. 2008, 1085–1091. doi:10.1002/ejoc.200700912
Return to citation in text: [1] -
Wender, P. A.; Dyckman, A. J. Org. Lett. 1999, 1, 2089–2092. doi:10.1021/ol991171f
Return to citation in text: [1] -
Wender, P. A.; Barzilay, C. M.; Dyckman, A. J. J. Am. Chem. Soc. 2001, 123, 179–180. doi:10.1021/ja0021159
Return to citation in text: [1] -
Salomon, R. G.; Salomon, M. F.; Kachinski, J. L. C. J. Am. Chem. Soc. 1977, 99, 1043–1054. doi:10.1021/ja00446a012
Return to citation in text: [1] -
Salomon, M. F.; Salomon, R. G. J. Chem. Soc., Chem. Commun. 1976, 89–90. doi:10.1039/C39760000089
Return to citation in text: [1]
| 45. | Wender, P. A.; Barzilay, C. M.; Dyckman, A. J. J. Am. Chem. Soc. 2001, 123, 179–180. doi:10.1021/ja0021159 |
| 43. | Revuelta, J.; Cicchi, S.; de Meijere, A.; Brandi, A. Eur. J. Org. Chem. 2008, 1085–1091. doi:10.1002/ejoc.200700912 |
| 44. | Wender, P. A.; Dyckman, A. J. Org. Lett. 1999, 1, 2089–2092. doi:10.1021/ol991171f |
| 1. | de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/ange.19790911104 |
| 2. | Salaün, J. In The Chemistry of the Cyclopropyl Group; Patai, S.; Rappoport, Z., Eds.; Wiley: New York, 1987; pp 809–878. |
| 3. | Salaün, J. Top. Curr. Chem. 2000, 207, 1–67. doi:10.1007/3-540-48255-5_1 |
| 10. | Brandi, A.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. Rev. 2003, 103, 1213–1269. doi:10.1021/cr010005u |
| 11. | Goti, A.; Anichini, B.; Brandi, A.; Kozhushkov, S. I.; Gratkowski, C.; de Meijere, A. J. Org. Chem. 1996, 61, 1665–1672. doi:10.1021/jo951838l |
| 8. | Brandi, A.; Cordero, F. M.; De Sarlo, F.; Goti, A.; Guarna, A. Synlett 1993, 1–8. doi:10.1055/s-1993-22329 |
| 9. | Goti, A.; Cordero, F. M.; Brandi, A. Top. Curr. Chem. 1996, 178, 1–97. doi:10.1007/3-540-60495-2_1 |
| 42. | Cicchi, S.; Corsi, M.; Goti, A. J. Org. Chem. 1999, 64, 7243–7245. doi:10.1021/jo990417r |
| 5. | Brandi, A.; Goti, A. Chem. Rev. 1998, 98, 589–636. doi:10.1021/cr940341t |
| 6. | de Meijere, A.; Kozhushkov, S. I. Eur. J. Org. Chem. 2000, 3809–3822. doi:10.1002/1099-0690(200012)2000:23<3809::AID-EJOC3809>3.0.CO;2-X |
| 7. | de Meijere, A.; Kozhushkov, S. I. Chem. Rev. 2000, 100, 93–142. doi:10.1021/cr960153y |
| 39. | Brandi, A.; Garro, S.; Guarna, A.; Goti, A.; Cordero, F.; De Sarlo, F. J. Org. Chem. 1988, 53, 2430–2434. doi:10.1021/jo00246a008 |
| 40. | Yamazaki, S. Bull. Chem. Soc. Jpn. 1997, 70, 877–883. doi:10.1246/bcsj.70.877 |
| 41. | Zhao, B.-X.; Yu, Y.; Eguchi, S. Org. Prep. Proced. Int. 1997, 29, 185–194. doi:10.1080/00304949709355182 |
| 4. | de Meijere, A., Ed. Houben−Weyl, Carbocyclic Three-Membered Ring Compounds; Thieme: Stuttgart, 1997; Vol. E17. |
| 13. | Zorn, C.; Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S. I.; de Meijere, A.; Citti, L. J. Org. Chem. 1999, 64, 7846–7855. doi:10.1021/jo990873f |
| 35. |
Brandi, A.; Cicchi, S.; Brandl, M.; Kozhushkov, S. I.; de Meijere, A. Synlett 2001, 433–435. doi:10.1055/s-2001-11396
Example for a study of the thermal VCP to cyclopentene rearrangement in such compounds. |
| 36. | Wender, P. A.; Takahashi, H.; Witulski, B. J. Am. Chem. Soc. 1995, 117, 4720–4721. doi:10.1021/ja00121a036 |
| 37. | Wegner, H. A.; de Meijere, A.; Wender, P. A. J. Am. Chem. Soc. 2005, 127, 6530–6531. doi:10.1021/ja043671w |
| 26. | Hayashi, M.; Ohmatsu, T.; Meng, Y.-P.; Saigo, K. Angew. Chem., Int. Ed. 1998, 110, 877–879. doi:10.1002/(SICI)1521-3773(19980403)37:6<837::AID-ANIE837>3.0.CO;2-R |
| 27. |
Zuo, G.; Louie, J. Angew. Chem., Int. Ed. 2004, 43, 2277–2279. doi:10.1002/anie.200353469
Angew. Chem. 2004, 116, 2327–2329. doi:10.1002/ange.200353469 |
| 28. | Hiroi, K.; Arinaga, Y. Tetrahedron Lett. 1994, 35, 153–156. doi:10.1016/0040-4039(94)88188-X |
| 29. | Morizawa, Y.; Oshima, K.; Nozaki, H. Tetrahedron Lett. 1982, 23, 2871–2874. doi:10.1016/S0040-4039(00)88436-1 |
| 30. | Davies, H. M. L.; Hu, B. J. Org. Chem. 1992, 57, 3186–3190. doi:10.1021/jo00037a041 |
| 31. | Hudlicky, T.; Koszyk, F. J.; Kutchan, T. M.; Sheth, J. P. J. Org. Chem. 1980, 45, 5020–5027. doi:10.1021/jo01313a003 |
| 32. | Buchert, M.; Reissig, H.-U. Liebigs Ann. 1996, 2007–2013. doi:10.1002/jlac.199619961210 |
| 33. | Davies, H. M. L.; Hu, B. Tetrahedron Lett. 1992, 33, 453–456. |
| 34. | Doyle, M. P.; van Leusen, D. J. Org. Chem. 1982, 47, 5326–5339. doi:10.1021/jo00148a020 |
| 38. | Sánchez-Izquierdo, F.; Blanco, P.; Busqué, F.; Alibés, R.; de March, P.; Figueredo, M.; Font, J.; Parella, T. Org. Lett. 2007, 9, 1769–1772. doi:10.1021/ol070486p |
| 18. | Neureiter, N. P. J. Org. Chem. 1959, 24, 2044–2046. doi:10.1021/jo01094a621 |
| 19. | Overberger, C. G.; Borchert, A. E. J. Am. Chem. Soc. 1960, 82, 1007–1008. doi:10.1021/ja01489a069 |
| 20. | Flowers, M. C.; Frey, H. M. J. Chem. Soc. 1961, 82, 3547–3548. |
| 21. | Hudlicky, T.; Becker, D. A.; Fan, R. L.; Kozhushkov, S. I. In Houben−Weyl; de Meijere, A., Ed.; Thieme: Stuttgart, 1997; Vol. E17c, pp 2538–2565. |
| 22. | Hudlicky, T.; Fan, R. L.; Reed, J. W.; Gadamasetti, K. G. Org. React. 1992, 41, 1–133. doi:10.1002/0471264180.or041.01 |
| 23. | Hudlicky, T.; Reed, J. W. In Comprehensive Organic Synthesis-Selectivity, Strategy & Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I.; Paquette, L. A., Eds.; Pergamon Press: Oxford, 1991; Vol. 5, pp 899–970. |
| 24. | Baldwin, J. E. Chem. Rev. 2003, 103, 1197–1212. doi:10.1021/cr010020z |
| 25. | Wang, S. C.; Tantillo, D. J. J. Organomet. Chem. 2006, 691, 4386–4392. doi:10.1016/j.jorganchem.2005.12.052 |
| 46. | Salomon, R. G.; Salomon, M. F.; Kachinski, J. L. C. J. Am. Chem. Soc. 1977, 99, 1043–1054. doi:10.1021/ja00446a012 |
| 47. | Salomon, M. F.; Salomon, R. G. J. Chem. Soc., Chem. Commun. 1976, 89–90. doi:10.1039/C39760000089 |
| 11. | Goti, A.; Anichini, B.; Brandi, A.; Kozhushkov, S. I.; Gratkowski, C.; de Meijere, A. J. Org. Chem. 1996, 61, 1665–1672. doi:10.1021/jo951838l |
| 12. | Goti, A.; Anichini, B.; Brandi, A.; de Meijere, A.; Citti, L.; Nevischi, S. Tetrahedron Lett. 1995, 36, 5811–5814. doi:10.1016/0040-4039(95)01103-O |
| 13. | Zorn, C.; Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S. I.; de Meijere, A.; Citti, L. J. Org. Chem. 1999, 64, 7846–7855. doi:10.1021/jo990873f |
| 14. | Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S. I.; de Meijere, A. Synlett 1997, 25–26. doi:10.1055/s-1997-684 |
| 15. | Marradi, M.; Brandi, A.; Magull, J.; Schill, H.; de Meijere, A. Eur. J. Org. Chem. 2006, 5485–5494. doi:10.1002/ejoc.200600417 |
| 16. | Zorn, C.; Goti, A.; Brandi, A.; Johnsen, K.; Noltemeyer, M.; Kozhushkov, S. I.; de Meijere, A. J. Org. Chem. 1999, 64, 755–763. doi:10.1021/jo981366l |
| 17. | Revuelta, J.; Cicchi, S.; Faggi, C.; Kozhushkov, S. I.; de Meijere, A.; Brandi, A. J. Org. Chem. 2006, 71, 2417–2423. doi:10.1021/jo052564x |
| 26. | Hayashi, M.; Ohmatsu, T.; Meng, Y.-P.; Saigo, K. Angew. Chem., Int. Ed. 1998, 110, 877–879. doi:10.1002/(SICI)1521-3773(19980403)37:6<837::AID-ANIE837>3.0.CO;2-R |
© 2011 Cordero et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)