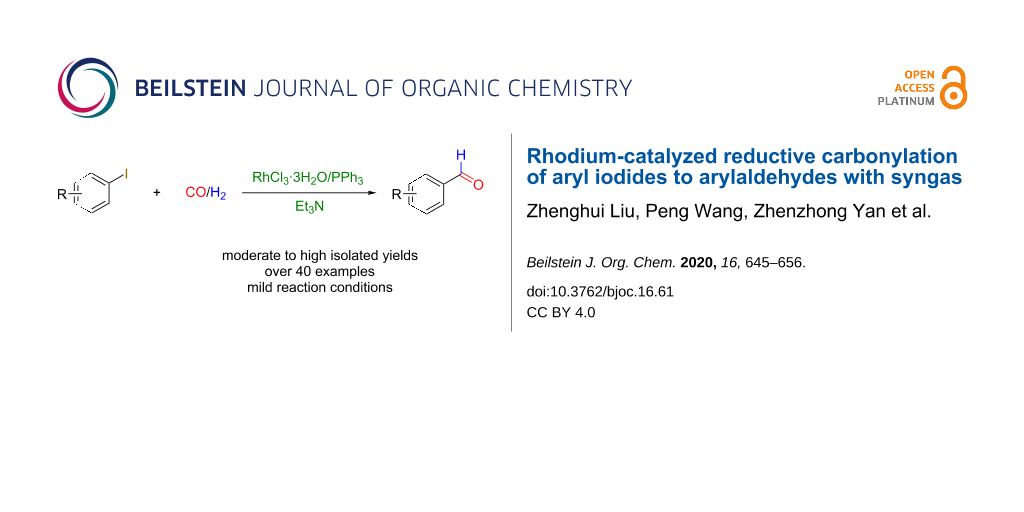
Abstract
The reductive carbonylation of aryl iodides to aryl aldehydes possesses broad application prospects. We present an efficient and facile Rh-based catalytic system composed of the commercially available Rh salt RhCl3·3H2O, PPh3 as phosphine ligand, and Et3N as the base, for the synthesis of arylaldehydes via the reductive carbonylation of aryl iodides with CO and H2 under relatively mild conditions with a broad substrate range affording the products in good to excellent yields. Systematic investigations were carried out to study the experimental parameters. We explored the optimal ratio of Rh salt and PPh3 ligand, substrate scope, carbonyl source and hydrogen source, and the reaction mechanism. Particularly, a scaled-up experiment indicated that the catalytic method could find valuable applications in industrial productions. The low gas pressure, cheap ligand and low metal dosage could significantly improve the practicability in both chemical researches and industrial applications.

Graphical Abstract
Introduction
The exploration of environmentally friendly and highly effective synthetic methods has been a significant goal of research [1-5]. In this aspect, effective catalytic systems and organometallic chemistry are suitable technologies to accomplish these goals. Carbonylation processes are important transformations in the refinement and reprocessing of readily available industrial raw materials into more functionalized products. These processes generally utilize carbon monoxide (CO), currently the most important C1 building block used in numerous industrial carbonylation processes [6-8] and widely applied in industrial productions [9-12]. Carbonylations are one of the industrial core technologies for transforming various bulk chemicals into useful products that are used in our daily life. Carbonylation reactions, together with polymerizations and oxidations, constitute the largest industrial applications in the field of homogeneous catalysis, and substantial value-added bulk and fine chemicals are available through this technology [13]. In spite of the existing plentiful progress in this conversion, the exploitation of advanced and more effective catalytic systems to the activity and to widen the range of substrates is crucial for new practical applications.
Syngas is a mixture of CO and H2, which is cheap, abundant and widely used in chemical industry productions [14-16]. In spite of its comprehensive utilization in industry, reactions involving CO are relatively seldom employed in fine chemicals syntheses. This could be due to the general difficulty of using gases as raw materials and the requirement of high-pressure equipment. In addition, relatively little attention has been paid to carbonylation chemistry using CO in academic research. Also, H2 as representative clean energy source is far more environmentally friendly than other frequently used hydrogen sources like hydrosilanes [17], tributyltin hydride (Bu3SnH) (often used in natural product syntheses) [18-20] and hydroboranes [21-23], since the only byproduct is water. The production, storage and use of H2 received much attention and plentiful achievements promoted the application of H2 into more and more chemistry researches and industrial productions [24-27].
Aromatic aldehydes are highly valuable organic compounds that are widely employed as indispensable building blocks in numerous areas of chemistry, especially for the preparation of biologically active molecules or their intermediates [28,29]. Generally, aromatic aldehydes are synthesized by Reimer–Tiemann, Gattermann–Koch, Vielsmeier–Haag, or Duff reactions and so forth. Unfortunately, these reactions usually use auxiliary reagents and thus generate large amounts of industrial waste and other side products. Particularly, the reductive carbonylation of aryl iodides to produce arylaldehydes with CO and H2 was seldom reported. Some homogeneous and heterogeneous catalytic systems based on palladium species using CO and H2 to complete the reductive carbonylation of aryl halogens to arylaldehydes have been developed. The homogeneous systems included Pd(OAc)2 with propyl di-tert-butylphosphinite ligand [30], Pd(acac)2 with dppm ligand [31], Pd(OAc)2 with CataCXium A ligand [32], and all of the three systems employed TMEDA as the base and toluene as the solvent. The heterogeneous systems contained: PdO/Co3O4 with K2CO3 [33], MCM-41-S-PdCl2 [34], and MCM-41-2P-PdCl2 [35]. However, the aforementioned systems often suffer from high toxicity of solvents, high pressure of gases or high reaction temperatures, which make these protocols inapplicable for large scale applications. Actually, in 2004, Eliseev et al. reported a catalytic system based on RhCl(CO)(PPh3)2 to achieve the conversion of iodobenzene to benzaldehyde in toluene using CO and H2 [36]. We sought for a commercially available Rh salt for this conversion at lower cost and higher potential for practical application.
In this work, we established a catalytic system composed of RhCl3·3H2O and PPh3, which allows the reductive carbonylation of aryl iodides using CO and H2 in the presence of Et3N as the base at 90 °C. In addition, the reported catalytic system demonstrates high catalytic activity affording the arylaldehydes in good to excellent yields, displays high functional-group tolerance, and broad substrate scope. In particular, the catalytic system could be applied in a scaled-up experiment and thus has potential for applications in industrial productions. The reaction mechanism study revealed that RhCl3·3H2O reacts with PPh3 to form RhCl(PPh3)3, which is able to activate C–I bonds in aryl iodides realizing the insertion of CO and hydrogenolysis with H2. The final trapping of HI by the base Et3N regenerates the catalyst to complete the reaction cycle. As far as we know, this is the first time that commercially available Rh salts with PPh3 as the ligand were utilized to complete the conversion of aryl iodides into arylaldehydes using CO and H2 with systematic researches. Considering the efficiency and generality, this catalytic system is expected to powerfully influence both laboratory research and chemical industry by offering a practical synthetic tool for the conversion of aryl iodides to arylaldehydes.
Results and Discussion
Effects of Rh species
Rhodium salts coordinated with proper ligands have been reported to be able to realize the activation of CO and H2 and thus might achieve the reductive carbonylation of aryl iodides to afford aromatic aldehydes [37-40]. Rh salts generally play vital roles in the catalytic results. Therefore, we tested 20 different Rh salts and the results are summarized in Table 1. The initial reaction conditions were set as PhI (1 mmol), Rh species (2.5 mol %), PPh3 (10 mol %), Et3N (1.2 mmol), DMA (2 mL), CO/H2 (5 bar:5 bar), 90 °C and 12 h. As can be seen from Table 1, most of the Rh salts gave unsatisfactory results with yields of benzaldehyde below 50%. However, three Rh salts provided yields over 50%, namely [RhCl(CO)2]2 (64%), RhCl3·3H2O (97%) and RhI3 (89%) (Table 1, entries 1, 18 and 20). Interestingly, RhCl3·3H2O and RhI3 performed well, whereas RhCl3 and RhBr2·2H2O afforded the product in very low yield (Table 1, entries 17–20). This strongly implied that not only the valence state of Rh and the species of anions were crucial for the conversion (even if all with halogen anions), but also structural differences (e.g., crystal water, RhCl3 vs RhCl3·3H2O) of analogous Rh salts played an important role in the catalytic synthesis of benzaldehyde. Among all tested rhodium salts, RhCl3·3H2O afforded benzaldehyde in the highest yield and was chosen as the most suitable Rh salt for the conversion. In addition, in spite of low yields, Rh species with valence states of 0, +1, +2 or +3 all promoted the reaction at least to some extent. It is worth noting that as a good leaving group, I− tends to leave in an alkaline environment, and dehalogenation and direct coupling products (namely, benzene and biphenyl) were detected as the main byproducts by GC–MS. This observation also explains why the conversions were always higher than the yields.
Table 1: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde: effects of the Rh speciesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-61-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Rh species | Conversion [%]b | Yield [%]b |
| 1 | [RhCl(CO)2]2 | 89 | 64 |
| 2 | RhCl(CO)(PPh3)2 | 37 | 31 |
| 3 | Rh(acac)(CO)2 | 21 | 15 |
| 4 | [RhCl(COD)]2 | 16 | 14 |
| 5 | Rh(MeCN)2(COD)BF4 | 15 | 12 |
| 6 | Rh(COD)BF4 | 27 | 21 |
| 7 | Rh(COD)2OTf | 21 | 18 |
| 8 | [Rh(OMe)(COD)]2 | 23 | 19 |
| 9 | Rh(OAc)2 | 71 | 33 |
| 10 | [Rh(CF3COO)]2 | 43 | 12 |
| 11 | [Rh(CH3(CH2)6CO2)2]2 | 29 | 22 |
| 12 | Rh(ethylene)2(acac) | 48 | 42 |
| 13 | [Rh(ethylene)2Cl]2 | 42 | 36 |
| 14 | Rh(norbornadiene)2BF4 | 25 | 10 |
| 15 | [Cp*RhCl2]2 | 51 | 46 |
| 16 | Rh/C | 38 | 21 |
| 17 | RhCl3 | 22 | 18 |
| 18 | RhCl3·3H2O | 100 | 97 |
| 19 | RhBr2·2H2O | 31 | 28 |
| 20 | RhI3 | 92 | 89 |
aStandard conditions: PhI (1 mmol), Rh species (2.5 mol %), PPh3 (10 mol %), Et3N (1.2 mmol), DMA (2 mL), CO/H2 (5 bar:5 bar), 90 °C, 12 h. bDetermined by GC using dodecane as an internal standard.
Effects of ligands
Ligands play a decisive role in adjusting the catalytic ability of metal cations [12,13,41-51]. Different ligands coordinating with the same metal cations could make a difference between full conversion with nearly quantitative yields and no reactions. To obtain the optimized conditions, 13 kinds of ligands were tested and their structures are included in Table 2. In accordance with the expectations, only PPh3 was effective and afforded benzaldehyde in 97% yield, whereas the majority of the other ligands did not afford any product (Table 2). Therefore, PPh3 was selected as the proper ligand employed in the subsequent reactions.
Table 2: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde: effects of the ligandsa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-61-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | Ligand | Conversion [%]b | Yield [%]b |
| 1 | PP3 | 8 | 0 |
| 2 | triphos | 5 | 0 |
| 3 | dpp-BINAP | 12 | 0 |
| 4 | dpp-OPh | 7 | 3 |
| 5 | dppb | 6 | 0 |
| 6 | dppe | 11 | 0 |
| 7 | P(PhF5)3 | 16 | 0 |
| 8 | P(4-FPh)3 | 13 | 0 |
| 9 | Cydpp | 8 | 2 |
| 10 | Bipy | 9 | 4 |
| 11 | DBU | 3 | 0 |
| 12 | Im | 2 | 0 |
| 13 | PPh3 | 100 | 97 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-61-i8.svg?max-width=637&scale=1.18182)
aStandard conditions: PhI (1 mmol), RhCl3·3H2O (2.5 mol %), ligand (10 mol %), Et3N (1.2 mmol), DMA (2 mL), CO/H2 (5 bar:5 bar), 90 °C, 12 h. bDetermined by GC using dodecane as an internal standard.
Effects of bases
As explored in the mechanism study, the acid HI is produced during the reaction process, since H+ and I− were generated from the reaction system. In order to make the reaction proceed continuously, HI produced needs to be removed effectively and in time. Five different bases were examined to assess their ability to bind HI. All of them were found suitable for the reaction and provided benzaldehyde with yields higher than 70%, except for TMEDA (53%, Figure 1a). The best results were obtained with Et3N, which was selected as the most appropriate base.
![[1860-5397-16-61-1]](/bjoc/content/figures/1860-5397-16-61-1.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 1: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde. a) Effects of the tested bases; b) effects of base amount; c) effects of tested solvents; d) temperature screen; e) yields at different times for reactions performed at 80, 90 and 100 °C (every point represents an average result of three parallel experiments). Conversions and yields were determined by GC using dodecane as an internal standard. Standard conditions: PhI (1 mmol), RhCl3·3H2O (2.5 mol %), PPh3 (10 mol %), base (1.2 mmol, if not stated otherwise), DMA (2 mL, if not stated otherwise), CO/H2 (5 bar:5 bar), temperature 90 °C (if not stated otherwise), time 12 h (if not stated otherwise).
Figure 1: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde. a) ...
The optimized base concentration was explored next and the results are shown in Figure 1b. Since the base held the post of absorber for acids produced during the reaction, sufficient amounts are necessary to ensure high yields. As shown in Figure 1b, amounts of Et3N less than 1 mmol led to lower yields of 33% for 0.5 mmol Et3N and 61% for 0.8 mmol Et3N, respectively. However, a slight excess of Et3N (1.2 mmol) allowed the reaction to be completed with a product yield of 97%. In addition, further increasing the amount of the base was not beneficial for the yield of benzaldehyde (1.5 mmol Et3N, 95% yield; 2 mmol Et3N, 92% yield; 3 mmol Et3N, 89% yield). Thus, the appropriate amount of Et3N was set at 1.2 mmol.
Effect of solvents
Solvents act as the reaction media and strongly influence the catalytic reactions [52,53]. We screened nine representative solvents in the catalytic reaction and the results are summarized in Figure 1c. Apparently, protic solvents like EtOH or MeOH and nonpolar solvents like THF or toluene were inappropriate for the reaction leading to yields lower than 10%. Medium polar solvents like MeCN or 1,4-dioxane afforded slightly higher yields but still below 15%. To our delight, polar solvents with high boiling points, NMP, DMI or DMA proved to be more suitable, and afforded benzaldehyde in 32%, 54% and 97% yield, respectively, likely owing to the high boiling point and better CO and H2 dissolution ability, highlighting a strong solvent dependency of the system [54,55]. Therefore, DMA was selected as the most suitable solvent for the reaction.
Effects of temperature and time
To further optimize the performance of the catalytic system, the effect of the reaction temperature was screened in the range of 50 °C to 100 °C. The results revealed that the reaction temperature had an important influence on the catalytic process and the product yield increased with increasing temperature. At low temperature the reaction did not proceed at all, however, at 90 °C or higher, a nearly quantitative yield of benzaldehyde was obtained (Figure 1d). Next, parallel experiments were conducted at three selected temperatures (80 °C, 90 °C, and 100 °C) to study the yields at different reaction times. The results revealed that equally high yields were obtained at 90 °C and 100 °, but the reaction was much faster at 100 °C than at 90 °C (Figure 1e). On the other hand, when performing the reaction at 80 °C, the product yield was lower and required longer times to be reached.
Effects of pressure of CO and H2
The influence of the pressures of CO and H2 was explored next. As expected, a low pressure of both, CO (1 atm) and H2 (1 atm), resulted in a lower yield of benzaldehyde (24% yield, Table 3, entry 1). When the pressures of both gases were increased to 2, 3, 4, or 5 bar, the yield of benzaldehyde accordingly increased to 51%, 62% 81%, and 97%, respectively (Table 3, entries 2–5). However, further increasing the pressures did not lead to higher yields (Table 3, entries 6 and 7). After that, we examined the proportions of the syngas components on the reaction and it was found that the optimized proportions of syngas were 5 bar CO and 5 bar H2 (1:1, (Table 3, entries 8–13).
Table 3: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde: effect of pressures of CO and H2a.
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-61-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry |
p(CO)
[bar] |
p(H2)
[bar] |
Conversion
[%]b |
Yield
[%]b |
| 1 | 1 | 1 | 32 | 24 |
| 2 | 2 | 2 | 63 | 51 |
| 3 | 3 | 3 | 74 | 62 |
| 4 | 4 | 4 | 92 | 81 |
| 5 | 5 | 5 | 100 | 97 |
| 6 | 6 | 6 | 100 | 97 |
| 7 | 7 | 7 | 100 | 97 |
| 8 | 2 | 4 | 72 | 63 |
| 9 | 4 | 2 | 66 | 58 |
| 10 | 3 | 6 | 85 | 72 |
| 11 | 6 | 3 | 74 | 68 |
| 12 | 4 | 6 | 99 | 94 |
| 13 | 6 | 4 | 95 | 89 |
aStandard conditions: PhI (1 mmol), RhCl3·3H2O (2.5 mol %), PPh3 (10 mol %), Et3N (1.2 mmol), DMA (2 mL), CO/H2, 90 °C, 12 h. bDetermined by GC using dodecane as an internal standard.
Scaled-up experiment
Noteworthy, when conducting the reaction at a larger scale (10 mmol), benzaldehyde was obtained with a high yield of 93%, indicating that our system could be suitable for industrial application (Scheme 1).
![[1860-5397-16-61-i1]](/bjoc/content/inline/1860-5397-16-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Scaled-up experiment of the reductive carbonylation of iodobenzene to benzaldehyde under the optimized conditions.
Scheme 1: Scaled-up experiment of the reductive carbonylation of iodobenzene to benzaldehyde under the optimi...
Optimal ratio of Rh salt and PPh3 ligand and active species participating in the catalytic process
In order to ascertain the optimized ratio of RhCl3·3H2O and the PPh3 ligand, a series of experiments with different concentrations and ratios of RhCl3·3H2O and PPh3 were conducted. At first, the concentration of RhCl3·3H2O was fixed at 1 mol %. When changing the amount of PPh3 from 2 mol % to 5 mol %, the yield gradually increased reaching a maximum yield of 74% at 4 mol % PPh3. Further increasing the amount of the ligand resulted in a decrease of the product yield (Table 4, entries 1–4). Next, the concentration of RhCl3·3H2O was increased to 2 mol % and 2.5 mol %, leading to the same results. In other words, the optimal ratio of RhCl3·3H2O and the PPh3 ligand was always 1:4 (Table 4, entries 5–12). No higher yields (Table 4, entries 13–15) could be obtained by increasing the loadings of RhCl3·3H2O and PPh3.
The effects of PPh3 dosage on the catalytic ability demonstrated that under otherwise identical conditions, a maximum yield of benzaldehyde (97%) was obtained with a 1:4 molar ratio of RhCl3·3H2O/PPh3, while higher or lower concentrations of PPh3 both led to decreased yields (Table 4). This indicated that in the active catalytic species the molar ratio of [Rh]:PPh3 was exactly 1:3 (not 1:4 because of the consumption of 1 equiv PPh3 during the redox process), which might be Rh(PPh3)3Cl. The mentioned redox reaction can be explained as shown in Scheme 2a. Initially, RhCl3·3H2O is reduced by 1 equiv PPh3 to form a Rh(I) species that subsequently combines with 3 equiv PPh3 forming Rh(PPh3)3Cl. During this process PPh3 is oxidized to O=PPh3, and HCl and H2O are released.
Table 4: Rhodium-catalyzed reductive carbonylation of iodobenzene with CO and H2 to afford benzaldehyde: effects of dosage of RhCl3·3H2O and PPh3a.
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-61-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry |
RhCl3·3H2O
[mol %] |
PPh3
[mol %] |
Conversion
[%]b |
Yield
[%]b |
| 1 | 1 | 2 | 35 | 28 |
| 2 | 1 | 3 | 51 | 47 |
| 3 | 1 | 4 | 78 | 74 |
| 4 | 1 | 5 | 72 | 70 |
| 5 | 2 | 4 | 34 | 31 |
| 6 | 2 | 6 | 65 | 62 |
| 7 | 2 | 8 | 92 | 88 |
| 8 | 2 | 10 | 88 | 84 |
| 9 | 2.5 | 5 | 47 | 42 |
| 10 | 2.5 | 7.5 | 81 | 73 |
| 11 | 2.5 | 10 | 100 | 97 |
| 12 | 2.5 | 12.5 | 100 | 92 |
| 13 | 3 | 12 | 100 | 97 |
| 14 | 4 | 16 | 100 | 97 |
| 15 | 5 | 20 | 100 | 97 |
aStandard conditions: PhI (1 mmol), RhCl3·3H2O, PPh3, Et3N (1.2 mmol), DMA (2 mL), CO/H2 (5 bar:5 bar), 90 °C, 12 h. bDetermined by GC using dodecane as an internal standard.
To test this assumption, we synthesized Rh(PPh3)3Cl according to the literature [56] on account of no commercial sources and employed it as the catalyst in the reaction instead of RhCl3·3H2O and PPh3 and a 93% yield of benzaldehyde was obtained under otherwise identical reaction conditions (Scheme 2b). This could be an evidence that after the reduction of one equivalent RhCl3 complexation with three molecular PPh3 takes place forming Rh(PPh3)3Cl, which participated in the subsequent catalytic process. Herein, Rh(III) is reduced to Rh(I), with concomitant oxidation of PPh3 to PPh3=O. A quite interesting fact is that Rh(PPh3)3Br and Rh(PPh3)3I afforded much lower yields of the aldehyde compared to Rh(PPh3)3Cl although they share the identical metal center and ligand (Scheme 2c and 2d). This indicated that the anion plays an important role in the catalytic process.
![[1860-5397-16-61-i2]](/bjoc/content/inline/1860-5397-16-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Catalytic species participating in the catalytic process.
Scheme 2: Catalytic species participating in the catalytic process.
Substrate scope
Next, the scope of the reductive carbonylation reaction was explored (shown in Scheme 3), after having identified the optimized conditions for benzaldehyde synthesis from iodobenzene. The results showed that electronic effects had little impact on the reaction. Both, substrates with electron-withdrawing or electron-donating groups afforded similar yields. However, steric effects played an important role in the catalytic process, i.e., the yields were influenced by the substituent group positions following the order of ortho < meta < para. The obtained isolated yields were slightly lower than the NMR yields owing to a loss of products during the workup process. Iodobenzene with no substituent group provided benzaldehyde (1) with 93% yield. For iodobenzene derivatives with electron-donating groups (Me, OMe), 83–95% yield of aldehydes 2–7 were obtained. As expected, the yields increased in the order of ortho < meta < para-substituted compounds. As for aryl iodides with halide substituents (F, Cl, Br), these were also compatible with the reaction system, affording 79−89% yields of the halogenated aromatic aldehydes 8–16. Iodobenzene derivatives with phenyl or tert-butyl groups displaying larger steric hindrance in the para-position still provided the corresponding aldehydes 17 and 18 in relatively high yields (85% and 81%, respectively). 2-Iodonaphthalene with smaller steric hindrance performed a little better than 1-iodonaphthalene (19, 84% and 20, 77%). However, starting iodides having methyl substituents in both ortho-positions afforded the corresponding aldehydes in much lower yields (21 61% and 22 60%). Also, iodobenzenes with an acetyl group in either ortho, meta, or para-position gave the products in satisfactory yields (23 79%, 24 81% and 25 87%). 1-Iodo-3,4-methylenedioxybenzene performed well providing aldehyde 26 with 92% yield. Aryl iodide with an acetamido group in the para-position gave a medium yield (27 82%). Aryl iodides containing one or two trifluoromethyl groups in their structure worked slightly less efficient producing arylaldehydes 28–30 with yields of 72–79%. It is worth noticing that a cyano group in the substrate stayed intact in the catalytic process giving aldehyde 31 with 77% yield. Also substrates comprising two halogen substituents performed well offering access to dihalogenated aldehydes 32–35 with yields between 76–81%. In addition, heterocyclic iodides were also amenable to the reductive carbonylation reaction and the corresponding aldehydes 36–42 were isolated with yields of 65−73%.
![[1860-5397-16-61-i3]](/bjoc/content/inline/1860-5397-16-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope for the Rh-catalyzed reductive carbonylation of aryl iodides using CO and H2. Reaction conditions: PhI (1 mmol), RhCl3·3H2O (2.5 mol %), PPh3 (10 mol %), Et3N (1.2 mmol), DMA (2 mL), CO (5 bar), H2 (5 bar), 90 °C, 12 h. Isolated yields of the products are given and the structures were determined by NMR. Detailed information is given in Supporting Information File 1.
Scheme 3: Substrate scope for the Rh-catalyzed reductive carbonylation of aryl iodides using CO and H2. React...
Isotope labeling experiments
Isotope labeling experiments were conducted to study the mechanism of the reductive carbonylation of aryl iodide with CO and H2 under our optimized conditions, using 13CO and D2 instead of CO and H2, respectively, as the sources of the carbonyl group and hydrogen in the formyl group. C6H513CHO, C6H5CDO, and C6H513CDO could be verified by the molecular ion peaks at m/z 107, 107, and 108 in the GC–MS spectrum of the reaction solution (for the spectrum, see Supporting Information File 1). This result confirmed that both CO and H2 participated in the formation of the formyl group in the product (Scheme 4a–c).
![[1860-5397-16-61-i4]](/bjoc/content/inline/1860-5397-16-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Reaction mechanism and role of each component
Based on the results from the labeling experiments a reaction mechanism for the reductive carbonylation of aryl iodides was proposed, as shown in Scheme 5 [57]. First, RhCl3·3H2O reacted with PPh3 to form Rh(PPh3)3Cl (A), followed by an oxidative addition of Rh(PPh3)3Cl (A) to the aryl iodide, producing the corresponding arylrhodium complex (B). Then, the coordination and insertion of CO led to the formation of benzoylrhodium complex (C). Next, metathesis with H2 afforded the aldehyde product. The base, Et3N neutralized the proton in the rhodium hydroiodide complex (D) and regenerated the active Rh species [11].
![[1860-5397-16-61-i5]](/bjoc/content/inline/1860-5397-16-61-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed reaction mechanism for the Rh-catalyzed reductive carbonylation of aryl iodides using CO and H2.
Scheme 5: Proposed reaction mechanism for the Rh-catalyzed reductive carbonylation of aryl iodides using CO a...
In the catalytic system, CO and H2 (or syngas) were used as the carbonyl and hydrogen sources, respectively. The catalyst RhCl3·3H2O and PPh3 reacted via a redox reaction to form Rh(PPh3)3Cl, which is the active catalytic species able to activate C–I bonds in the aryl iodides for the insertion of CO in the next step. The base Et3N neutralized the proton in the intermediate rhodium hydroiodide (D) complex and regenerated the active Rh species completing the catalytic cycle.
Conclusion
An efficient and facile Rh-based catalytic system composed of a commercially available Rh salt, RhCl3·3H2O, a phosphine ligand PPh3, and a base Et3N, was evaluated for the synthesis of arylaldehydes via the reductive carbonylation of aryl iodides using CO as carbonyl source and H2 as hydrogen source under relatively mild conditions with a broad substrate range. The low gas pressure, cost-effective ligand and low metal dosage significantly improved the practicability of the system for industrial productions. Another advantage of the method includes the use of cheap and abundant syngas (mixtures of CO and H2) in the catalytic system as an effective and convenient formyl source at relatively low pressures, which further enhanced the possibility of practical application of the proposed system.
Experimental
Materials
Rhodium species ([RhCl(CO)2]2, RhCl(CO)(PPh3)2, Rh(acac)(CO)2, [RhCl(COD)]2, Rh(MeCN)2(COD)BF4, Rh(COD)BF4, Rh(COD)2OTf, [Rh(OMe)(COD)]2, Rh(OAc)2, [Rh(CF3COO)]2, [Rh(CH3(CH2)6CO2)2]2, Rh(acac)3, Rh(ethylene)2(acac), [Rh(ethylene)2Cl]2, Rh(norbornadiene)2BF4, [Cp*RhCl2]2, Rh/C, and RhCl3·3H2O), ligands containing nitrogen or phosphorus (PP3, triphos, dpp-BINAP, dpp-OPh, dppb, dppe, P(PhF5)3, P(4-FPh)3, Cydpp, Bipy, DBU, Im, and PPh3 (their structures are shown in Table 2)), bases (Et3N, 1,4-diaza[2.2.2]bicyclooctane (DABCO), N,N-diisopropylethylamine (DIPEA), N,N,N',N'-tetramethylethylenediamine (TMEDA) and 1,2,2,6,6-pentamethylpiperidine (PMP)), and solvents (N,N-dimethylacetamide (DMA), 1,3-dimethyl-2-imidazolidinone (DMI), N-methyl-2-pyrrolidinone (NMP), tetrahydrofuran (THF), 1,4-dioxane, ethanol, methanol, acetonitrile, and toluene) together with aryl iodides and other reagents were purchased from commercial sources (namely J&K Scientific Ltd. and Innochem Science & Technology Co., Ltd.) and used without further purification. CO and H2 with high purity (99.99%) were supplied by Beijing Analytical Instrument Factory.
Instrumentation
1H NMR spectra in solution were recorded in CDCl3 using the residual CHCl3 as internal reference (7.26 ppm) on a Bruker 400 spectrometer. 1H NMR peaks were labeled as singlet (s), doublet (d), triplet (t), and multiplet (m). The coupling constants, J, are reported in hertz (Hz). 13C NMR spectra in solution were recorded at 101 MHz in CDCl3 using the solvent as internal reference (77.0 ppm). GC analysis was performed on Agilent 4890D with a FID detector and a nonpolar capillary column (DB-5) (30 m × 0.25 mm × 0.25 μm). The column oven was temperature-programmed with a 2 min initial hold at 50 °C, followed by heating to 265 °C at a rate of 10 °C /min and kept at 265 °C for 10 min. High purity nitrogen was used as the carrier gas.
General procedure for reductive carbonylation of aryl iodides with CO and H2
All reactions were carried out in an 80 mL Teflon-lined stainless steel reactor equipped with a magnetic stirring bar. Typically, in a glovebox, the aryl iodides (1.0 mmol), RhI3 (0.025 mmol), PPh3 (0.1 mmol), Et3N (1.2 mmol), and DMA (2 mL) were loaded into the reactor. Then, the autoclave was screwed up, charged with CO and H2 to a total pressure of 10 bar (1:1) and transferred to an oil bath preheated at 90 °C, which was controlled by a Haake-D3 temperature controller. After completion of the reaction, the reactor was cooled in iced water and the gas carefully vented. The conversion and yield of the aryl iodides and arylaldehydes were determined by GC analysis using dodecane as an internal standard. For yield determination of the other products, the reaction mixture was first analyzed by GC–MS to determine the structures of the aromatic aldehyde products. Then, CH2Cl2 (5 mL) was added to the reaction mixture, after which deionized water (10 mL) was added to extract the solvent DMA for 5 times. The organic layer was dried over anhydrous Na2SO4, concentrated by rotary evaporation and finally purified by column chromatography on silica gel using n-hexane/ethyl acetate as eluent to obtain the pure products and isolated yields.
Procedures for the preparations of RhCl(PPh3)3, RhBr(PPh3)3 and RhI(PPh3)3
Preparation of RhCl(PPh3)3: A round-bottomed flask filled with anhydrous ethanol (60 mL) and triphenylphosphine (10 mmol) was put in an oil bath at 70 °C under a nitrogen atmosphere. Then, hydrated rhodium(III) chloride (1.5 mmol) dissolved in anhydrous ethanol (10 mL) was added to the solution under stirring and the resulting solution was kept at gentle reflux for 2.5 h. Afterwards, the hot mixture was filtered. Finally, the solid was washed with ether and dried under vacuum overnight to afford RhCl(PPh3)3 as maroon powder. The preparations of RhBr(PPh3)3 and RhI(PPh3)3 were similar only with different halide ions.
Supporting Information
| Supporting Information File 1: MS spectra of isotope-labeling experiments and characterization of products. | ||
| Format: PDF | Size: 4.4 MB | Download |
References
-
Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 4114–4133. doi:10.1002/anie.200900013
Return to citation in text: [1] -
Balme, G.; Bossharth, E.; Monteiro, N. Eur. J. Org. Chem. 2003, 4101–4111. doi:10.1002/ejoc.200300378
Return to citation in text: [1] -
Stambouli, A. B.; Traversa, E. Renewable Sustainable Energy Rev. 2002, 6, 433–455. doi:10.1016/s1364-0321(02)00014-x
Return to citation in text: [1] -
Tsarevsky, N. V.; Matyjaszewski, K. Chem. Rev. 2007, 107, 2270–2299. doi:10.1021/cr050947p
Return to citation in text: [1] -
Zhu, C.; Guo, S.; Fang, Y.; Dong, S. ACS Nano 2010, 4, 2429–2437. doi:10.1021/nn1002387
Return to citation in text: [1] -
Peng, J.-B.; Wu, X.-F. Angew. Chem., Int. Ed. 2018, 57, 1152–1160. doi:10.1002/anie.201709807
Return to citation in text: [1] -
Friis, S. D.; Lindhardt, A. T.; Skrydstrup, T. Acc. Chem. Res. 2016, 49, 594–605. doi:10.1021/acs.accounts.5b00471
Return to citation in text: [1] -
Franke, R.; Selent, D.; Börner, A. Chem. Rev. 2012, 112, 5675–5732. doi:10.1021/cr3001803
Return to citation in text: [1] -
Schoenberg, A.; Heck, R. F. J. Am. Chem. Soc. 1974, 96, 7761–7764. doi:10.1021/ja00832a024
Return to citation in text: [1] -
Klaus, S.; Neumann, H.; Zapf, A.; Strübing, D.; Hübner, S.; Almena, J.; Riermeier, T.; Groß, P.; Sarich, M.; Krahnert, W.-R.; Rossen, K.; Beller, M. Angew. Chem., Int. Ed. 2006, 45, 154–158. doi:10.1002/anie.200502697
Return to citation in text: [1] -
Sergeev, A. G.; Spannenberg, A.; Beller, M. J. Am. Chem. Soc. 2008, 130, 15549–15563. doi:10.1021/ja804997z
Return to citation in text: [1] [2] -
Dong, K.; Sang, R.; Fang, X.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2017, 56, 5267–5271. doi:10.1002/anie.201700317
Return to citation in text: [1] [2] -
Dong, K.; Fang, X.; Gülak, S.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Nat. Commun. 2017, 8, 14117. doi:10.1038/ncomms14117
Return to citation in text: [1] [2] -
Gong, J.; Yue, H.; Zhao, Y.; Zhao, S.; Zhao, L.; Lv, J.; Wang, S.; Ma, X. J. Am. Chem. Soc. 2012, 134, 13922–13925. doi:10.1021/ja3034153
Return to citation in text: [1] -
Spivey, J. J.; Egbebi, A. Chem. Soc. Rev. 2007, 36, 1514–1528. doi:10.1039/b414039g
Return to citation in text: [1] -
Zhong, L.; Yu, F.; An, Y.; Zhao, Y.; Sun, Y.; Li, Z.; Lin, T.; Lin, Y.; Qi, X.; Dai, Y.; Gu, L.; Hu, J.; Jin, S.; Shen, Q.; Wang, H. Nature 2016, 538, 84–87. doi:10.1038/nature19786
Return to citation in text: [1] -
Corey, J. Y. Chem. Rev. 2016, 116, 11291–11435. doi:10.1021/acs.chemrev.5b00559
Return to citation in text: [1] -
Gao, M.; Wang, Y.-C.; Yang, K.-R.; He, W.; Yang, X.-L.; Yao, Z.-J. Angew. Chem., Int. Ed. 2018, 57, 13313–13318. doi:10.1002/anie.201808517
Return to citation in text: [1] -
Picazo, E.; Morrill, L. A.; Susick, R. B.; Moreno, J.; Smith, J. M.; Garg, N. K. J. Am. Chem. Soc. 2018, 140, 6483–6492. doi:10.1021/jacs.8b03404
Return to citation in text: [1] -
Shi, H.; Michaelides, I. N.; Darses, B.; Jakubec, P.; Nguyen, Q. N. N.; Paton, R. S.; Dixon, D. J. J. Am. Chem. Soc. 2017, 139, 17755–17758. doi:10.1021/jacs.7b10956
Return to citation in text: [1] -
Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Angew. Chem., Int. Ed. 2016, 55, 7296–7343. doi:10.1002/anie.201507458
Return to citation in text: [1] -
Klankermayer, J.; Leitner, W. Science 2015, 350, 629–630. doi:10.1126/science.aac7997
Return to citation in text: [1] -
Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A. V.; Wezendonk, T. A.; Makkee, M.; Gascon, J.; Kapteijn, F. Chem. Rev. 2017, 117, 9804–9838. doi:10.1021/acs.chemrev.6b00816
Return to citation in text: [1] -
Chen, P.; Wu, X.; Lin, J.; Tan, K. L. Science 1999, 285, 91–93. doi:10.1126/science.285.5424.91
Return to citation in text: [1] -
Kubas, G. J. Chem. Rev. 2007, 107, 4152–4205. doi:10.1021/cr050197j
Return to citation in text: [1] -
Le Goff, A.; Artero, V.; Jousselme, B.; Tran, P. D.; Guillet, N.; Metaye, R.; Fihri, A.; Palacin, S.; Fontecave, M. Science 2009, 326, 1384–1387. doi:10.1126/science.1179773
Return to citation in text: [1] -
Yu, X.; Yang, Z.; Guo, S.; Liu, Z.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Chem. Commun. 2018, 54, 7633–7636. doi:10.1039/c8cc03346c
Return to citation in text: [1] -
Yu, B.; Yang, Z.; Zhao, Y.; Hao, L.; Zhang, H.; Gao, X.; Han, B.; Liu, Z. Chem. – Eur. J. 2016, 22, 1097–1102. doi:10.1002/chem.201504320
Return to citation in text: [1] -
Yu, B.; Zhao, Y.; Zhang, H.; Xu, J.; Hao, L.; Gao, X.; Liu, Z. Chem. Commun. 2014, 50, 2330–2333. doi:10.1039/c3cc49365b
Return to citation in text: [1] -
Neumann, H.; Kadyrov, R.; Wu, X.-F.; Beller, M. Chem. – Asian J. 2012, 7, 2213–2216. doi:10.1002/asia.201200462
Return to citation in text: [1] -
Singh, A. S.; Bhanage, B. M.; Nagarkar, J. M. Tetrahedron Lett. 2011, 52, 2383–2386. doi:10.1016/j.tetlet.2011.02.097
Return to citation in text: [1] -
Klaus, S.; Neumann, H.; Zapf, A.; Strübing, D.; Hübner, S.; Almena, J.; Riermeier, T.; Groß, P.; Sarich, M.; Krahnert, W.-R.; Rossen, K.; Beller, M. Angew. Chem., Int. Ed. 2006, 45, 154–158. doi:10.1002/anie.200502697
Return to citation in text: [1] -
Hamasaki, A.; Yasutake, Y.; Norio, T.; Ishida, T.; Akita, T.; Ohashi, H.; Yokoyama, T.; Honma, T.; Tokunaga, M. Appl. Catal., A 2014, 469, 146–152. doi:10.1016/j.apcata.2013.09.043
Return to citation in text: [1] -
Hao, W.; Ding, G.; Cai, M. Catal. Commun. 2014, 51, 53–57. doi:10.1016/j.catcom.2014.03.027
Return to citation in text: [1] -
Jiang, J.; Wang, P.; Cai, M. J. Chem. Res. 2014, 38, 218–222. doi:10.3184/174751914x13934116089943
Return to citation in text: [1] -
Eliseev, O. L.; Bondarenko, T. N.; Myshenkova, T. N.; Lapidus, A. L. Mendeleev Commun. 2014, 24, 229–230. doi:10.1016/j.mencom.2014.06.014
Return to citation in text: [1] -
Chen, B.; Wu, X.-F. Org. Lett. 2019, 21, 2899–2902. doi:10.1021/acs.orglett.9b00930
Return to citation in text: [1] -
Chien, C.-W.; Teng, Y.-H. G.; Honda, T.; Ojima, I. J. Org. Chem. 2018, 83, 11623–11644. doi:10.1021/acs.joc.8b01608
Return to citation in text: [1] -
Dai, P.; Ogunlana, A. A.; Bao, X. J. Org. Chem. 2018, 83, 12734–12743. doi:10.1021/acs.joc.8b02178
Return to citation in text: [1] -
Liu, Z.; Yang, Z.; Yu, B.; Yu, X.; Zhang, H.; Zhao, Y.; Yang, P.; Liu, Z. Org. Lett. 2018, 20, 5130–5134. doi:10.1021/acs.orglett.8b02027
Return to citation in text: [1] -
Choi, G.; Hong, S. H. Angew. Chem. 2018, 130, 6274–6278. doi:10.1002/ange.201801524
Return to citation in text: [1] -
Senthamarai, T.; Murugesan, K.; Schneidewind, J.; Kalevaru, N. V.; Baumann, W.; Neumann, H.; Kamer, P. C. J.; Beller, M.; Jagadeesh, R. V. Nat. Commun. 2018, 9, 4123. doi:10.1038/s41467-018-06416-6
Return to citation in text: [1] -
Prichatz, C.; Trincado, M.; Tan, L.; Casas, F.; Kammer, A.; Junge, H.; Beller, M.; Grützmacher, H. ChemSusChem 2018, 11, 3092–3095. doi:10.1002/cssc.201801072
Return to citation in text: [1] -
Ren, X.; Zheng, Z.; Zhang, L.; Wang, Z.; Xia, C.; Ding, K. Angew. Chem., Int. Ed. 2017, 56, 310–313. doi:10.1002/anie.201608628
Return to citation in text: [1] -
Junge, K.; Wendt, B.; Cingolani, A.; Spannenberg, A.; Wei, Z.; Jiao, H.; Beller, M. Chem. – Eur. J. 2018, 24, 1046–1052. doi:10.1002/chem.201705201
Return to citation in text: [1] -
Pedrajas, E.; Sorribes, I.; Guillamón, E.; Junge, K.; Beller, M.; Llusar, R. Chem. – Eur. J. 2017, 23, 13205–13212. doi:10.1002/chem.201702783
Return to citation in text: [1] -
Wang, L.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2018, 57, 6910–6914. doi:10.1002/anie.201802384
Return to citation in text: [1] -
Dong, K.; Sang, R.; Liu, J.; Razzaq, R.; Franke, R.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2017, 56, 6203–6207. doi:10.1002/anie.201701950
Return to citation in text: [1] -
Liu, Z.; Yang, Z.; Ke, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. New J. Chem. 2018, 42, 13933–13937. doi:10.1039/c8nj03047b
Return to citation in text: [1] -
Liu, Z.; Yang, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Org. Lett. 2017, 19, 5228–5231. doi:10.1021/acs.orglett.7b02462
Return to citation in text: [1] -
Liu, Z.; Yang, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Adv. Synth. Catal. 2017, 359, 4278–4283. doi:10.1002/adsc.201701044
Return to citation in text: [1] -
Liu, Z.; Yang, Z.; Wang, P.; Yu, X.; Wu, Y.; Wang, H.; Liu, Z. ACS Sustainable Chem. Eng. 2019, 7, 18236–18241. doi:10.1021/acssuschemeng.9b04803
Return to citation in text: [1] -
Liu, Z.; Wang, P.; Ou, H.; Yan, Z.; Chen, S.; Tan, X.; Yu, D.; Zhao, X.; Mu, T. RSC Adv. 2020, 10, 7698–7707. doi:10.1039/c9ra10422d
Return to citation in text: [1] -
Liu, Y.; Sun, Z.; Huang, C.; Tu, T. Chem. – Asian J. 2017, 12, 355–360. doi:10.1002/asia.201601537
Return to citation in text: [1] -
Al-Shaal, M. G.; Wright, W. R. H.; Palkovits, R. Green Chem. 2012, 14, 1260–1263. doi:10.1039/c2gc16631c
Return to citation in text: [1] -
Zhou, D.; Gao, Y.; Liu, B.; Tan, Q.; Xu, B. Org. Lett. 2017, 19, 4628–4631. doi:10.1021/acs.orglett.7b02254
Return to citation in text: [1] -
Jessop, P. G.; Ikariya, T.; Noyori, R. Chem. Rev. 1995, 95, 259–272. doi:10.1021/cr00034a001
Return to citation in text: [1]
| 12. | Dong, K.; Sang, R.; Fang, X.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2017, 56, 5267–5271. doi:10.1002/anie.201700317 |
| 13. | Dong, K.; Fang, X.; Gülak, S.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Nat. Commun. 2017, 8, 14117. doi:10.1038/ncomms14117 |
| 41. | Choi, G.; Hong, S. H. Angew. Chem. 2018, 130, 6274–6278. doi:10.1002/ange.201801524 |
| 42. | Senthamarai, T.; Murugesan, K.; Schneidewind, J.; Kalevaru, N. V.; Baumann, W.; Neumann, H.; Kamer, P. C. J.; Beller, M.; Jagadeesh, R. V. Nat. Commun. 2018, 9, 4123. doi:10.1038/s41467-018-06416-6 |
| 43. | Prichatz, C.; Trincado, M.; Tan, L.; Casas, F.; Kammer, A.; Junge, H.; Beller, M.; Grützmacher, H. ChemSusChem 2018, 11, 3092–3095. doi:10.1002/cssc.201801072 |
| 44. | Ren, X.; Zheng, Z.; Zhang, L.; Wang, Z.; Xia, C.; Ding, K. Angew. Chem., Int. Ed. 2017, 56, 310–313. doi:10.1002/anie.201608628 |
| 45. | Junge, K.; Wendt, B.; Cingolani, A.; Spannenberg, A.; Wei, Z.; Jiao, H.; Beller, M. Chem. – Eur. J. 2018, 24, 1046–1052. doi:10.1002/chem.201705201 |
| 46. | Pedrajas, E.; Sorribes, I.; Guillamón, E.; Junge, K.; Beller, M.; Llusar, R. Chem. – Eur. J. 2017, 23, 13205–13212. doi:10.1002/chem.201702783 |
| 47. | Wang, L.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2018, 57, 6910–6914. doi:10.1002/anie.201802384 |
| 48. | Dong, K.; Sang, R.; Liu, J.; Razzaq, R.; Franke, R.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2017, 56, 6203–6207. doi:10.1002/anie.201701950 |
| 49. | Liu, Z.; Yang, Z.; Ke, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. New J. Chem. 2018, 42, 13933–13937. doi:10.1039/c8nj03047b |
| 50. | Liu, Z.; Yang, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Org. Lett. 2017, 19, 5228–5231. doi:10.1021/acs.orglett.7b02462 |
| 51. | Liu, Z.; Yang, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Adv. Synth. Catal. 2017, 359, 4278–4283. doi:10.1002/adsc.201701044 |
| 36. | Eliseev, O. L.; Bondarenko, T. N.; Myshenkova, T. N.; Lapidus, A. L. Mendeleev Commun. 2014, 24, 229–230. doi:10.1016/j.mencom.2014.06.014 |
| 37. | Chen, B.; Wu, X.-F. Org. Lett. 2019, 21, 2899–2902. doi:10.1021/acs.orglett.9b00930 |
| 38. | Chien, C.-W.; Teng, Y.-H. G.; Honda, T.; Ojima, I. J. Org. Chem. 2018, 83, 11623–11644. doi:10.1021/acs.joc.8b01608 |
| 39. | Dai, P.; Ogunlana, A. A.; Bao, X. J. Org. Chem. 2018, 83, 12734–12743. doi:10.1021/acs.joc.8b02178 |
| 40. | Liu, Z.; Yang, Z.; Yu, B.; Yu, X.; Zhang, H.; Zhao, Y.; Yang, P.; Liu, Z. Org. Lett. 2018, 20, 5130–5134. doi:10.1021/acs.orglett.8b02027 |
| 1. | Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 4114–4133. doi:10.1002/anie.200900013 |
| 2. | Balme, G.; Bossharth, E.; Monteiro, N. Eur. J. Org. Chem. 2003, 4101–4111. doi:10.1002/ejoc.200300378 |
| 3. | Stambouli, A. B.; Traversa, E. Renewable Sustainable Energy Rev. 2002, 6, 433–455. doi:10.1016/s1364-0321(02)00014-x |
| 4. | Tsarevsky, N. V.; Matyjaszewski, K. Chem. Rev. 2007, 107, 2270–2299. doi:10.1021/cr050947p |
| 5. | Zhu, C.; Guo, S.; Fang, Y.; Dong, S. ACS Nano 2010, 4, 2429–2437. doi:10.1021/nn1002387 |
| 14. | Gong, J.; Yue, H.; Zhao, Y.; Zhao, S.; Zhao, L.; Lv, J.; Wang, S.; Ma, X. J. Am. Chem. Soc. 2012, 134, 13922–13925. doi:10.1021/ja3034153 |
| 15. | Spivey, J. J.; Egbebi, A. Chem. Soc. Rev. 2007, 36, 1514–1528. doi:10.1039/b414039g |
| 16. | Zhong, L.; Yu, F.; An, Y.; Zhao, Y.; Sun, Y.; Li, Z.; Lin, T.; Lin, Y.; Qi, X.; Dai, Y.; Gu, L.; Hu, J.; Jin, S.; Shen, Q.; Wang, H. Nature 2016, 538, 84–87. doi:10.1038/nature19786 |
| 34. | Hao, W.; Ding, G.; Cai, M. Catal. Commun. 2014, 51, 53–57. doi:10.1016/j.catcom.2014.03.027 |
| 13. | Dong, K.; Fang, X.; Gülak, S.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Nat. Commun. 2017, 8, 14117. doi:10.1038/ncomms14117 |
| 35. | Jiang, J.; Wang, P.; Cai, M. J. Chem. Res. 2014, 38, 218–222. doi:10.3184/174751914x13934116089943 |
| 9. | Schoenberg, A.; Heck, R. F. J. Am. Chem. Soc. 1974, 96, 7761–7764. doi:10.1021/ja00832a024 |
| 10. | Klaus, S.; Neumann, H.; Zapf, A.; Strübing, D.; Hübner, S.; Almena, J.; Riermeier, T.; Groß, P.; Sarich, M.; Krahnert, W.-R.; Rossen, K.; Beller, M. Angew. Chem., Int. Ed. 2006, 45, 154–158. doi:10.1002/anie.200502697 |
| 11. | Sergeev, A. G.; Spannenberg, A.; Beller, M. J. Am. Chem. Soc. 2008, 130, 15549–15563. doi:10.1021/ja804997z |
| 12. | Dong, K.; Sang, R.; Fang, X.; Franke, R.; Spannenberg, A.; Neumann, H.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2017, 56, 5267–5271. doi:10.1002/anie.201700317 |
| 32. | Klaus, S.; Neumann, H.; Zapf, A.; Strübing, D.; Hübner, S.; Almena, J.; Riermeier, T.; Groß, P.; Sarich, M.; Krahnert, W.-R.; Rossen, K.; Beller, M. Angew. Chem., Int. Ed. 2006, 45, 154–158. doi:10.1002/anie.200502697 |
| 11. | Sergeev, A. G.; Spannenberg, A.; Beller, M. J. Am. Chem. Soc. 2008, 130, 15549–15563. doi:10.1021/ja804997z |
| 6. | Peng, J.-B.; Wu, X.-F. Angew. Chem., Int. Ed. 2018, 57, 1152–1160. doi:10.1002/anie.201709807 |
| 7. | Friis, S. D.; Lindhardt, A. T.; Skrydstrup, T. Acc. Chem. Res. 2016, 49, 594–605. doi:10.1021/acs.accounts.5b00471 |
| 8. | Franke, R.; Selent, D.; Börner, A. Chem. Rev. 2012, 112, 5675–5732. doi:10.1021/cr3001803 |
| 33. | Hamasaki, A.; Yasutake, Y.; Norio, T.; Ishida, T.; Akita, T.; Ohashi, H.; Yokoyama, T.; Honma, T.; Tokunaga, M. Appl. Catal., A 2014, 469, 146–152. doi:10.1016/j.apcata.2013.09.043 |
| 24. | Chen, P.; Wu, X.; Lin, J.; Tan, K. L. Science 1999, 285, 91–93. doi:10.1126/science.285.5424.91 |
| 25. | Kubas, G. J. Chem. Rev. 2007, 107, 4152–4205. doi:10.1021/cr050197j |
| 26. | Le Goff, A.; Artero, V.; Jousselme, B.; Tran, P. D.; Guillet, N.; Metaye, R.; Fihri, A.; Palacin, S.; Fontecave, M. Science 2009, 326, 1384–1387. doi:10.1126/science.1179773 |
| 27. | Yu, X.; Yang, Z.; Guo, S.; Liu, Z.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Chem. Commun. 2018, 54, 7633–7636. doi:10.1039/c8cc03346c |
| 30. | Neumann, H.; Kadyrov, R.; Wu, X.-F.; Beller, M. Chem. – Asian J. 2012, 7, 2213–2216. doi:10.1002/asia.201200462 |
| 56. | Zhou, D.; Gao, Y.; Liu, B.; Tan, Q.; Xu, B. Org. Lett. 2017, 19, 4628–4631. doi:10.1021/acs.orglett.7b02254 |
| 21. | Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Angew. Chem., Int. Ed. 2016, 55, 7296–7343. doi:10.1002/anie.201507458 |
| 22. | Klankermayer, J.; Leitner, W. Science 2015, 350, 629–630. doi:10.1126/science.aac7997 |
| 23. | Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A. V.; Wezendonk, T. A.; Makkee, M.; Gascon, J.; Kapteijn, F. Chem. Rev. 2017, 117, 9804–9838. doi:10.1021/acs.chemrev.6b00816 |
| 31. | Singh, A. S.; Bhanage, B. M.; Nagarkar, J. M. Tetrahedron Lett. 2011, 52, 2383–2386. doi:10.1016/j.tetlet.2011.02.097 |
| 57. | Jessop, P. G.; Ikariya, T.; Noyori, R. Chem. Rev. 1995, 95, 259–272. doi:10.1021/cr00034a001 |
| 18. | Gao, M.; Wang, Y.-C.; Yang, K.-R.; He, W.; Yang, X.-L.; Yao, Z.-J. Angew. Chem., Int. Ed. 2018, 57, 13313–13318. doi:10.1002/anie.201808517 |
| 19. | Picazo, E.; Morrill, L. A.; Susick, R. B.; Moreno, J.; Smith, J. M.; Garg, N. K. J. Am. Chem. Soc. 2018, 140, 6483–6492. doi:10.1021/jacs.8b03404 |
| 20. | Shi, H.; Michaelides, I. N.; Darses, B.; Jakubec, P.; Nguyen, Q. N. N.; Paton, R. S.; Dixon, D. J. J. Am. Chem. Soc. 2017, 139, 17755–17758. doi:10.1021/jacs.7b10956 |
| 52. | Liu, Z.; Yang, Z.; Wang, P.; Yu, X.; Wu, Y.; Wang, H.; Liu, Z. ACS Sustainable Chem. Eng. 2019, 7, 18236–18241. doi:10.1021/acssuschemeng.9b04803 |
| 53. | Liu, Z.; Wang, P.; Ou, H.; Yan, Z.; Chen, S.; Tan, X.; Yu, D.; Zhao, X.; Mu, T. RSC Adv. 2020, 10, 7698–7707. doi:10.1039/c9ra10422d |
| 17. | Corey, J. Y. Chem. Rev. 2016, 116, 11291–11435. doi:10.1021/acs.chemrev.5b00559 |
| 28. | Yu, B.; Yang, Z.; Zhao, Y.; Hao, L.; Zhang, H.; Gao, X.; Han, B.; Liu, Z. Chem. – Eur. J. 2016, 22, 1097–1102. doi:10.1002/chem.201504320 |
| 29. | Yu, B.; Zhao, Y.; Zhang, H.; Xu, J.; Hao, L.; Gao, X.; Liu, Z. Chem. Commun. 2014, 50, 2330–2333. doi:10.1039/c3cc49365b |
| 54. | Liu, Y.; Sun, Z.; Huang, C.; Tu, T. Chem. – Asian J. 2017, 12, 355–360. doi:10.1002/asia.201601537 |
| 55. | Al-Shaal, M. G.; Wright, W. R. H.; Palkovits, R. Green Chem. 2012, 14, 1260–1263. doi:10.1039/c2gc16631c |
© 2020 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)