Abstract
Enantiomerically pure tertiary thiols provide a major synthetic challenge, and despite the importance of chiral sulfur-containing compounds in biological and medicinal chemistry, surprisingly few effective methods are suitable for the asymmetric synthesis of tertiary thiols. This review details the most practical of the methods available.

Graphical Abstract
Introduction
Organosulfur compounds play key roles in many biological structures and functions: two of the 21 proteinogenic amino acids contain sulfur, and seven of the 10 best-selling drugs in the US in 2009 were organosulfur compounds (Figure 1) [1]. Glutathione plays a crucial role in primary metabolism, and (R)-thioterpineol (limonenethiol or “grapefruit mercaptan”) is an important and extremely powerful flavour compound, providing the distinctive taste of grapefruit (Figure 2). The consequent need to prepare and manipulate enantiomerically pure organosulfur species has powered the development of asymmetric synthetic methods leading to various classes of organic sulfur compounds, with chirality residing at sulfur, at carbon, or at both [2-8].
![[1860-5397-7-68-1]](/bjoc/content/figures/1860-5397-7-68-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Seven out of the ten top selling drugs in the USA in 2009 contain sulfur. Figures in italics are total retail sales in dollars [1].
Figure 1: Seven out of the ten top selling drugs in the USA in 2009 contain sulfur. Figures in italics are to...
![[1860-5397-7-68-2]](/bjoc/content/figures/1860-5397-7-68-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Naturally occurring organosulfur compounds glutathione and (R)-thioterpineol.
Figure 2: Naturally occurring organosulfur compounds glutathione and (R)-thioterpineol.
This field of asymmetric organosulfur chemistry is particularly well developed in connection with sulfur(IV) and sulfur(VI) species with chirality at sulfur – namely sulfoxides, sulfinates, sulfimines and sulfilimines [9]. Chiral sulfoxides and sulfonium ylids have themselves been extensively used as tools for asymmetric synthesis [3]. With regard to chiral sulfur(II) compounds – namely thiols and thioethers (sulfides) with chirality at carbon – methods available for their asymmetric preparation are abundant, and usually rely on stereospecific substitution reactions [9]. These reactions are well suited to the construction of secondary thiol derivatives. By contrast, few methods are suitable for the asymmetric preparation of simple tertiary thiols 1. While the asymmetric synthesis of simple tertiary alcohols is generally achieved by controlling enantiofacial selectivity in nucleophilic attack on a prochiral ketone [10], comparable approaches to tertiary thiols 1 are not practical due to the instability of thioketones, their tendency to undergo nucleophilic attack at sulfur rather than carbon, and their intolerable stench [11]. As a result, enantiomerically pure tertiary thiols are a remarkably difficult class of compounds to make, despite the simplicity of their structure. This review will cover the methods available for the asymmetric synthesis of chiral tertiary thiols and their sulfur(II) derivatives.
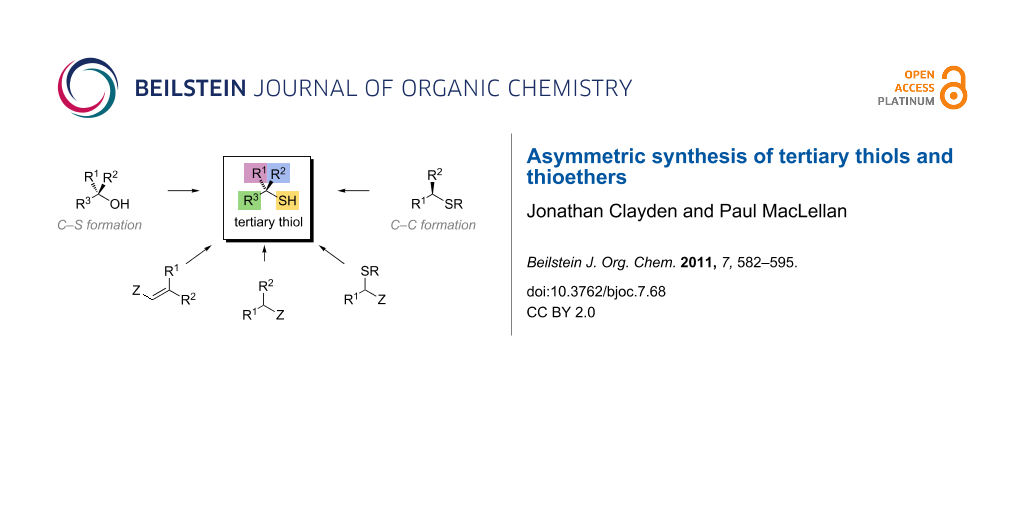
Two general approaches to the preparation of a chiral tertiary thiol 1 may be envisaged, entailing disconnection of either a C–S or a C–C bond (Figure 3). The thiol could be installed by stereoselective attack of a sulfur-centred nucleophile on a substituted carbon centre (C–S bond formation). Alternatively, stereoselective alkylation, arylation or acylation of a secondary sulfur-based substrate could generate the quaternary centre (C–C bond formation).
![[1860-5397-7-68-3]](/bjoc/content/figures/1860-5397-7-68-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Methods for the synthesis of chiral tertiary thiol 1.
Figure 3: Methods for the synthesis of chiral tertiary thiol 1.
Review
1 Carbon–sulfur bond formation
1.1 SN2 displacement of a leaving group
Stereospecific nucleophilic attack on substituted carbon atoms is a simple and versatile way to construct stereocentres next to heteroatoms with overall inversion of stereochemistry. Sulfur nucleophiles are commonly used very effectively to accomplish reactions of this type [9]. However, SN2 displacements are very sensitive to steric crowding at the reaction centre: SN1 substitution and elimination reactions are almost always favoured over the SN2 pathway in quaternary electrophiles, limiting the use of SN2 reactions for the synthesis of tertiary thiols.
1.1.1 Sulfonate leaving groups
The efficiency of sulfonates as leaving groups has allowed the preparation of certain families of tertiary thiols and thioethers. For example, SN2 displacement of a mesylate leaving group by thiophenol can be accomplished using α-hydroxy esters 2 (Scheme 1) [12].
![[1860-5397-7-68-i1]](/bjoc/content/inline/1860-5397-7-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of thioethers 4 from α-hydroxy esters.
Scheme 1: Preparation of thioethers 4 from α-hydroxy esters.
The presence of the α-ester group of 3 promotes SN2 reaction in two ways: The electron-withdrawing nature of the substituent inhibits SN1 dissociation and carbocation formation, and the planar ester group poses minimal steric hindrance towards approach of the nucleophile.
Yields of substitution in α-aryl-α-hydroxy esters 5 are poor with a significantly decreased enantiomeric ratio in the product 6 (Scheme 2). The low yields can be attributed to the formation of β-phenylthioesters 7 by elimination followed by conjugate addition. Even in the presence of the adjacent carbonyl group, competing SN1 dissociation of the benzylic leaving group leads to a loss of enantiomeric purity in some cases. The use of α,α-dialkyl hydroxy esters 8 is more successful: Thioethers 10 are formed in high yield and with almost complete stereospecificity (Scheme 3).
![[1860-5397-7-68-i2]](/bjoc/content/inline/1860-5397-7-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Nucleophilic substitution in α-aryl-α-hydroxy esters.
Scheme 2: Nucleophilic substitution in α-aryl-α-hydroxy esters.
![[1860-5397-7-68-i3]](/bjoc/content/inline/1860-5397-7-68-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of α,α-dialkylthioethers.
Scheme 3: Preparation of α,α-dialkylthioethers.
Using the same principles of low steric bulk and electronic inhibition of the SN1 reaction pathway, α-(sulfonyloxy)nitriles, easily prepared from cyanohydrins, can also be made to undergo SN2 reaction with sulfur nucleophiles [13]. However, substitution of the quaternary α-(sulfonyloxy)nitrile 11 is very slow, and conversion of 11 to 12 with potassium thioacetate in DMF proceeds in only 35% yield after 72 hours. SN2 displacement of the leaving group by thioacetate in toluene proceeds faster at 55 °C (Scheme 4).
![[1860-5397-7-68-i4]](/bjoc/content/inline/1860-5397-7-68-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of α-cyanothioacetate 12.
Scheme 4: Preparation of α-cyanothioacetate 12.
The synthesis of the unnatural enantiomer of the natural product spirobrassinin has been achieved by substitution at a quaternary centre by a sulfur nucleophile [14]. Intramolecular cyclisation of a dithiocarbamate 13 allows isolation of 14 with complete inversion of stereochemistry, although no yield for this reaction was reported (Scheme 5).
![[1860-5397-7-68-i5]](/bjoc/content/inline/1860-5397-7-68-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of (R)-(+)-spirobrassinin.
Scheme 5: Synthesis of (R)-(+)-spirobrassinin.
Peregrina and co-workers have developed a general method for the synthesis of α-functionalised β-amino acids [15-17] employing nucleophilic ring opening of cyclic sulfamidate 15 via an invertive SN2 mechanism (Scheme 6) [16].
![[1860-5397-7-68-i6]](/bjoc/content/inline/1860-5397-7-68-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Opening of cyclic sulfamidates with thiol nucleophiles.
Scheme 6: Opening of cyclic sulfamidates with thiol nucleophiles.
A variety of nucleophiles are capable of ring opening the sulfamidate 15 in good yields, and simple functional group transformations allow isolation of the functionalised β-amino acids. Sulfur-based nucleophiles generally undergo reaction with sulfamidate 15 cleanly to give products 16 with complete inversion of configuration [16,17]. Acid catalysed cleavage of a thioether or thioacetate yields tert-thiols 17 in good yield.
1.1.2 Epoxide ring opening
SN2 displacements from quaternary electrophiles require specific structural features to avoid competing racemisation: Excellent leaving groups and electron withdrawing, non-aromatic and small substituents at the reactive centre all favour invertive substitution. Under basic conditions, epoxides also undergo invertive reactions, even at quaternary centres. Sulfur-based nucleophiles have been employed in nucleophilic ring opening of epoxides in hindered systems [18-23], with most examples of the latter occurring at quaternary carbon atoms which are part of ring systems [18-22], and in particular in steroids [18-20]. For example, the thiol-containing androgen 19 is prepared in 71% yield by epoxide ring opening of 18 with potassium hydrogen sulfide (Scheme 7) [18].
![[1860-5397-7-68-i7]](/bjoc/content/inline/1860-5397-7-68-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Epoxide ring opening by a sulfur nucleophile was also employed as the key step in the synthesis of (+)-BE-52440A (22) [22] (Scheme 8). Dimerisation of nanaomycin derivative 21 through a bridging sulfide involves a double regioselective invertive epoxide opening.
![[1860-5397-7-68-i8]](/bjoc/content/inline/1860-5397-7-68-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
1.1.3 Mitsunobu reactions
The Mitsunobu reaction offers an operationally straightforward method for activating simple alcohols to invertive substitution, and it is widely used for constructing new stereodefined carbon–heteroatom bonds [24,25]. The Mitsunobu reaction proceeds by reaction of a phosphine and DIAD or DEAD with an alcohol 23 to form an O-phosphinite leaving group (Scheme 9). SN2 substitution to yield 24 is accompanied by formation of a phosphine oxide.
![[1860-5397-7-68-i9]](/bjoc/content/inline/1860-5397-7-68-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
There are many examples of the use of sulfur nucleophiles in the Mitsunobu reaction [25-27]: The reaction is successful with a wide range of primary and secondary alcohols, but Mitsunobu-type reactions are very sensitive to steric bulk at the electrophilic carbon atom. For example, reaction of alcohol 25 with a phenol under standard Mitsunobu conditions at 50 °C for 16 hours provides only a trace amount of the desired chiral ether 26, while reaction at 100 °C allows isolation of 26 in moderate yield (Scheme 10) [28]. A gem-diethyl analogue of 25 failed to give any substitution at all, the major product being the result of elimination.
![[1860-5397-7-68-i10]](/bjoc/content/inline/1860-5397-7-68-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Mitsunobu substitution at a quaternary centre.
Scheme 10: Mitsunobu substitution at a quaternary centre.
Despite the proven reluctance of tertiary alcohols to undergo Mitsunobu reactions, there is one example of the displacement of a tertiary alcohol by a thiol under Mitsunobu conditions in the literature. La Clair reported the synthesis of the initially assigned structure of natural product hexacyclinol (27, Figure 4) [29].
![[1860-5397-7-68-4]](/bjoc/content/figures/1860-5397-7-68-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Initially assigned structure of hexacyclinol.
Figure 4: Initially assigned structure of hexacyclinol.
The reported synthesis entailed Mitsunobu reaction at a very hindered quaternary centre, accomplishing invertive substitution of 28 with thiophenol under mild reaction conditions to yield 29 in 94% yield (Scheme 11).
![[1860-5397-7-68-i11]](/bjoc/content/inline/1860-5397-7-68-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
The paucity of literature precedents for Mitsunobu reactions of tertiary alcohols brings into sharp focus the remarkable success of this reaction under such mild conditions. However, this reaction and other unusual steps in the synthesis attracted scepticism among synthetic chemists [30], and many aspects of this synthesis were called into question when a revised structure of hexacyclinol was proposed by Rychnovsky [31]. A successful synthesis of the revised structure matched the published data for the natural compound [32].
A modification of the Mitsunobu reaction, that does allow the formation of sulfur-substituted tertiary carbon atoms with inversion of stereochemistry, was reported by Mukaiyama and co-workers [33,34]. The method employs benzoquinone derivatives instead of azodicarboxylates as the oxidising agent. A series of phosphinites 31 was prepared by treatment of alcohols 30 with chlorodiphenylphosphine in the presence of triethylamine and DMAP. SN2 substitution of the phosphinites proceeds by oxidative activation of the leaving group with a 1,4-benzoquinone derivative, DBBQ (35) being most effective (Scheme 12). Addition of a thiol nucleophile to adduct 32 results in SN2 inversion and isolation of the enantiomerically pure (94:6–99:1 er) tertiary thioether 33 and by-product 34. Highest yields were obtained with BtzSH (36) and BoxSH (37).
![[1860-5397-7-68-i12]](/bjoc/content/inline/1860-5397-7-68-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Thioethers 33 prepared from phosphinites 31.
Scheme 12: Thioethers 33 prepared from phosphinites 31.
The reaction proceeds well with many hindered substrates incorporating aromatic, alkyl and ester substituents with excellent stereospecificity. Enantiomerically pure thiols can also be made from the product: Aromatic thioether 38 is reduced with lithium aluminium hydride to give 39 in 95% yield (Scheme 13).
![[1860-5397-7-68-i13]](/bjoc/content/inline/1860-5397-7-68-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Preparation of enantiomerically pure thiol 39.
Scheme 13: Preparation of enantiomerically pure thiol 39.
The methodology was later refined to allow substitution of alcohols in 1 step. Phenoxydiphenylphosphine was used to prepare the phosphinite intermediate and an azide was used as the oxidising agent instead of a benzoquinone derivative (Scheme 14) [35-37].
![[1860-5397-7-68-i14]](/bjoc/content/inline/1860-5397-7-68-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Thioethers prepared by a modified Mitsunobu reaction.
Scheme 14: Thioethers prepared by a modified Mitsunobu reaction.
These conditions allow the efficient preparation of chiral tertiary thioether products 40 from hindered tertiary alcohols. However, the yield of the simple alkyl-substituted thioether 40e was a disappointing 17%. As with other methods, the preparation of simple optically pure chiral tertiary thiols by Mitsunobu-type procedures is not straightforward. Mitsunobu reactions also suffer from atom inefficiency due to the stoichiometric quantities of phosphorus-containing by-products produced by the reaction.
1.2 Conjugate addition
The carbon–sulfur bond of enantiomerically pure tertiary organosulfur compounds 42 may be constructed by facially selective addition of a sulfur nucleophile to the disubstituted terminus of a conjugated alkene 41 (Scheme 15).
![[1860-5397-7-68-i15]](/bjoc/content/inline/1860-5397-7-68-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Nucleophilic conjugate addition.
Scheme 15: Nucleophilic conjugate addition.
There are several examples of such facially selective addition of thiols to substituted sp2 carbon atoms, including intramolecular conjugate addition in steroidal systems [38,39] and intermolecular addition directed by substrate stereochemistry [40,41] or a chiral auxiliary [42,43]. Despite these examples, there are very few general procedures for the preparation of chiral tertiary thiols and thioethers by this method. This may be attributed to three difficulties: The low reactivity of β,β-disubstituted Michael receptors, the difficulty in controlling π-facial stereoselectivity, and the equilibrium of stereoisomers through an addition/elimination mechanism.
The reduced reactivity of β,β-disubstituted Michael receptors towards nucleophilic addition is demonstrated by the catalytic asymmetric additions of thiols to α,β-unsaturated ketones [44]. (R)-LaNa3tris(binaphthoxide) (LSB) catalyses asymmetric addition to cyclic enones (Scheme 16), and products of addition 43 are isolated in good yields and enantiomeric ratios.
![[1860-5397-7-68-i16]](/bjoc/content/inline/1860-5397-7-68-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Asymmetric addition to cyclic enones.
Scheme 16: Asymmetric addition to cyclic enones.
However, addition to the substituted substrate 44 is more difficult. A higher loading of the catalyst and higher temperature are required to produce the enantioenriched thioether 45 even in moderate yield and acceptable er (Scheme 17).
![[1860-5397-7-68-i17]](/bjoc/content/inline/1860-5397-7-68-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Despite the poor yield obtained with this substrate, similar conditions lead to catalytic kinetic resolution of the enantiomers of hindered enone 46 by addition, oxidation and elimination of a sulfenic acid under basic conditions (Scheme 18) [45].
![[1860-5397-7-68-i18]](/bjoc/content/inline/1860-5397-7-68-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Catalytic kinetic resolution of the enantiomers of enone 46.
Scheme 18: Catalytic kinetic resolution of the enantiomers of enone 46.
Xiao and co-workers developed an organocatalytic process for the addition of thiols to nitroalkenes [46]. Using thiourea organocatalyst 48, conjugate addition of a variety of thiols to a range of nitroalkenes 49 proceeds to give 50 and hence 51 in good yield and good enantioselectivity (Scheme 19).
![[1860-5397-7-68-i19]](/bjoc/content/inline/1860-5397-7-68-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Organocatalytic conjugate addition to nitroalkenes 49.
Scheme 19: Organocatalytic conjugate addition to nitroalkenes 49.
α-Arylthio-β-amino acid 54 was prepared from Michael adduct 52 in three steps (Scheme 20) in good yield and with full conservation of enantiomeric purity.
![[1860-5397-7-68-i20]](/bjoc/content/inline/1860-5397-7-68-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Preparation of β-amino acid 54.
Scheme 20: Preparation of β-amino acid 54.
Palomo and co-workers postulated that poor reactivity and facial selectivity in conjugate additions with sulfur could be minimised by an intramolecular approach [47,48]. A Lewis acid-promoted sulfur migration within N-enoyl oxazolidine-2-thione substrates 56 followed by hydrolysis gave optically pure tertiary thiols 57 directly (Scheme 21), with only the electron rich para-methoxyphenyl substituent performing poorly.
![[1860-5397-7-68-i21]](/bjoc/content/inline/1860-5397-7-68-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Sulfur migration within oxazolidine-2-thiones 56.
Scheme 21: Sulfur migration within oxazolidine-2-thiones 56.
As with stereospecific nucleophilic displacement, only specific substrates are tolerated in conjugate additions, and further functionality is always required to activate the electrophilic alkene towards attack by the sulfur nucleophile.
2 Carbon–carbon bond formation
2.1 Electrophilic addition to α-thioenolates
Diastereoselective alkylation of α-thioenolates has been used to prepare several types of enantiomerically pure tertiary thioethers [49-54]. The method of “self-regeneration of stereocentres” developed by Seebach [55] employs α-thiocarboxylic acids 58 condensed with pivaldehyde to generate 1,3-oxathiolan-4-ones 59 (Scheme 22) [49,50]. The cis diastereomer is formed preferentially (2:1–8:1 selectivity), and can usually be purified by crystallisation. Enolate 60 is formed by deprotonation with a lithium base and treated with an electrophile. Electrophilic addition takes place diastereoselectively anti to the bulky tert-butyl group, producing oxathiolanone products 61 in excellent diastereoselectivities.
![[1860-5397-7-68-i22]](/bjoc/content/inline/1860-5397-7-68-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Preparation of thiols 62 by self-regeneration of stereocentres.
Scheme 22: Preparation of thiols 62 by self-regeneration of stereocentres.
Enantiopure tertiary thiols 62 can be liberated from the oxathiolanone products by hydrolysis, and the method was employed by Townsend and co-workers in the synthesis of (5R)-thiolactomycin (66, Scheme 23) [51]. Diastereoselective addition of an aldehyde to oxathiolanone 64 affords 65 in 81% yield. Further synthetic manipulations allowed preparation of (5R)-thiolactomycin (66) in >99:1 er.
![[1860-5397-7-68-i23]](/bjoc/content/inline/1860-5397-7-68-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of (5R)-thiolactomycin.
Scheme 23: Synthesis of (5R)-thiolactomycin.
2.2 Electrophilic attack on α-thioorganolithiums
Formation of carbon–carbon bonds adjacent to heteroatoms by deprotonation with an organolithium base and subsequent reaction with an electrophile has become an important and versatile method, especially when chiral ligands may be used to govern the enantioselectivity of the reaction pathway [56]. Tertiary organosulfur compounds can be made in this way by lithiation of a chiral secondary thiol derivative (Scheme 24).
![[1860-5397-7-68-i24]](/bjoc/content/inline/1860-5397-7-68-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Preparation of tertiary thiols and thioethers via α-thioorganolithiums.
Scheme 24: Preparation of tertiary thiols and thioethers via α-thioorganolithiums.
Three conditions must be met if this method is to be used for preparation of enantiomerically pure tertiary thiol derivatives:
1) The substrate 67 must be available in an enantiomerically pure form,
2) The lithiated intermediate 68 must be configurationally stable, and
3) Electrophilic addition to give 69 must proceed with either complete retention or complete inversion of stereochemistry.
2.2.1 Configurational stability in α-thioorganolithiums
In contrast to α-oxy and α-aminoorganolithiums, simple α-thioorganolithium compounds show high configurational lability even at –80 °C [57]. Beak noted configurational instability during the diastereoselective methylation of α-thioorganolithium 71 (Scheme 25) [58]. Equilibration of the diastereoisomers of 71 favoured axial methylation of the lithiated intermediate to give 72.
![[1860-5397-7-68-i25]](/bjoc/content/inline/1860-5397-7-68-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Diastereoselective methylation of organolithium 71.
Scheme 25: Diastereoselective methylation of organolithium 71.
Studies by both Hoffmann and co-workers [59,60] and by Reich and co-workers [61] showed that the rate determining step for racemisation of α-thio, α-seleno and α-telluroorganolithiums is rotation about the C–S, Se or Te bond [62]. Simple inversion in α-thio-substituted carbanions has a barrier as low as 0.5 kcal·mol−1 [63] making inversion itself unlikely to comprise the rate determining step of the racemisation. Consistent with this explanation, the barrier to racemisation increases with greater steric bulk in the chalcogen substituent.
2.2.2 Lithiation and alkylation of thiocarbamates
Hoppe and co-workers applied the observations of Hoffmann and Reich in comprehensive studies on configurationally stable α-lithiothiocarbamates. Stereoselective deprotonations of thiocarbamate 73 in the presence of (−)-sparteine 74 (Scheme 26) were attempted [64].
![[1860-5397-7-68-i26]](/bjoc/content/inline/1860-5397-7-68-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Addition to lithiated thiocarbamate 75.
Scheme 26: Addition to lithiated thiocarbamate 75.
Trapping the lithiated intermediate 75 with electrophiles gave products with poor enantioselectivities, but the results gave no indication of the configurational stability of the intermediate organolithium 75. To establish the factors governing enantioselectivity in the electrophilic substitution of 73, Hoppe made use of an extremely high kinetic H/D isotope effect observed for deprotonation in similar compounds [65]. Reaction of a test substrate demonstrated an isotope effect of kH/kD ≥ 100. A racemic mixture of 77 was lithiated in the presence of (−)-sparteine and quenched with trimethylsilyl chloride (Scheme 27). Deuterated thiocarbamate 78 containing >99% deuterium was isolated from the reaction with an enantiomeric ratio of 67:33. The high deuterium content of the product indicated that both enantiomers of 77 must have been deprotonated, and thus that the lithio derivative of thiocarbamate 77 must be configurationally labile since the product is non-racemic.
![[1860-5397-7-68-i27]](/bjoc/content/inline/1860-5397-7-68-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Configurational lability in unhindered α-lithiothiocarbamates.
Scheme 27: Configurational lability in unhindered α-lithiothiocarbamates.
In order to induce greater configurational stability, thiocarbamate 79, with increased steric bulk, was prepared. A sample of 79 with an enantiomeric ratio of 73:27 was lithiated with sec-butyllithium in diethyl ether and TMEDA at −78 °C for 2.5 hours and quenched with MeOD (Scheme 28). Deuterated thiocarbamate 78 was isolated with an enantiomeric ratio of 72:28, indicating almost full conservation of enantiomeric purity and hence demonstrating that the more congested organolithium 80 is now configurationally stable under the reaction conditions. Enantiomerically pure tertiary thiols can be liberated from the substitution products by cleavage of the carbamoyl functionality, providing a rare asymmetric route to simple unfunctionalised tertiary thiols.
![[1860-5397-7-68-i28]](/bjoc/content/inline/1860-5397-7-68-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Configurational stability in bulky α-lithiothiocarbamates.
Scheme 28: Configurational stability in bulky α-lithiothiocarbamates.
The lithio derivative of benzylic thiocarbamate 81 was also configurationally stable in diethyl ether and TMEDA at −78 °C [66,67]. Reaction with a series of electrophiles allowed isolation of functionalised thiocarbamates 83 in excellent yield and enantiomeric ratio (Scheme 29). Benzylic organolithiums are notorious for the lack of consistency with which they undergo electrophilic substitution [56], and 82 is remarkable in that all electrophiles, except protonating agents, react with complete inversion of configuration.
![[1860-5397-7-68-i29]](/bjoc/content/inline/1860-5397-7-68-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Asymmetric functionalisation of secondary benzylic thiocarbamates.
Scheme 29: Asymmetric functionalisation of secondary benzylic thiocarbamates.
Enantiopure cyclohexenyl thiocarbamates also form configurationally stable α-thioallyllithium species [68-70]. Lithiation of thiocarbamate 85 followed by methylation results in the isolation of regioisomeric products arising from electrophilic substitution at either end of the lithioallyl system (Scheme 30).
![[1860-5397-7-68-i30]](/bjoc/content/inline/1860-5397-7-68-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Methylation of lithioallyl thiocarbamates.
Scheme 30: Methylation of lithioallyl thiocarbamates.
In the case of the secondary thiocarbamate, the γ-substituted product 87 is preferentially formed, whilst with an N,N-diisopropylthiocarbamate selective formation of the α-substituted product 89 (Scheme 31) is observed.
![[1860-5397-7-68-i31]](/bjoc/content/inline/1860-5397-7-68-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Asymmetric preparation of tertiary allylic thiols.
Scheme 31: Asymmetric preparation of tertiary allylic thiols.
Reduction of the functionalised thiocarbamate products 89 with lithium aluminium hydride affords enantiomerically pure thiols 90 in excellent yields and enantiomeric ratios.
2.2.3 Lithiation and rearrangement of thiocarbamates
Carbon–carbon bond formation in configurationally stable α-thioorganolithiums allows access to non-racemic tertiary thiols of varying structure and complexity. Hoppe’s extensive studies of lithiated thiocarbamates have continued to display the synthetic utility of these species in the formation of secondary thiol derivatives. These were prepared by diastereoselective electrophilic additions in proline-derived systems [71,72] and asymmetric alkylation of thiocarbamates in the presence of a chiral ligand [73,74]. Stereospecific functionalisation of configurationally stable lithiated thiocarbamates with electrophiles is general for a range of structures, but until very recently it has not been applicable to arylation reactions. However, we have discovered [75] a variant of a rearrangement reaction of lithiated ureas that we first reported in 2007 [76] in which a lithiated thiocarbamate undergoes an intramolecular aryl transfer. Treatment of an N-aryl S-benzyl thiocarbamate 92 leads to formation of a lithio derivative 93 comparable with those reported by Hoppe (Scheme 32). As in the case of lithiated N-aryl ureas [76-80] and N-aryl carbamates [81,82], the N-aryl ring is susceptible to attack by the anionic centre, and migration of the ring to the position α to sulfur occurs, presumably via an intermediate related to 94. After work up the product is a tertiary thiocarbamate 95, and simple mildly basic hydrolysis leads to the tertiary thiol 96. The aryl migration is applicable to a range of products 96 (Table 1) and amounts to an intramolecular arylation, allowing the formation of otherwise inaccessible doubly benzylic tertiary thiols in enantiomerically enriched form from benzylic alcohols 91. Similar to the rearrangement of lithiated ureas [76] (but interestingly in contrast to the rearrangement of lithiated carbamates [81,82]), aryl migration proceeds with retention of stereochemistry. Current work is continuing with the aim of expanding the utility of this new mode of reactivity displayed by thiocarbamates.
![[1860-5397-7-68-i32]](/bjoc/content/inline/1860-5397-7-68-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Asymmetric preparation of thiols 96 by aryl migration in lithiated thiocarbamates.
Scheme 32: Asymmetric preparation of thiols 96 by aryl migration in lithiated thiocarbamates.
Table 1: Scope of the thiocarbamate rearrangement [75].
| SM, er | Ar1 | Ar2 | R |
95
yield (%) |
95
er |
|---|---|---|---|---|---|
| (S)-92a, 98:2 | Ph | 4-MeC6H4 | Me | 83 | 96:4 |
| (S)-92b, 97:3 | Ph | 2-OMeC6H4 | Me | 89 | 91:9 |
| (S)-92c, 98:2 | Ph | 4-ClC6H4 | Me | 94 | 96:4 |
| (S)-92d, 98:2 | Ph | 3-ClC6H4 | Me | 69 | 96:4 |
| (S)-92e, 98:2 | Ph | 4-CNC6H4 | Me | 78 | 97:3 |
| (S)-92f, 99:1 | Ph | 1-naphthyl | Me | 98 | 96:4 |
| (S)-92g, 98:2 | Ph | 3-FC6H4 | Me | 0 | – |
| (R)-92h, 99:1 | Ph | 4-MeC6H4 | n-Pr | 74 | 98:2 |
| (R)-92i, 96:4 | 3-CF3 | Ph | Me | 85 | 67:33 |
| (±)-92j, 50:50 | 4-OMe | 4-MeC6H4 | Me | 86 | 50:50 |
| (R)-92k, 79:21 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-68-i33.svg?max-width=637&scale=1.0)
|
87 | 74:26 | ||
Conclusion
Of the methods available for the synthesis of tertiary thiols, the majority (for example, those involving epoxide opening or electrophilic attack on thio-substituted enolates) lead to thiols carrying further functionality in some form. Tertiary thiols without further functionality are still challenging, with organolithium-based alkylations and arylation (by rearrangement) of thiocarbamates offering the best prospects. Further developments in this area – particularly with regard to carbolithiation reactions [79] – are to be expected.
References
-
2009 Top 200 Branded Drugs by Retail Dollars. Drug Topics, 2010; http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard//drugtopics/252010/674961/article.pdf.
Return to citation in text: [1] [2] -
Cremlyn, R. J. An Introduction to Organosulfur Chemistry; John Wiley & Sons Ltd.: Chichester, U.K., 1996.
Return to citation in text: [1] -
Toru, T.; Bolm, C., Eds. Organosulfur Chemistry in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2008.
Return to citation in text: [1] [2] -
Fernández, I.; Khiar, N. Chem. Rev. 2003, 103, 3651–3706. doi:10.1021/cr990372u
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2006, 62, 5559–5601. doi:10.1016/j.tet.2006.03.093
Return to citation in text: [1] -
Mellah, M.; Voituriez, A.; Schulz, E. Chem. Rev. 2007, 107, 5133–5205. doi:10.1021/cr068440h
Return to citation in text: [1] -
Carreño, M. C.; Hernández-Torres, G.; Ribagorda, M.; Urbano, A. Chem. Commun. 2009, 6129–6144. doi:10.1039/B908043K
Return to citation in text: [1] -
Wojaczyńska, E.; Wojaczyński, J. Chem. Rev. 2010, 110, 4303–4356. doi:10.1021/cr900147h
Return to citation in text: [1] -
Procter, D. J. J. Chem. Soc., Perkin Trans. 1 2001, 335–354. doi:10.1039/b002081h
Return to citation in text: [1] [2] [3] -
Shibasaki, M.; Kanai, M. Chem. Rev. 2008, 108, 2853–2873. doi:10.1021/cr078340r
Return to citation in text: [1] -
Voss, J. J. Sulfur Chem. 2009, 30, 167–207. doi:10.1080/17415990802673017
Return to citation in text: [1] -
Weaver, J. D.; Morris, D. K.; Tunge, J. A. Synlett 2010, 470–474. doi:10.1055/s-0029-1219186
Return to citation in text: [1] -
Effenberger, F.; Gaupp, S. Tetrahedron: Asymmetry 1999, 10, 1765–1775. doi:10.1016/S0957-4166(99)00154-8
Return to citation in text: [1] -
Monde, K.; Taniguchi, T.; Miura, N.; Nishimura, S.-I.; Harada, N.; Dukor, R. K.; Nafie, L. A. Tetrahedron Lett. 2003, 44, 6017–6020. doi:10.1016/S0040-4039(03)01513-2
Return to citation in text: [1] -
Avenoza, A.; Busto, J. H.; Corzana, F.; Jiménez-Osés, G.; Peregrina, J. M. Chem. Commun. 2004, 980–981. doi:10.1039/b400282b
Return to citation in text: [1] -
Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. J. Org. Chem. 2006, 71, 1692–1695. doi:10.1021/jo051632c
Return to citation in text: [1] [2] [3] -
Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. Org. Lett. 2006, 8, 2855–2858. doi:10.1021/ol060993r
Return to citation in text: [1] [2] -
Bednarski, P. J.; Nelson, S. D. J. Med. Chem. 1989, 32, 203–213. doi:10.1021/jm00121a037
Return to citation in text: [1] [2] [3] [4] -
Kitagawa, I.; Ueda, Y.; Kawasaki, T.; Mosettig, E. J. Org. Chem. 1963, 28, 2228–2232. doi:10.1021/jo01044a018
Return to citation in text: [1] [2] [3] -
Komeno, T.; Kishi, M. Tetrahedron 1971, 27, 1517–1526. doi:10.1016/S0040-4020(01)98017-2
Return to citation in text: [1] [2] [3] -
Caine, D.; Crews, E.; Salvino, J. M. Tetrahedron Lett. 1983, 24, 2083–2086. doi:10.1016/S0040-4039(00)81850-X
Return to citation in text: [1] [2] -
Tatsuta, K.; Suzuki, Y.; Toriumi, T.; Furuya, Y.; Hosokawa, S. Tetrahedron Lett. 2007, 48, 8018–8021. doi:10.1016/j.tetlet.2007.09.039
Return to citation in text: [1] [2] [3] -
López, I.; Rodríguez, S.; Izquierdo, J.; González, F. V. J. Org. Chem. 2007, 72, 6614–6617. doi:10.1021/jo0709955
Return to citation in text: [1] -
Mitsunobu, O. Synthesis 1981, 1–28. doi:10.1055/s-1981-29317
Return to citation in text: [1] -
Kumara Swamy, K. C.; Bhuvan Kumar, N. N.; Balaraman, E.; Pavan Kumar, K. V. P. Chem. Rev. 2009, 109, 2551–2651. doi:10.1021/cr800278z
Return to citation in text: [1] [2] -
Walker, K. A. M. Tetrahedron Lett. 1977, 18, 4475–4478. doi:10.1016/S0040-4039(01)83541-3
Return to citation in text: [1] -
Kotsuki, H.; Matsumoto, K.; Nishizawa, H. Tetrahedron Lett. 1991, 32, 4155–4158. doi:10.1016/S0040-4039(00)79890-X
Return to citation in text: [1] -
Shi, Y.-J.; Hughes, D. L.; McNamara, J. M. Tetrahedron Lett. 2003, 44, 3609–3611. doi:10.1016/S0040-4039(03)00728-7
Return to citation in text: [1] -
La Clair, J. J. Angew. Chem., Int. Ed. 2006, 45, 2769–2773. doi:10.1002/anie.200504033
Return to citation in text: [1] -
Marris, E. Nature 2006, 442, 492–493. doi:10.1038/442492c
Return to citation in text: [1] -
Rychnovsky, S. D. Org. Lett. 2006, 8, 2895–2898. doi:10.1021/ol0611346
Return to citation in text: [1] -
Porco, J. A., Jr.; Su, S.; Lei, X.; Bardhan, S.; Rychnovsky, S. D. Angew. Chem., Int. Ed. 2006, 45, 5790–5792. doi:10.1002/anie.200602854
Return to citation in text: [1] -
Ikegai, K.; Pluempanupat, W.; Mukaiyama, T. Chem. Lett. 2005, 34, 638–639. doi:10.1246/cl.2005.638
Return to citation in text: [1] -
Ikegai, K.; Pluempanupat, W.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 780–790. doi:10.1246/bcsj.79.780
Return to citation in text: [1] -
Kuroda, K.; Maruyama, Y.; Hayashi, Y.; Mukaiyama, T. Chem. Lett. 2008, 37, 836–837. doi:10.1246/cl.2008.836
Return to citation in text: [1] -
Kuroda, K.; Maruyama, Y.; Hayashi, Y.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2009, 82, 381–392. doi:10.1246/bcsj.82.381
Return to citation in text: [1] -
Mukaiyama, T.; Kuroda, K.; Maruyama, Y. Heterocycles 2010, 80, 63–82. doi:10.3987/REV-09-SR(S)1
Return to citation in text: [1] -
Childers, W. E.; Furth, P. S.; Shih, M.-J.; Robinson, C. H. J. Org. Chem. 1988, 53, 5947–5951. doi:10.1021/jo00260a026
Return to citation in text: [1] -
Lesuisse, D.; Gourvest, J.-F.; Benslimane, O.; Canu, F.; Delaisi, C.; Doucet, B.; Hartmann, C.; Lefrançois, J.-M.; Tric, B.; Mansuy, D.; Philibert, D.; Teutsch, G. J. Med. Chem. 1996, 39, 757–772. doi:10.1021/jm950539l
Return to citation in text: [1] -
Hanessian, S.; Reddy, B. Tetrahedron 1999, 55, 3427–3443. doi:10.1016/S0040-4020(98)01152-1
Return to citation in text: [1] -
McDevitt, J. P.; Lansbury, P. T., Jr. J. Am. Chem. Soc. 1996, 118, 3818–3828. doi:10.1021/ja9525622
Return to citation in text: [1] -
Ohata, K.; Terashima, S. Tetrahedron Lett. 2006, 47, 2787–2791. doi:10.1016/j.tetlet.2006.02.058
Return to citation in text: [1] -
Ohata, K.; Terashima, S. Bioorg. Med. Chem. Lett. 2007, 17, 4070–4074. doi:10.1016/j.bmcl.2007.04.067
Return to citation in text: [1] -
Emori, E.; Arai, T.; Sasai, H.; Shibasaki, M. J. Am. Chem. Soc. 1998, 120, 4043–4044. doi:10.1021/ja980397v
Return to citation in text: [1] -
Emori, E.; Iida, T.; Shibasaki, M. J. Org. Chem. 1999, 64, 5318–5320. doi:10.1021/jo9904922
Return to citation in text: [1] -
Lu, H.-H.; Zhang, F.-G.; Meng, X.-G.; Duan, S.-W.; Xiao, W.-J. Org. Lett. 2009, 11, 3946–3949. doi:10.1021/ol901572x
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; Dias, F.; López, R.; Linden, A. Angew. Chem., Int. Ed. 2004, 43, 3307–3310. doi:10.1002/anie.200453889
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; López, R.; González, P. B.; Gómez-Bengoa, E.; Saá, J. M.; Linden, A. J. Am. Chem. Soc. 2006, 128, 15236–15247. doi:10.1021/ja0654027
Return to citation in text: [1] -
Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 3664–3671. doi:10.1021/jo00369a020
Return to citation in text: [1] [2] -
Strijtveen, B.; Kellogg, R. M. Tetrahedron 1987, 43, 5039–5054. doi:10.1016/S0040-4020(01)87682-1
Return to citation in text: [1] [2] -
McFadden, J. M.; Frehywot, G. L.; Townsend, C. A. Org. Lett. 2002, 4, 3859–3862. doi:10.1021/ol026685k
Return to citation in text: [1] [2] -
Alibés, R.; Bayón, P.; de March, P.; Figueredo, M.; Font, J.; Marjanet, G. Org. Lett. 2006, 8, 1617–1620. doi:10.1021/ol060173e
Return to citation in text: [1] -
Arpin, A.; Manthorpe, J. M.; Gleason, J. L. Org. Lett. 2006, 8, 1359–1362. doi:10.1021/ol060106k
Return to citation in text: [1] -
Tiong, E. A.; Gleason, J. L. Org. Lett. 2009, 11, 1725–1728. doi:10.1021/ol802643k
Return to citation in text: [1] -
Seebach, D.; Naef, R.; Calderari, G. Tetrahedron 1984, 40, 1313–1324. doi:10.1016/S0040-4020(01)82417-0
Return to citation in text: [1] -
Clayden, J. Organolithiums: Selectivity for Synthesis; Tetrahedron Organic Chemistry Series, Vol. 23; Pergamon: Oxford, U.K., 2002.
Return to citation in text: [1] [2] -
McDougal, P. G.; Condon, B. D.; Laffosse, M. D., Jr.; Lauro, A. M.; VanDerveer, D. Tetrahedron Lett. 1988, 29, 2547–2550. doi:10.1016/S0040-4039(00)86108-0
Return to citation in text: [1] -
Beak, P.; Becker, P. D. J. Org. Chem. 1982, 47, 3855–3861. doi:10.1021/jo00141a010
Return to citation in text: [1] -
Ruhland, T.; Dress, R. K.; Hoffmann, R. W. Angew. Chem., Int. Ed. Engl. 1993, 32, 1467–1468. doi:10.1002/anie.199314671
Return to citation in text: [1] -
Hoffmann, R. W.; Dress, R. K.; Ruhland, T.; Wenzel, A. Chem. Ber. 1995, 128, 861–870. doi:10.1002/cber.19951280903
Return to citation in text: [1] -
Reich, H. J.; Dykstra, R. R. Angew. Chem., Int. Ed. Engl. 1993, 32, 1469–1470. doi:10.1002/anie.199314691
Return to citation in text: [1] -
Aggarwal, V. K. Angew. Chem., Int. Ed. Engl. 1994, 33, 175–177. doi:10.1002/anie.199401751
Return to citation in text: [1] -
Wiberg, K. B.; Castejon, H. J. Am. Chem. Soc. 1994, 116, 10489–10497. doi:10.1021/ja00102a016
Return to citation in text: [1] -
Kaiser, B.; Hoppe, D. Angew. Chem., Int. Ed. Engl. 1995, 34, 323–325. doi:10.1002/anie.199503231
Return to citation in text: [1] -
Hoppe, D.; Paetow, M.; Hintze, F. Angew. Chem., Int. Ed. Engl. 1993, 32, 394–396. doi:10.1002/anie.199303941
Return to citation in text: [1] -
Hoppe, D.; Kaiser, B.; Stratmann, O.; Fröhlich, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 2784–2786. doi:10.1002/anie.199727841
Return to citation in text: [1] -
Stratmann, O.; Kaiser, B.; Fröhlich, R.; Meyer, O.; Hoppe, D. Chem.–Eur. J. 2001, 7, 423–435. doi:10.1002/1521-3765(20010119)7:2<423::AID-CHEM423>3.0.CO;2-Y
Return to citation in text: [1] -
Marr, F.; Fröhlich, R.; Hoppe, D. Org. Lett. 1999, 1, 2081–2083. doi:10.1021/ol991134o
Return to citation in text: [1] -
Marr, F.; Hoppe, D. Org. Lett. 2002, 4, 4217–4220. doi:10.1021/ol0266828
Return to citation in text: [1] -
Marr, F.; Fröhlich, R.; Wibbeling, B.; Diedrich, C.; Hoppe, D. Eur. J. Org. Chem. 2002, 2970–2988. doi:10.1002/1099-0690(200209)2002:17<2970::AID-EJOC2970>3.0.CO;2-J
Return to citation in text: [1] -
Sonawane, R. P.; Fröhlich, R.; Hoppe, D. Chem. Commun. 2006, 3101–3103. doi:10.1039/b604029b
Return to citation in text: [1] -
Sonawane, R. P.; Mück-Lichtenfeld, C.; Fröhlich, R.; Bergander, K.; Hoppe, D. Chem.–Eur. J. 2007, 13, 6419–6429. doi:10.1002/chem.200601853
Return to citation in text: [1] -
Sonawane, R. P.; Fröhlich, R.; Hoppe, D. Adv. Synth. Catal. 2006, 348, 1847–1854. doi:10.1002/adsc.200606177
Return to citation in text: [1] -
Lange, H.; Bergander, K.; Frölich, R.; Kehr, S.; Nakamura, S.; Shibata, N.; Toru, T.; Hoppe, D. Chem.–Asian J. 2008, 3, 88–101. doi:10.1002/asia.200700262
Return to citation in text: [1] -
MacLellan, P.; Clayden, J. Chem. Commun. 2011, 47, 3395–3397. doi:10.1039/C0CC04912C
Return to citation in text: [1] [2] -
Clayden, J.; Dufour, J.; Grainger, D. M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129, 7488–7489. doi:10.1021/ja071523a
Return to citation in text: [1] [2] [3] -
Clayden, J.; Hennecke, U. Org. Lett. 2008, 10, 3567–3570. doi:10.1021/ol801332n
Return to citation in text: [1] -
Bach, R.; Clayden, J.; Hennecke, U. Synlett 2009, 421–424. doi:10.1055/s-0028-1087543
Return to citation in text: [1] -
Clayden, J.; Donnard, M.; Lefranc, J.; Minassi, A.; Tetlow, D. J. J. Am. Chem. Soc. 2010, 132, 6624–6625. doi:10.1021/ja1007992
Return to citation in text: [1] [2] -
Tetlow, D. J.; Hennecke, U.; Raftery, J.; Waring, M. J.; Clarke, D. S.; Clayden, J. Org. Lett. 2010, 12, 5442–5445. doi:10.1021/ol102155h
Return to citation in text: [1] -
Clayden, J.; Farnaby, W.; Grainger, D. M.; Hennecke, U.; Mancinelli, M.; Tetlow, D. J.; Hillier, I. H.; Vincent, M. A. J. Am. Chem. Soc. 2009, 131, 3410–3411. doi:10.1021/ja808959e
Return to citation in text: [1] [2] -
Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-Ondozabal, H.; Clayden, J. Org. Lett. 2010, 12, 2222–2225. doi:10.1021/ol100627c
Return to citation in text: [1] [2]
| 33. | Ikegai, K.; Pluempanupat, W.; Mukaiyama, T. Chem. Lett. 2005, 34, 638–639. doi:10.1246/cl.2005.638 |
| 34. | Ikegai, K.; Pluempanupat, W.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 780–790. doi:10.1246/bcsj.79.780 |
| 35. | Kuroda, K.; Maruyama, Y.; Hayashi, Y.; Mukaiyama, T. Chem. Lett. 2008, 37, 836–837. doi:10.1246/cl.2008.836 |
| 36. | Kuroda, K.; Maruyama, Y.; Hayashi, Y.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2009, 82, 381–392. doi:10.1246/bcsj.82.381 |
| 37. | Mukaiyama, T.; Kuroda, K.; Maruyama, Y. Heterocycles 2010, 80, 63–82. doi:10.3987/REV-09-SR(S)1 |
| 38. | Childers, W. E.; Furth, P. S.; Shih, M.-J.; Robinson, C. H. J. Org. Chem. 1988, 53, 5947–5951. doi:10.1021/jo00260a026 |
| 39. | Lesuisse, D.; Gourvest, J.-F.; Benslimane, O.; Canu, F.; Delaisi, C.; Doucet, B.; Hartmann, C.; Lefrançois, J.-M.; Tric, B.; Mansuy, D.; Philibert, D.; Teutsch, G. J. Med. Chem. 1996, 39, 757–772. doi:10.1021/jm950539l |
| 49. | Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 3664–3671. doi:10.1021/jo00369a020 |
| 50. | Strijtveen, B.; Kellogg, R. M. Tetrahedron 1987, 43, 5039–5054. doi:10.1016/S0040-4020(01)87682-1 |
| 51. | McFadden, J. M.; Frehywot, G. L.; Townsend, C. A. Org. Lett. 2002, 4, 3859–3862. doi:10.1021/ol026685k |
| 52. | Alibés, R.; Bayón, P.; de March, P.; Figueredo, M.; Font, J.; Marjanet, G. Org. Lett. 2006, 8, 1617–1620. doi:10.1021/ol060173e |
| 53. | Arpin, A.; Manthorpe, J. M.; Gleason, J. L. Org. Lett. 2006, 8, 1359–1362. doi:10.1021/ol060106k |
| 54. | Tiong, E. A.; Gleason, J. L. Org. Lett. 2009, 11, 1725–1728. doi:10.1021/ol802643k |
| 55. | Seebach, D.; Naef, R.; Calderari, G. Tetrahedron 1984, 40, 1313–1324. doi:10.1016/S0040-4020(01)82417-0 |
| 46. | Lu, H.-H.; Zhang, F.-G.; Meng, X.-G.; Duan, S.-W.; Xiao, W.-J. Org. Lett. 2009, 11, 3946–3949. doi:10.1021/ol901572x |
| 47. | Palomo, C.; Oiarbide, M.; Dias, F.; López, R.; Linden, A. Angew. Chem., Int. Ed. 2004, 43, 3307–3310. doi:10.1002/anie.200453889 |
| 48. | Palomo, C.; Oiarbide, M.; López, R.; González, P. B.; Gómez-Bengoa, E.; Saá, J. M.; Linden, A. J. Am. Chem. Soc. 2006, 128, 15236–15247. doi:10.1021/ja0654027 |
| 44. | Emori, E.; Arai, T.; Sasai, H.; Shibasaki, M. J. Am. Chem. Soc. 1998, 120, 4043–4044. doi:10.1021/ja980397v |
| 45. | Emori, E.; Iida, T.; Shibasaki, M. J. Org. Chem. 1999, 64, 5318–5320. doi:10.1021/jo9904922 |
| 40. | Hanessian, S.; Reddy, B. Tetrahedron 1999, 55, 3427–3443. doi:10.1016/S0040-4020(98)01152-1 |
| 41. | McDevitt, J. P.; Lansbury, P. T., Jr. J. Am. Chem. Soc. 1996, 118, 3818–3828. doi:10.1021/ja9525622 |
| 42. | Ohata, K.; Terashima, S. Tetrahedron Lett. 2006, 47, 2787–2791. doi:10.1016/j.tetlet.2006.02.058 |
| 43. | Ohata, K.; Terashima, S. Bioorg. Med. Chem. Lett. 2007, 17, 4070–4074. doi:10.1016/j.bmcl.2007.04.067 |
| 49. | Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 3664–3671. doi:10.1021/jo00369a020 |
| 50. | Strijtveen, B.; Kellogg, R. M. Tetrahedron 1987, 43, 5039–5054. doi:10.1016/S0040-4020(01)87682-1 |
| 51. | McFadden, J. M.; Frehywot, G. L.; Townsend, C. A. Org. Lett. 2002, 4, 3859–3862. doi:10.1021/ol026685k |
| 56. | Clayden, J. Organolithiums: Selectivity for Synthesis; Tetrahedron Organic Chemistry Series, Vol. 23; Pergamon: Oxford, U.K., 2002. |
| 64. | Kaiser, B.; Hoppe, D. Angew. Chem., Int. Ed. Engl. 1995, 34, 323–325. doi:10.1002/anie.199503231 |
| 65. | Hoppe, D.; Paetow, M.; Hintze, F. Angew. Chem., Int. Ed. Engl. 1993, 32, 394–396. doi:10.1002/anie.199303941 |
| 62. | Aggarwal, V. K. Angew. Chem., Int. Ed. Engl. 1994, 33, 175–177. doi:10.1002/anie.199401751 |
| 63. | Wiberg, K. B.; Castejon, H. J. Am. Chem. Soc. 1994, 116, 10489–10497. doi:10.1021/ja00102a016 |
| 59. | Ruhland, T.; Dress, R. K.; Hoffmann, R. W. Angew. Chem., Int. Ed. Engl. 1993, 32, 1467–1468. doi:10.1002/anie.199314671 |
| 60. | Hoffmann, R. W.; Dress, R. K.; Ruhland, T.; Wenzel, A. Chem. Ber. 1995, 128, 861–870. doi:10.1002/cber.19951280903 |
| 61. | Reich, H. J.; Dykstra, R. R. Angew. Chem., Int. Ed. Engl. 1993, 32, 1469–1470. doi:10.1002/anie.199314691 |
| 57. | McDougal, P. G.; Condon, B. D.; Laffosse, M. D., Jr.; Lauro, A. M.; VanDerveer, D. Tetrahedron Lett. 1988, 29, 2547–2550. doi:10.1016/S0040-4039(00)86108-0 |
| 58. | Beak, P.; Becker, P. D. J. Org. Chem. 1982, 47, 3855–3861. doi:10.1021/jo00141a010 |
| 56. | Clayden, J. Organolithiums: Selectivity for Synthesis; Tetrahedron Organic Chemistry Series, Vol. 23; Pergamon: Oxford, U.K., 2002. |
| 68. | Marr, F.; Fröhlich, R.; Hoppe, D. Org. Lett. 1999, 1, 2081–2083. doi:10.1021/ol991134o |
| 69. | Marr, F.; Hoppe, D. Org. Lett. 2002, 4, 4217–4220. doi:10.1021/ol0266828 |
| 70. | Marr, F.; Fröhlich, R.; Wibbeling, B.; Diedrich, C.; Hoppe, D. Eur. J. Org. Chem. 2002, 2970–2988. doi:10.1002/1099-0690(200209)2002:17<2970::AID-EJOC2970>3.0.CO;2-J |
| 66. | Hoppe, D.; Kaiser, B.; Stratmann, O.; Fröhlich, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 2784–2786. doi:10.1002/anie.199727841 |
| 67. | Stratmann, O.; Kaiser, B.; Fröhlich, R.; Meyer, O.; Hoppe, D. Chem.–Eur. J. 2001, 7, 423–435. doi:10.1002/1521-3765(20010119)7:2<423::AID-CHEM423>3.0.CO;2-Y |
| 1. | 2009 Top 200 Branded Drugs by Retail Dollars. Drug Topics, 2010; http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard//drugtopics/252010/674961/article.pdf. |
| 3. | Toru, T.; Bolm, C., Eds. Organosulfur Chemistry in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2008. |
| 16. | Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. J. Org. Chem. 2006, 71, 1692–1695. doi:10.1021/jo051632c |
| 17. | Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. Org. Lett. 2006, 8, 2855–2858. doi:10.1021/ol060993r |
| 76. | Clayden, J.; Dufour, J.; Grainger, D. M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129, 7488–7489. doi:10.1021/ja071523a |
| 9. | Procter, D. J. J. Chem. Soc., Perkin Trans. 1 2001, 335–354. doi:10.1039/b002081h |
| 18. | Bednarski, P. J.; Nelson, S. D. J. Med. Chem. 1989, 32, 203–213. doi:10.1021/jm00121a037 |
| 19. | Kitagawa, I.; Ueda, Y.; Kawasaki, T.; Mosettig, E. J. Org. Chem. 1963, 28, 2228–2232. doi:10.1021/jo01044a018 |
| 20. | Komeno, T.; Kishi, M. Tetrahedron 1971, 27, 1517–1526. doi:10.1016/S0040-4020(01)98017-2 |
| 21. | Caine, D.; Crews, E.; Salvino, J. M. Tetrahedron Lett. 1983, 24, 2083–2086. doi:10.1016/S0040-4039(00)81850-X |
| 22. | Tatsuta, K.; Suzuki, Y.; Toriumi, T.; Furuya, Y.; Hosokawa, S. Tetrahedron Lett. 2007, 48, 8018–8021. doi:10.1016/j.tetlet.2007.09.039 |
| 23. | López, I.; Rodríguez, S.; Izquierdo, J.; González, F. V. J. Org. Chem. 2007, 72, 6614–6617. doi:10.1021/jo0709955 |
| 1. | 2009 Top 200 Branded Drugs by Retail Dollars. Drug Topics, 2010; http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard//drugtopics/252010/674961/article.pdf. |
| 15. | Avenoza, A.; Busto, J. H.; Corzana, F.; Jiménez-Osés, G.; Peregrina, J. M. Chem. Commun. 2004, 980–981. doi:10.1039/b400282b |
| 16. | Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. J. Org. Chem. 2006, 71, 1692–1695. doi:10.1021/jo051632c |
| 17. | Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. Org. Lett. 2006, 8, 2855–2858. doi:10.1021/ol060993r |
| 76. | Clayden, J.; Dufour, J.; Grainger, D. M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129, 7488–7489. doi:10.1021/ja071523a |
| 77. | Clayden, J.; Hennecke, U. Org. Lett. 2008, 10, 3567–3570. doi:10.1021/ol801332n |
| 78. | Bach, R.; Clayden, J.; Hennecke, U. Synlett 2009, 421–424. doi:10.1055/s-0028-1087543 |
| 79. | Clayden, J.; Donnard, M.; Lefranc, J.; Minassi, A.; Tetlow, D. J. J. Am. Chem. Soc. 2010, 132, 6624–6625. doi:10.1021/ja1007992 |
| 80. | Tetlow, D. J.; Hennecke, U.; Raftery, J.; Waring, M. J.; Clarke, D. S.; Clayden, J. Org. Lett. 2010, 12, 5442–5445. doi:10.1021/ol102155h |
| 2. | Cremlyn, R. J. An Introduction to Organosulfur Chemistry; John Wiley & Sons Ltd.: Chichester, U.K., 1996. |
| 3. | Toru, T.; Bolm, C., Eds. Organosulfur Chemistry in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2008. |
| 4. | Fernández, I.; Khiar, N. Chem. Rev. 2003, 103, 3651–3706. doi:10.1021/cr990372u |
| 5. | Pellissier, H. Tetrahedron 2006, 62, 5559–5601. doi:10.1016/j.tet.2006.03.093 |
| 6. | Mellah, M.; Voituriez, A.; Schulz, E. Chem. Rev. 2007, 107, 5133–5205. doi:10.1021/cr068440h |
| 7. | Carreño, M. C.; Hernández-Torres, G.; Ribagorda, M.; Urbano, A. Chem. Commun. 2009, 6129–6144. doi:10.1039/B908043K |
| 8. | Wojaczyńska, E.; Wojaczyński, J. Chem. Rev. 2010, 110, 4303–4356. doi:10.1021/cr900147h |
| 16. | Avenoza, A.; Busto, J. H.; Jiménez-Osés, G.; Peregrina, J. M. J. Org. Chem. 2006, 71, 1692–1695. doi:10.1021/jo051632c |
| 81. | Clayden, J.; Farnaby, W.; Grainger, D. M.; Hennecke, U.; Mancinelli, M.; Tetlow, D. J.; Hillier, I. H.; Vincent, M. A. J. Am. Chem. Soc. 2009, 131, 3410–3411. doi:10.1021/ja808959e |
| 82. | Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-Ondozabal, H.; Clayden, J. Org. Lett. 2010, 12, 2222–2225. doi:10.1021/ol100627c |
| 9. | Procter, D. J. J. Chem. Soc., Perkin Trans. 1 2001, 335–354. doi:10.1039/b002081h |
| 13. | Effenberger, F.; Gaupp, S. Tetrahedron: Asymmetry 1999, 10, 1765–1775. doi:10.1016/S0957-4166(99)00154-8 |
| 75. | MacLellan, P.; Clayden, J. Chem. Commun. 2011, 47, 3395–3397. doi:10.1039/C0CC04912C |
| 14. | Monde, K.; Taniguchi, T.; Miura, N.; Nishimura, S.-I.; Harada, N.; Dukor, R. K.; Nafie, L. A. Tetrahedron Lett. 2003, 44, 6017–6020. doi:10.1016/S0040-4039(03)01513-2 |
| 76. | Clayden, J.; Dufour, J.; Grainger, D. M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129, 7488–7489. doi:10.1021/ja071523a |
| 10. | Shibasaki, M.; Kanai, M. Chem. Rev. 2008, 108, 2853–2873. doi:10.1021/cr078340r |
| 71. | Sonawane, R. P.; Fröhlich, R.; Hoppe, D. Chem. Commun. 2006, 3101–3103. doi:10.1039/b604029b |
| 72. | Sonawane, R. P.; Mück-Lichtenfeld, C.; Fröhlich, R.; Bergander, K.; Hoppe, D. Chem.–Eur. J. 2007, 13, 6419–6429. doi:10.1002/chem.200601853 |
| 9. | Procter, D. J. J. Chem. Soc., Perkin Trans. 1 2001, 335–354. doi:10.1039/b002081h |
| 12. | Weaver, J. D.; Morris, D. K.; Tunge, J. A. Synlett 2010, 470–474. doi:10.1055/s-0029-1219186 |
| 73. | Sonawane, R. P.; Fröhlich, R.; Hoppe, D. Adv. Synth. Catal. 2006, 348, 1847–1854. doi:10.1002/adsc.200606177 |
| 74. | Lange, H.; Bergander, K.; Frölich, R.; Kehr, S.; Nakamura, S.; Shibata, N.; Toru, T.; Hoppe, D. Chem.–Asian J. 2008, 3, 88–101. doi:10.1002/asia.200700262 |
| 18. | Bednarski, P. J.; Nelson, S. D. J. Med. Chem. 1989, 32, 203–213. doi:10.1021/jm00121a037 |
| 18. | Bednarski, P. J.; Nelson, S. D. J. Med. Chem. 1989, 32, 203–213. doi:10.1021/jm00121a037 |
| 19. | Kitagawa, I.; Ueda, Y.; Kawasaki, T.; Mosettig, E. J. Org. Chem. 1963, 28, 2228–2232. doi:10.1021/jo01044a018 |
| 20. | Komeno, T.; Kishi, M. Tetrahedron 1971, 27, 1517–1526. doi:10.1016/S0040-4020(01)98017-2 |
| 21. | Caine, D.; Crews, E.; Salvino, J. M. Tetrahedron Lett. 1983, 24, 2083–2086. doi:10.1016/S0040-4039(00)81850-X |
| 22. | Tatsuta, K.; Suzuki, Y.; Toriumi, T.; Furuya, Y.; Hosokawa, S. Tetrahedron Lett. 2007, 48, 8018–8021. doi:10.1016/j.tetlet.2007.09.039 |
| 18. | Bednarski, P. J.; Nelson, S. D. J. Med. Chem. 1989, 32, 203–213. doi:10.1021/jm00121a037 |
| 19. | Kitagawa, I.; Ueda, Y.; Kawasaki, T.; Mosettig, E. J. Org. Chem. 1963, 28, 2228–2232. doi:10.1021/jo01044a018 |
| 20. | Komeno, T.; Kishi, M. Tetrahedron 1971, 27, 1517–1526. doi:10.1016/S0040-4020(01)98017-2 |
| 79. | Clayden, J.; Donnard, M.; Lefranc, J.; Minassi, A.; Tetlow, D. J. J. Am. Chem. Soc. 2010, 132, 6624–6625. doi:10.1021/ja1007992 |
| 81. | Clayden, J.; Farnaby, W.; Grainger, D. M.; Hennecke, U.; Mancinelli, M.; Tetlow, D. J.; Hillier, I. H.; Vincent, M. A. J. Am. Chem. Soc. 2009, 131, 3410–3411. doi:10.1021/ja808959e |
| 82. | Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-Ondozabal, H.; Clayden, J. Org. Lett. 2010, 12, 2222–2225. doi:10.1021/ol100627c |
| 75. | MacLellan, P.; Clayden, J. Chem. Commun. 2011, 47, 3395–3397. doi:10.1039/C0CC04912C |
| 32. | Porco, J. A., Jr.; Su, S.; Lei, X.; Bardhan, S.; Rychnovsky, S. D. Angew. Chem., Int. Ed. 2006, 45, 5790–5792. doi:10.1002/anie.200602854 |
| 29. | La Clair, J. J. Angew. Chem., Int. Ed. 2006, 45, 2769–2773. doi:10.1002/anie.200504033 |
| 25. | Kumara Swamy, K. C.; Bhuvan Kumar, N. N.; Balaraman, E.; Pavan Kumar, K. V. P. Chem. Rev. 2009, 109, 2551–2651. doi:10.1021/cr800278z |
| 26. | Walker, K. A. M. Tetrahedron Lett. 1977, 18, 4475–4478. doi:10.1016/S0040-4039(01)83541-3 |
| 27. | Kotsuki, H.; Matsumoto, K.; Nishizawa, H. Tetrahedron Lett. 1991, 32, 4155–4158. doi:10.1016/S0040-4039(00)79890-X |
| 28. | Shi, Y.-J.; Hughes, D. L.; McNamara, J. M. Tetrahedron Lett. 2003, 44, 3609–3611. doi:10.1016/S0040-4039(03)00728-7 |
| 22. | Tatsuta, K.; Suzuki, Y.; Toriumi, T.; Furuya, Y.; Hosokawa, S. Tetrahedron Lett. 2007, 48, 8018–8021. doi:10.1016/j.tetlet.2007.09.039 |
| 24. | Mitsunobu, O. Synthesis 1981, 1–28. doi:10.1055/s-1981-29317 |
| 25. | Kumara Swamy, K. C.; Bhuvan Kumar, N. N.; Balaraman, E.; Pavan Kumar, K. V. P. Chem. Rev. 2009, 109, 2551–2651. doi:10.1021/cr800278z |
© 2011 Clayden and MacLellan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)