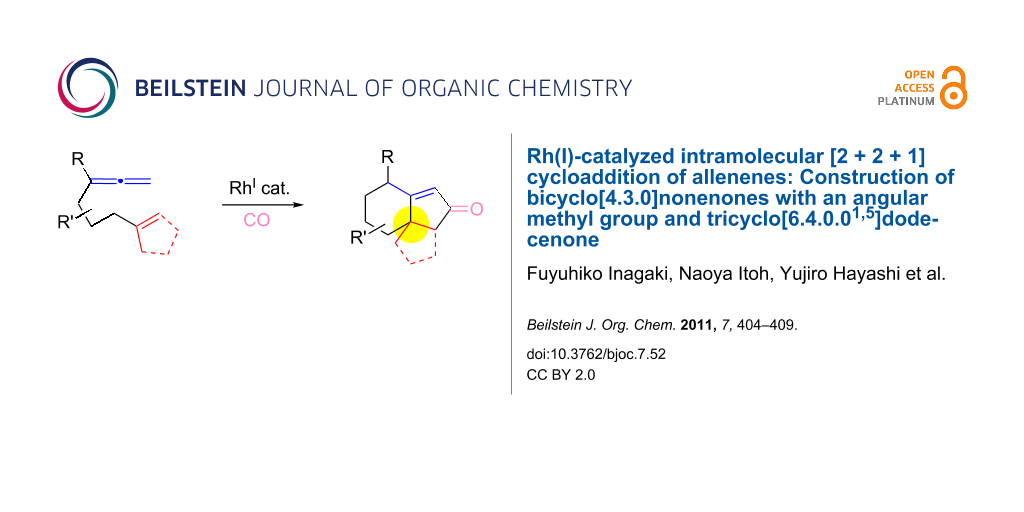
Abstract
The [RhCl(CO)dppp]2-catalyzed intramolecular carbonylative [2 + 2 + 1] cycloaddition of allenenes was developed to prepare bicyclo[4.3.0]nonenones possessing a methyl group at the ring junction, which is difficult to achieve by the Pauson–Khand reaction of the corresponding enynes. This method also provided a new procedure for the construction of the tricyclo[6.4.0.01,5]dodecenone framework in a satisfactory yield.

Graphical Abstract
Introduction
The Co2(CO)8-mediated Pauson–Khand reaction (PKR) [1,2] is well recognized as a formal [2 + 2 + 1] cycloaddition of three components, the alkyne π-bond, the alkene π-bond and carbon monoxide (CO), for producing cyclopentenone derivatives. The intramolecular PKR [3-9] provides a powerful method for the construction of bicyclo[3.3.0]oct-1-en-3-one and bicyclo[4.3.0]non-1(9)-en-8-one frameworks. In contrast to the fact that the 2-alkyl (or phenyl)-1-hepten-6-ynes also efficiently afford 5-alkyl (or phenyl)bicyclo[3.3.0]oct-1-en-3-ones [10-19], the one-methylene homologated substrates furnish the corresponding bicyclo[4.3.0] skeletons with an angular substituent in rather lower yields [20-22].
Recent efforts in our laboratory have developed the [RhCl(CO)2]2-catalyzed [2 + 2 + 1] cycloaddition of 3-phenylsulfonyl-1,2,7-octatrienes and lead to bicyclo[4.3.0]non-1(9)-en-8-one derivatives, in which the distal double bond of the allenyl moiety serves exclusively as the π-component [23]. This method could not only be applicable for the construction of the larger bicyclo[5.3.0]dec-1(10)-en-9-one skeleton, but also provides a new entry for the preparation of three types of trans-2-phenylsulfonylbicyclo[4.3.0]non-1(9)-en-8-one derivatives 2a–c in moderate to high yields with an alkyl group (Me, (CH2)2OMe) at the ring junction from 7-alkyl-3-phenylsulfonyl-1,2,7-octatrienes 1a–c. The formation of 2 can be rationalized by the initial formation of the 6-alkyl-2-phenylsulfonylbicyclo[4.3.0]non-1-en-8-one derivative 3, followed by subsequent isomerization to the α,β-unsaturated ketones 2. By taking advangage of this newly developed reaction, we have completed the first total syntheses of two tricyclic sesquiterpenes 6a,b, isolated from Jatropha neopauciflora, from the methoxycarbonylallenene 4 via the bicyclo[4.3.0]nonenone derivative 5 [24]. The key step in these total syntheses was carried out as follows: The allenene 4, derived from D-tartrate, was heated under reflux in the presence of 10 mol % of [RhCO(dppp)2]Cl [25] under a CO atmosphere to produce exclusively the bicyclo[4.3.0]derivative 5 with the desired stereochemistry in 74% yield (Scheme 1). Our continuous interest in this field prompted us to optimize the reaction conditions in order to make our method more useful. This paper describes the Rh(I)-catalyzed [2 + 2 + 1] cycloaddition of allenenes for the facile preparation of the bicyclo[4.3.0]nonenone derivatives possessing an angular methyl group, and for the construction of the tricyclo[6.4.0.01,5]dodecenone skeleton.
![[1860-5397-7-52-i1]](/bjoc/content/inline/1860-5397-7-52-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Rh(I)-catalyzed Pauson–Khand type reaction of 1 and 4.
Scheme 1: Rh(I)-catalyzed Pauson–Khand type reaction of 1 and 4.
Results and Discussion
In previous studies, we investigated the carbonylative [2 + 2 + 1] cycloaddition reaction of two allenenes 1 and 4 with either a SO2Ph or CO2Me substituent on the proximal double bond of the allene group in the presence of several Rh(I) catalysts ([RhCl(CO)2]2, [RhCO(dppp)2]Cl or [RhCl(CO)dppp]2). We have now examined the similar ring-closing reaction using an alternative allenene 7a with a methyl group on the allenyl group for further evaluation of these reactions. According to the conditions described for the formation of 2, a solution of 7a in toluene was heated at 120 °C for 4 h in the presence of 5 mol % of [RhCl(CO)2]2 under 5 atm of CO to afford the desired 4,4-bis(methoxycarbonyl)-2,6-dimethylbicyclo[4.3.0]non-1-en-8-one (8a) in 38% yield (Table 1, entry 1). A similar reaction under 1 atm of CO gave 8a in a better yield (Table 1, entry 2). These results are in sharp contrast to the previous findings, where increasing the CO pressure from 1 to 5 atm led to an improvement in the chemical yield (see reference [23], Table 1). The alternative condition ([RhCO(dppp)2]Cl (10 mol %) under a CO atmosphere) provided the ring-closing product 8a in 75% yield (Table 1, entry 3). It has already been shown that the Rh(I)-catalyzed Pauson–Khand type reaction (PKTR) of enynes under a low CO pressure [26,27] leads to better results. Thus, the ring-closing reaction of 7a was performed under an atmosphere consisting of 0.2 atm of CO and 0.8 atm of Ar, or of 0.05 atm of CO and 0.95 atm of Ar to produce 8a in 71% and 74% yields, respectively (Table 1, entries 4 and 5). Next, the reaction with [RhCl(CO)dppp]2 in the presence or absence of a silver salt [27-34] was examined (Table 1, entries 6–13). A significant improvement (8a, 93%) was observed when the ring-closing reaction of 7a was carried out in the presence of 5 mol % of [RhCl(CO)dppp]2 and 12 mol % of AgBF4 under an atmosphere consisting of 0.05 atm of CO and 0.95 atm of Ar at 120 °C (Table 1, entry 11). Almost all of the reactions gave 2,6-dimethylbicyclo[4.3.0]non-1-en-8-one 8a, and the α,β-unsaturated ketone 9a could not be detected except for entry 9. This observation obviously differs from the PKTR products 2 and 5 having an electron withdrawing group instead of a methyl group (Scheme 1).
Table 1: Rh(I)-catalyzed PKTR of 7a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-52-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry | Rh(I) cat. (mol %) | additive (mol %) | CO (atm) | time (h) | yield of 8a (%) |
|---|---|---|---|---|---|
| 1 | [RhCl(CO)2]2 (5) | — | 5 | 4 | 38 |
| 2 | [RhCl(CO)2]2 (5) | — | 1 | 16 | 54 |
| 3 | [RhCO(dppp)2]Cl (10) | — | 1 | 17 | 75b |
| 4 | [RhCO(dppp)2]Cl (10) | — | 0.2c | 17 | 71 |
| 5 | [RhCO(dppp)2]Cl (10) | — | 0.05d | 6 | 74 |
| 6 | [RhCl(CO)dppp]2 (5) | — | 1 | 19 | 68 |
| 7 | [RhCl(CO)dppp]2 (5) | — | 0.2c | 11 | 79 |
| 8 | [RhCl(CO)dppp]2 (5) | — | 0.1e | 7 | 88 |
| 9 | [RhCl(CO)dppp]2 (5) | — | 0.05d | 8 | 51f |
| 10 | [RhCl(CO)dppp]2 (5) | AgBF4 (12) | 0.1e | 1.5 | 78 |
| 11 | [RhCl(CO)dppp]2 (5) | AgBF4 (12) | 0.05d | 1.5 | 93 |
| 12 | [RhCl(CO)dppp]2 (5) | AgSbF6 (12) | 0.05d | 1.5 | 79 |
| 13 | [RhCl(CO)dppp]2 (5) | AgCl (12) | 0.05d | 6 | 84 |
aBath temperature. b8a was obtained in 18% yield under 5 atm of CO. cThe reaction was conducted under 0.2 atm of CO and 0.8 atm of Ar. dThe reaction was conducted under 0.05 atm of CO and 0.95 atm of Ar. eThe reaction was conducted under 0.1 atm of CO and 0.9 atm of Ar. fCompound 9a was obtained in 31%.
We next investigated the scope of this Rh(I)-catalyzed PKTR using several substrates under the condition of entry 11 in Table 1. The nitrogen congener 7b produced the corresponding azabicyclic compound 8b in 56% yield, although a prolonged reaction time was required (Table 2, entry 1). The sulfonylallenene 7c, which has a simple methylene tether, provided the cyclopentenone derivative 9c in 55% yield as expected from the results given in Scheme 1 (Table 2, entry 2). As noted above, compounds 2a,b could be obtained in the presence of [RhCl(CO)2]2: [RhCl(CO)dppp]2 also worked as well as the previous conditions (Table 2, entries 3 and 4). The oxygen congener 7d produced the corresponding oxabicyclic compound 9d in 49% yield (Table 2, entry 5). The methoxycarbonyl functionality on the allenyl moiety 7e was found not to interfere with the ring-closing reaction and afforded 9e in 63% yield (Table 2, entry 6). On the other hand, 7f provided the oxabicyclic derivative 9f in a low yield (Table 2, entry 7). The geminal disubstituent effect (Thorpe–Ingold-type effect) of 1a and 7a might be responsible for the high yields of the ring-closed products.
Table 2: Rh(I)-catalyzed PKTR of 7-methyl-1,2,7-trienes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-52-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | allenene | product | time (h) | yield (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-52-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-52-i6.svg?max-width=637&scale=1.0)
|
6.5 | 56 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-52-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-52-i8.svg?max-width=637&scale=1.0)
|
3 | 55 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-52-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-52-i10.svg?max-width=637&scale=1.0)
|
1 | 88a,b |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-52-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-52-i12.svg?max-width=637&scale=1.0)
|
2 | 66 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-52-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-52-i14.svg?max-width=637&scale=1.0)
|
3 | 49 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-52-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-52-i16.svg?max-width=637&scale=1.0)
|
5 | 63 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-52-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-52-i18.svg?max-width=637&scale=1.0)
|
4.5 | 22 |
aThe reaction was carried out without AgBF4 under a CO atmosphere. b2a was obtained in 68% yield in the presence of 12 mol % of AgBF4.
The Rh(I)-catalyzed PKTR of the non-substituted allenene 7g was investigated as a control experiment under the several conditions (e.g., conditions of entries 3, 6, 10 and 11 in Table 1) but resulted in the formation of the intractable mixtures (Scheme 2). These results in combination with the successful examples shown in Table 2 indicated that the substituent on the proximal double bond of the allene might be mandatory. Removal of the allylic sulfone group in 2a was achieved by tributyltin hydride in the presence of AIBN to give 9g in 91% yield [35-41]. Thus, the phenylsulfonyl group of 2a can be regarded as a hydrogen surrogate and this procedure provides a new method for the preparation of 9g.
![[1860-5397-7-52-i2]](/bjoc/content/inline/1860-5397-7-52-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of 4,4-bis(methoxycarbonyl)-6-methylbicyclo[4.3.0]non-1(9)-en-8-one (9g).
Scheme 2: Formation of 4,4-bis(methoxycarbonyl)-6-methylbicyclo[4.3.0]non-1(9)-en-8-one (9g).
The intramolecular Rh(I)-catalyzed PKTR of allenenes is applicable to the more complex substrate as shown in Table 3 [42-44]. Exposure of the cyclopentene 10 to the standard conditions in Table 2 did not lead to the desired tricyclo[6.4.0.01,5]dodec-7-en-6-one 11, instead the spiro[4.5]deca-1,6-diene derivative 12 was formed exclusively in 87% yield (Table 3, entry 1). After screening several reaction conditions, the tricyclic derivative 11 was obtained in 73% yield along with 12 (24%) when the reaction was carried out in the absence of AgBF4 under a CO atmosphere (Table 3, entry 2). The [RhCl(CO)dppp]2-catalyzed reaction of 10 under a N2 atmosphere with or without AgBF4 produced the spiro product 12 in 93% and 91% yield, respectively, whereas no spiro product 12 was obtained under thermal conditions without a Rh(I)-catalyst (Table 3, entries 3–5). Therefore, the formation of 12 must be rationalized by the Rh(I)-catalyzed cycloisomerization. The oxidative addition of the Rh(I)-catalyst to an alkene group and the distal double bond of the allenyl moiety would form a rhodabicyclo[4.3.0]nonene intermediate, which would collapse to the spiro[4.5]deca-1,6-diene system via β-elimination and subsequent reductive elimination [31,32].
Table 3: Rh(I)-catalyzed cyclization of 6-(cyclopent-1-enyl)-1,2-hexadiene 10.
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-52-i19.svg?max-width=637&scale=1.0)
|
|||||
| entry | Rh(I) cat. (5 mol %) | additive (mol %) | CO or N2 (atm) | yield of 11 (%) | yield of 12 (%) |
|---|---|---|---|---|---|
| 1 | [RhCl(CO)dppp]2 | AgBF4 (12) | CO (0.05)a | — | 87 |
| 2 | [RhCl(CO)dppp]2 | — | CO (1) | 73 | 24 |
| 3 | [RhCl(CO)dppp]2 | AgBF4 (12) | N2 (1) | — | 93 |
| 4 | [RhCl(CO)dppp]2 | — | N2 (1) | — | 91 |
| 5 | — | — | N2 (1) | — | — |
aThe reaction was conducted under 0.05 atm of CO and 0.95 atm of Ar.
In summary, we have developed the intramolecular Rh(I)-catalyzed PKTR between 1,1-disubstituted allene and 1,1-disubstituted alkene functionalities, which leads to the facile formation of bicyclo[4.3.0]nonenone derivatives with an angular methyl group. This method was also successfully applied to the construction of the more complex tricyclo[6.4.0.01,5]dodecenone skeleton.
Supporting Information
Full experimental details, compound characterization data, 1H NMR spectra and 13C NMR spectra for all new compounds described.
| Supporting Information File 1: Experimental, characterization data and spectra. | ||
| Format: PDF | Size: 9.4 MB | Download |
References
-
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc. D 1971, 36a. doi:10.1039/C2971000036A
Return to citation in text: [1] -
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/P19730000977
Return to citation in text: [1] -
Geis, O.; Schmalz, H.-G. Angew. Chem., Int. Ed. 1998, 37, 911–914. doi:10.1002/(SICI)1521-3773(19980420)37:7<911::AID-ANIE911>3.0.CO;2-O
Return to citation in text: [1] -
Brummond, K. M.; Kent, J. L. Tetrahedron 2000, 56, 3263–3283. doi:10.1016/S0040-4020(00)00148-4
Return to citation in text: [1] -
Gibson, S. E.; Stevenazzi, A. Angew. Chem., Int. Ed. 2003, 42, 1800–1810. doi:10.1002/anie.200200547
Return to citation in text: [1] -
Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2004, 33, 32–42. doi:10.1039/b300976a
Return to citation in text: [1] -
Boñaga, L. V. R.; Krafft, M. E. Tetrahedron 2004, 60, 9795–9833. doi:10.1016/j.tet.2004.06.072
Return to citation in text: [1] -
Shibata, T. Adv. Synth. Catal. 2006, 348, 2328–2336. doi:10.1002/adsc.200600328
Return to citation in text: [1] -
Lee, H.-W.; Kwong, F.-K. Eur. J. Org. Chem. 2010, 789–811. doi:10.1002/ejoc.200900892
Return to citation in text: [1] -
Jeong, N.; Hwang, S. H.; Lee, Y.; Chung, Y. K. J. Am. Chem. Soc. 1994, 116, 3159–3160. doi:10.1021/ja00086a070
Return to citation in text: [1] -
Pagenkopf, B. L.; Livinghouse, T. J. Am. Chem. Soc. 1996, 118, 2285–2286. doi:10.1021/ja9535975
Return to citation in text: [1] -
Koga, Y.; Kobayashi, T.; Narasaka, K. Chem. Lett. 1998, 27, 249–250. doi:10.1246/cl.1998.249
Return to citation in text: [1] -
Sugihara, T.; Yamaguchi, M. J. Am. Chem. Soc. 1998, 120, 10782–10783. doi:10.1021/ja982635s
Return to citation in text: [1] -
Hicks, F. A.; Kablaoui, N. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 5881–5898. doi:10.1021/ja990682u
Return to citation in text: [1] -
Krafft, M. E.; Boñaga, L. V. R.; Hirosawa, C. Tetrahedron Lett. 1999, 40, 9171–9175. doi:10.1016/S0040-4039(99)01960-7
Return to citation in text: [1] -
Hiroi, K.; Watanabe, T.; Kawagishi, R.; Abe, I. Tetrahedron: Asymmetry 2000, 11, 797–808. doi:10.1016/S0957-4166(99)00570-4
Return to citation in text: [1] -
Fletcher, A. J.; Christie, S. D. R. J. Chem. Soc., Perkin Trans. 1 2000, 1657–1668. doi:10.1039/A903168E
Return to citation in text: [1] -
Krafft, M. E.; Wright, J. A.; Boñaga, L. V. R. Synlett 2005, 71–74. doi:10.1055/s-2004-836053
Return to citation in text: [1] -
Tang, Y.; Deng, L.; Zhang, Y.; Dong, G.; Chen, J.; Yang, Z. Org. Lett. 2005, 7, 593–595. doi:10.1021/ol047651a
Return to citation in text: [1] -
Bolton, G. L.; Hodges, J. C.; Rubin, J. R. Tetrahedron 1997, 53, 6611–6634. doi:10.1016/S0040-4020(97)00220-2
Return to citation in text: [1] -
Ishizaki, M.; Satoh, H.; Hoshino, O. Chem. Lett. 2002, 31, 1040–1041. doi:10.1246/cl.2002.1040
Return to citation in text: [1] -
Ishizaki, M.; Satoh, H.; Hoshino, O.; Nishitani, K.; Hara, H. Heterocycles 2004, 63, 827–844. doi:10.3987/COM-03-9993
Return to citation in text: [1] -
Inagaki, F.; Mukai, C. Org. Lett. 2006, 8, 1217–1220. doi:10.1021/ol0600990
Return to citation in text: [1] [2] -
Hayashi, Y.; Miyakoshi, N.; Kitagaki, S.; Mukai, C. Org. Lett. 2008, 10, 2385–2388. doi:10.1021/ol800633a
Return to citation in text: [1] -
Sanger, A. R. J. Chem. Soc., Dalton Trans. 1977, 120–129. doi:10.1039/DT9770000120
Return to citation in text: [1] -
Kobayashi, T.; Koga, Y.; Narasaka, K. J. Organomet. Chem. 2001, 624, 73–87. doi:10.1016/S0022-328X(00)00835-4
Return to citation in text: [1] -
Kim, D. E.; Kim, I. S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Jeong, N. J. Org. Chem. 2008, 73, 7985–7989. doi:10.1021/jo801236c
Return to citation in text: [1] [2] -
Jeong, N.; Lee, S.; Sung, B. K. Organometallics 1998, 17, 3642–3644. doi:10.1021/om980410k
Return to citation in text: [1] -
Jeong, N.; Sung, B. K.; Choi, Y. K. J. Am. Chem. Soc. 2000, 122, 6771–6772. doi:10.1021/ja0007049
Return to citation in text: [1] -
Mukai, C.; Nomura, I.; Yamanishi, K.; Hanaoka, M. Org. Lett. 2002, 4, 1755–1758. doi:10.1021/ol020051w
Return to citation in text: [1] -
Mukai, C.; Nomura, I.; Kitagaki, S. J. Org. Chem. 2003, 68, 1376–1385. doi:10.1021/jo020649h
Return to citation in text: [1] [2] -
Mukai, C.; Inagaki, F.; Yoshida, T.; Kitagaki, S. Tetrahedron Lett. 2004, 45, 4117–4121. doi:10.1016/j.tetlet.2004.03.160
Return to citation in text: [1] [2] -
Mukai, C.; Inagaki, F.; Yoshida, T.; Yoshitani, K.; Hara, Y.; Kitagaki, S. J. Org. Chem. 2005, 70, 7159–7171. doi:10.1021/jo050770z
Return to citation in text: [1] -
Mukai, C.; Hirose, T.; Teramoto, S.; Kitagaki, S. Tetrahedron 2005, 61, 10983–10994. doi:10.1016/j.tet.2005.08.088
Return to citation in text: [1] -
Nájera, C.; Yus, M. Tetrahedron 1999, 55, 10547–10658. doi:10.1016/S0040-4020(99)00600-6
Return to citation in text: [1] -
Simpkins, N. S. Sulphones in Organic Synthesis; Pergamon Press: Oxford, 1993.
Return to citation in text: [1] -
Watanabe, Y.; Ueno, Y.; Araki, T.; Endo, T.; Okawara, M. Tetrahedron Lett. 1986, 27, 215–218. doi:10.1016/S0040-4039(00)83980-5
Return to citation in text: [1] -
Miyakoshi, N.; Ohgaki, Y.; Masui, K.; Mukai, C. Heterocycles 2007, 74, 185–189. doi:10.3987/COM-07-S(W)46
Return to citation in text: [1] -
Aburano, D.; Inagaki, F.; Tomonaga, S.; Mukai, C. J. Org. Chem. 2009, 74, 5590–5594. doi:10.1021/jo901141t
Return to citation in text: [1] -
Inagaki, F.; Narita, S.; Hasegawa, T.; Kitagaki, S.; Mukai, C. Angew. Chem., Int. Ed. 2009, 48, 2007–2011. doi:10.1002/anie.200806029
Return to citation in text: [1] -
Kawamura, T.; Inagaki, F.; Narita, S.; Takahashi, Y.; Hirata, S.; Kitagaki, S.; Mukai, C. Chem.–Eur. J. 2010, 16, 5173–5183. doi:10.1002/chem.200903568
Return to citation in text: [1] -
Magnus, P.; Fielding, M. R.; Wells, C.; Lynch, V. Tetrahedron Lett. 2002, 43, 947–950. doi:10.1016/S0040-4039(01)02301-2
Return to citation in text: [1] -
Rosillo, M.; Arnáiz, E.; Abdi, D.; Blanco-Urgoiti, J.; Domínguez, G.; Pérez-Castells, J. Eur. J. Org. Chem. 2008, 3917–3927. doi:10.1002/ejoc.200800332
Return to citation in text: [1] -
Inagaki, F.; Kinebuchi, M.; Miyakoshi, N.; Mukai, C. Org. Lett. 2010, 12, 1800–1803. doi:10.1021/ol100412f
Return to citation in text: [1]
| 1. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc. D 1971, 36a. doi:10.1039/C2971000036A |
| 2. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/P19730000977 |
| 20. | Bolton, G. L.; Hodges, J. C.; Rubin, J. R. Tetrahedron 1997, 53, 6611–6634. doi:10.1016/S0040-4020(97)00220-2 |
| 21. | Ishizaki, M.; Satoh, H.; Hoshino, O. Chem. Lett. 2002, 31, 1040–1041. doi:10.1246/cl.2002.1040 |
| 22. | Ishizaki, M.; Satoh, H.; Hoshino, O.; Nishitani, K.; Hara, H. Heterocycles 2004, 63, 827–844. doi:10.3987/COM-03-9993 |
| 10. | Jeong, N.; Hwang, S. H.; Lee, Y.; Chung, Y. K. J. Am. Chem. Soc. 1994, 116, 3159–3160. doi:10.1021/ja00086a070 |
| 11. | Pagenkopf, B. L.; Livinghouse, T. J. Am. Chem. Soc. 1996, 118, 2285–2286. doi:10.1021/ja9535975 |
| 12. | Koga, Y.; Kobayashi, T.; Narasaka, K. Chem. Lett. 1998, 27, 249–250. doi:10.1246/cl.1998.249 |
| 13. | Sugihara, T.; Yamaguchi, M. J. Am. Chem. Soc. 1998, 120, 10782–10783. doi:10.1021/ja982635s |
| 14. | Hicks, F. A.; Kablaoui, N. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 5881–5898. doi:10.1021/ja990682u |
| 15. | Krafft, M. E.; Boñaga, L. V. R.; Hirosawa, C. Tetrahedron Lett. 1999, 40, 9171–9175. doi:10.1016/S0040-4039(99)01960-7 |
| 16. | Hiroi, K.; Watanabe, T.; Kawagishi, R.; Abe, I. Tetrahedron: Asymmetry 2000, 11, 797–808. doi:10.1016/S0957-4166(99)00570-4 |
| 17. | Fletcher, A. J.; Christie, S. D. R. J. Chem. Soc., Perkin Trans. 1 2000, 1657–1668. doi:10.1039/A903168E |
| 18. | Krafft, M. E.; Wright, J. A.; Boñaga, L. V. R. Synlett 2005, 71–74. doi:10.1055/s-2004-836053 |
| 19. | Tang, Y.; Deng, L.; Zhang, Y.; Dong, G.; Chen, J.; Yang, Z. Org. Lett. 2005, 7, 593–595. doi:10.1021/ol047651a |
| 31. | Mukai, C.; Nomura, I.; Kitagaki, S. J. Org. Chem. 2003, 68, 1376–1385. doi:10.1021/jo020649h |
| 32. | Mukai, C.; Inagaki, F.; Yoshida, T.; Kitagaki, S. Tetrahedron Lett. 2004, 45, 4117–4121. doi:10.1016/j.tetlet.2004.03.160 |
| 3. | Geis, O.; Schmalz, H.-G. Angew. Chem., Int. Ed. 1998, 37, 911–914. doi:10.1002/(SICI)1521-3773(19980420)37:7<911::AID-ANIE911>3.0.CO;2-O |
| 4. | Brummond, K. M.; Kent, J. L. Tetrahedron 2000, 56, 3263–3283. doi:10.1016/S0040-4020(00)00148-4 |
| 5. | Gibson, S. E.; Stevenazzi, A. Angew. Chem., Int. Ed. 2003, 42, 1800–1810. doi:10.1002/anie.200200547 |
| 6. | Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2004, 33, 32–42. doi:10.1039/b300976a |
| 7. | Boñaga, L. V. R.; Krafft, M. E. Tetrahedron 2004, 60, 9795–9833. doi:10.1016/j.tet.2004.06.072 |
| 8. | Shibata, T. Adv. Synth. Catal. 2006, 348, 2328–2336. doi:10.1002/adsc.200600328 |
| 9. | Lee, H.-W.; Kwong, F.-K. Eur. J. Org. Chem. 2010, 789–811. doi:10.1002/ejoc.200900892 |
| 26. | Kobayashi, T.; Koga, Y.; Narasaka, K. J. Organomet. Chem. 2001, 624, 73–87. doi:10.1016/S0022-328X(00)00835-4 |
| 27. | Kim, D. E.; Kim, I. S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Jeong, N. J. Org. Chem. 2008, 73, 7985–7989. doi:10.1021/jo801236c |
| 35. | Nájera, C.; Yus, M. Tetrahedron 1999, 55, 10547–10658. doi:10.1016/S0040-4020(99)00600-6 |
| 36. | Simpkins, N. S. Sulphones in Organic Synthesis; Pergamon Press: Oxford, 1993. |
| 37. | Watanabe, Y.; Ueno, Y.; Araki, T.; Endo, T.; Okawara, M. Tetrahedron Lett. 1986, 27, 215–218. doi:10.1016/S0040-4039(00)83980-5 |
| 38. | Miyakoshi, N.; Ohgaki, Y.; Masui, K.; Mukai, C. Heterocycles 2007, 74, 185–189. doi:10.3987/COM-07-S(W)46 |
| 39. | Aburano, D.; Inagaki, F.; Tomonaga, S.; Mukai, C. J. Org. Chem. 2009, 74, 5590–5594. doi:10.1021/jo901141t |
| 40. | Inagaki, F.; Narita, S.; Hasegawa, T.; Kitagaki, S.; Mukai, C. Angew. Chem., Int. Ed. 2009, 48, 2007–2011. doi:10.1002/anie.200806029 |
| 41. | Kawamura, T.; Inagaki, F.; Narita, S.; Takahashi, Y.; Hirata, S.; Kitagaki, S.; Mukai, C. Chem.–Eur. J. 2010, 16, 5173–5183. doi:10.1002/chem.200903568 |
| 42. | Magnus, P.; Fielding, M. R.; Wells, C.; Lynch, V. Tetrahedron Lett. 2002, 43, 947–950. doi:10.1016/S0040-4039(01)02301-2 |
| 43. | Rosillo, M.; Arnáiz, E.; Abdi, D.; Blanco-Urgoiti, J.; Domínguez, G.; Pérez-Castells, J. Eur. J. Org. Chem. 2008, 3917–3927. doi:10.1002/ejoc.200800332 |
| 44. | Inagaki, F.; Kinebuchi, M.; Miyakoshi, N.; Mukai, C. Org. Lett. 2010, 12, 1800–1803. doi:10.1021/ol100412f |
| 25. | Sanger, A. R. J. Chem. Soc., Dalton Trans. 1977, 120–129. doi:10.1039/DT9770000120 |
| 24. | Hayashi, Y.; Miyakoshi, N.; Kitagaki, S.; Mukai, C. Org. Lett. 2008, 10, 2385–2388. doi:10.1021/ol800633a |
| 27. | Kim, D. E.; Kim, I. S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Jeong, N. J. Org. Chem. 2008, 73, 7985–7989. doi:10.1021/jo801236c |
| 28. | Jeong, N.; Lee, S.; Sung, B. K. Organometallics 1998, 17, 3642–3644. doi:10.1021/om980410k |
| 29. | Jeong, N.; Sung, B. K.; Choi, Y. K. J. Am. Chem. Soc. 2000, 122, 6771–6772. doi:10.1021/ja0007049 |
| 30. | Mukai, C.; Nomura, I.; Yamanishi, K.; Hanaoka, M. Org. Lett. 2002, 4, 1755–1758. doi:10.1021/ol020051w |
| 31. | Mukai, C.; Nomura, I.; Kitagaki, S. J. Org. Chem. 2003, 68, 1376–1385. doi:10.1021/jo020649h |
| 32. | Mukai, C.; Inagaki, F.; Yoshida, T.; Kitagaki, S. Tetrahedron Lett. 2004, 45, 4117–4121. doi:10.1016/j.tetlet.2004.03.160 |
| 33. | Mukai, C.; Inagaki, F.; Yoshida, T.; Yoshitani, K.; Hara, Y.; Kitagaki, S. J. Org. Chem. 2005, 70, 7159–7171. doi:10.1021/jo050770z |
| 34. | Mukai, C.; Hirose, T.; Teramoto, S.; Kitagaki, S. Tetrahedron 2005, 61, 10983–10994. doi:10.1016/j.tet.2005.08.088 |
© 2011 Inagaki et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)