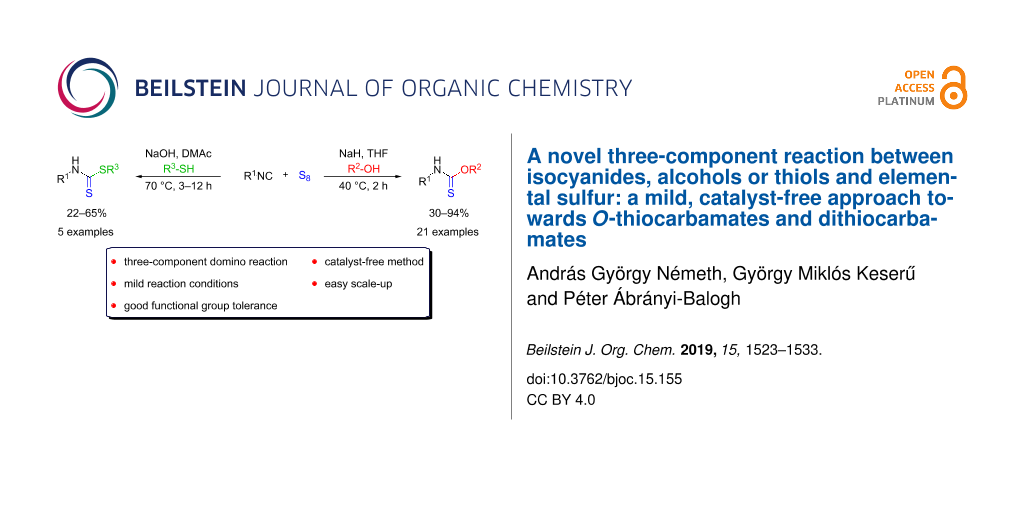
Abstract
A new multicomponent reaction has been developed between isocyanides, sulfur and alcohols or thiols under mild reaction conditions to afford O-thiocarbamates and dithiocarbamates in moderate to good yields. The one-pot reaction cascade involves the formation of an isothiocyanate intermediate, thus a catalyst-free synthesis of isothiocyanates, as valuable building blocks from isocyanides and sulfur is proposed, as well. The synthetic procedure suits the demand of a modern organic chemist, as it tolerates a wide range of functional groups, it is atom economic and easily scalable.

Graphical Abstract
Introduction
O-Thiocarbamates belong to a class of important biologically active molecules, used mainly as fungicides [1-3] in agricultural and pharmaceutical fields. In particular, recently antitumor [4], anesthetic [5] and enzyme inhibitory effects were discovered, including HIV-1 reverse transcriptase inhibition activity [6-11]. Moreover, their utilization as highly regio- and stereoselective organocatalysts in specific types of chemical tranformations [12-17] was introduced, as well. More recently, O-thiocarbamates have been used as H2S donors in biological systems [18] and as intermediates in pharmaceutically significant organic syntheses [19,20]. The dithiocarbamate structural moiety can be found in biologically active molecules widely applied as fungicides, herbicides, pesticides [21-25] and in some cases as enzyme inhibitors [26] or antitumor agents [27]. These species are also used as valuable synthetic intermediates [28] and chemosensors for mercury and silver [29,30].
The general methods for the synthesis of O-thiocarbamates and dithiocarbamates traditionally rely on substitution reactions of the corresponding halogenated precursors, including thiophosgene [31-33], thiocarbamoyl chlorides [34-37], chlorothionoformates or chlorodithioformates [38-41] providing the appropriate thiocarbamate analogues in good yields (Scheme 1). However, these methods suffer from the formation of toxic, malodorous and/or extremely corrosive byproducts generated by the elimination of the halogen atoms. One should note that the application of these halogenated thiocarbonic acid derivatives might be dangerous and require thorough precaution. Considering dithiocarbamates, a number of methods are based on the reaction of amines and the readily available, but toxic and volatile carbon disulfide [42-45]. Greener methods for the synthesis of thiocarbamates and dithiocarbamates have been developed such as the addition of the amine component to potassium thiocyanate [46,47] or isothiocyanate [48-52] showing better atom economy. Nonetheless, only a few examples can be found in the literature starting from thiocyanates, and regarding the isothiocyanates the preparation of the reagent is required as an additional reaction step before.
![[1860-5397-15-155-i1]](/bjoc/content/inline/1860-5397-15-155-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to O-thiocarbamates and dithiocarbamates.
Scheme 1: Synthetic routes to O-thiocarbamates and dithiocarbamates.
The synthesis of isothiocyanates generally relies on the reaction between thiophosgene and amines, thus involves the use of a highly toxic reagent with narrow functional group compatibility [53-56]. Various thiocarbonyl transfer reagents have been developed in the last decades to overcome these drawbacks, such as thiocarbonyl-diimidazole or di-2-pyridyl thionocarbonate [57,58]. Decomposition of dithiocarbamate salts or thiocarbamates with various reagents offers a good alternative [59-62] as well, however, this approach first requires the synthesis of the appropriate precursor. Nitrile oxides react with thiourea to afford isothiocyanate and harmless urea [63-65], but one should note that the instability of the nitrile oxides leads to many byproducts, turning this approach less attractive. The synthesis of isothiocyanates starting from isonitriles involves sulfur-containing reagents such as thallium thiocarboxylates or thiols in the presence of radical initiators [66-68]. All the previously reported methods for the synthesis of O-thiocarbamates, dithiocarbamates and isothiocyanates start from toxic and/or unstable reagents, generate halogen waste or have narrow functional group tolerance. The bench-stable, environmentally benign, cheap and nontoxic elemental sulfur offers an alternative starting material to integrate sulfur into the product [69]. For a single molecule, Tan and co-workers showed isothiocyanate might be formed from an isocyanide by elemental sulfur in the presence of a base in low yield [70]. In certain cases, sulfur can be trapped by in situ generated carbenes to afford O-thiocarbamates [71,72]. Thioureas and S-thiocarbamates are also accessible through multicomponent reactions starting from isocyanides and sulfur [73-75]. The cumbersome synthesis of isothiocyanates from isocyanides and sulfur [76] can be enhanced using various catalysts such as selenium, molybdenum, copper, rhodium [77-82] or tellurium [83] providing the isothiocyanates in excellent yields. These approaches on the other hand suffer from the use of heavy metals, toxic chalcogens and/or long reaction times. More recently, a novel three-component method has been published starting from readily available amines and sodium bromodifluoracetate [84], but this synthetic route provides halogenated waste, as well. As a continuation of our interest in the development of multicomponent reactions [85-87] and reactions involving sulfur [88], herein, we describe a novel synthesis of O-thiocarbamates and dithiocarbamates via a three-component reaction of elemental sulfur, isocyanides and alcohols or thiols (Scheme 1). Moreover, during the investigation of the reaction mechanism, we have identified and improved a catalyst-free method for the preparation of isothiocyanates.
Results and Discussion
The model reaction of 2,6-dimethylphenyl isocyanide (1a), elemental sulfur (S8) and methanol (2a) was employed to screen for the optimal reaction conditions (Table 1). The reactions were followed by TLC and HPLC–MS. Based on preliminary experiments in our laboratory, the reaction was performed in tetrahydrofuran (THF) at 40 °C for 1 h using a 1.5 equiv excess of sodium hydride as the base, S8 and the alcohol component (Table 1, entry 1) resulting in the desired thiocarbamate 3a in 58% yield. During the purification procedure, the change of the stationary phase for the column chromatography from aluminium oxide to silica, resulted in an increased yield of 72% (Table 1, entry 2). After the optimization of the purification process, the excess of the reagents and the role of the base were studied. Increasing the molar excess of all reagents to 2 equiv provided 3a in 91% yield (Table 1, entry 3), however, the yield was decreased by the use of a larger excess (Table 1, entry 4). Reducing the amount of the reagents was not helpful (Table 1, entry 5), and one can see that 2.5 equiv of sulfur and methanol did not increase the yields either (Table 1, entry 6). A longer reaction time (2 h), however, enhanced the product yield from 72% (Table 1, entry 2) to 84% (Table 1, entry 7). Thus we have combined this reaction time with the elevated molar excess of the reagents resulting in thiocarbamate 3a in 94% yield (Table 1, entry 8). The advantageous effect of heating was supported by the decreased yield (72%) obtained when performing the reaction at ambient temperature (Table 1, entry 9). Next, the effect of different solvents was investigated, showing that acetonitrile (MeCN) and 2-methyltetrahydrofuran (MeTHF) proved to be suitable alternatives to THF (Table 1, entries 13 and 15) that might be advantageous considering the wide application of these solvents in industrial production [89,90]. On the contrary, the use of dioxane, methyl tert-butyl ether (MTBE), toluene or dichloromethane (DCM) was unfavorable, providing the thiocarbamate in 67%, 29%, 12% and 30% yields, respectively (Table 1, entries 10, 11, 12, and 14). Notably, in the lack of inert atmosphere, the yield decreased to 72%, and unidentified byproducts were detected that might be explained with the decomposition or side reactions of the isocyanide component under air. In the case of using other bases, such as caesium carbonate (Cs2CO3), diisopropylethylamine (DIPEA) or sodium ethoxide (NaOEt), only the isothiocyanate intermediate of the reaction was isolated (Table 1, entries 17, 18, and 20). However, using diazabicycloundecene (DBU) as the base allowed the formation of the desired product, but only in 39% yield (Table 1, entry 19). Without any basic additive, no reaction occurred (Table 1, entry 21) and the starting compounds were recovered.
Table 1: Optimization of the reaction conditions for the synthesis of O-thiocarbamates.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-155-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Base | Time [h] |
Molar excess
2a/S8/base |
Yield [%]a,b |
| 1 | NaH | THF | 1 | 1.5:1.5:1.5 | 58c |
| 2 | NaH | THF | 1 | 1.5:1.5:1.5 | 72 |
| 3 | NaH | THF | 1 | 2:2:2 | 91 |
| 4 | NaH | THF | 1 | 2.5:2.5:2.5 | 80d |
| 5 | NaH | THF | 1 | 1.5:1.5:2 | 61 |
| 6 | NaH | THF | 1 | 2.5:2.5:2 | 88 |
| 7 | NaH | THF | 2 | 1.5:1.5:1.5 | 84 |
| 8 | NaH | THF | 2 | 2:2:2 | 94d |
| 9 | NaH | THF | 2 | 2:2:2 | 72e |
| 10 | NaH | dioxane | 2 | 2:2:2 | 67 |
| 11 | NaH | MTBE | 2 | 2:2:2 | 29 |
| 12 | NaH | toluene | 2 | 2:2:2 | 12d |
| 13 | NaH | MeCN | 2 | 2:2:2 | 92d |
| 14 | NaH | DCM | 2 | 2:2:2 | 30 |
| 15 | NaH | MeTHF | 2 | 2:2:2 | 87d |
| 16 | NaH | THF | 2 | 2:2:2 | 72f |
| 17 | Cs2CO3 | THF | 2 | 2:2:2 | 0 (26)g |
| 18 | DIPEA | THF | 2 | 2:2:2 | 0 (30)g |
| 19 | DBU | THF | 2 | 2:2:2 | 39 |
| 20 | NaOEt | THF | 2 | 2:2:2 | 0 (53)g |
| 21 | – | THF | 2 | 2:2:2 | n.r. |
aReaction conditions: 1a (1 mmol), S8, 2a, base, solvent (3 mL), time, under argon atmosphere at 40 °C. bIsolated yields. cFlash column chromatography performed on aluminium oxide as stationary phase. dAverage of two runs. eRoom temperature. fLack of inert atmosphere. gYield of isothiocyanate intermediate. n.r. = no reaction.
With the optimized reaction conditions in hand, the generality and substrate scope of the reaction using different isocyanides were investigated (Scheme 2). Considering the yield of various aromatic isonitriles, no significant difference was noticed between electron-withdrawing and electron-donating substituents (3c and 3d, respectively). The ortho-iodo-substituted 3b was obtained in an excellent yield (94%) demonstrating that no steric hindrance occurs during the reaction. Taking into account aliphatic derivatives, O-methyl cyclohexylcarbamothioate (3e) was formed in good yield (82%), however, O-methyl tert-butylcarbamothioate (3f), partly due to the volatile nature of the isocyanide and the thiocarbamate as well, was obtained only in 54% yield.
![[1860-5397-15-155-i2]](/bjoc/content/inline/1860-5397-15-155-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of isocyanides. aReaction conditions: 1 (1 mmol), S8 (2 mmol), 2a (2mmol), NaH (2 mmol), THF (3 mL), 2 h, under argon atmosphere at 40 °C. Yields refer to isolated yields.
Scheme 2: Substrate scope of isocyanides. aReaction conditions: 1 (1 mmol), S8 (2 mmol), 2a (2mmol), NaH (2 m...
In order to further explore the scope of the reaction, different alcohols were tested (Scheme 3). Regarding the compounds 3g–i it can be noticed that the yield drops from the primary alcohol towards the tertiary one (85%, 52% and 45%, respectively) that might be attributed either to steric hindrance or the growing instability of the conjugate base of the secondary and tertiary alcohol, respectively. The present method provided the allylic derivative 3j in 72% yield, however, applying ethylene glycol resulted in 3k in 34% yield only. In the latter case no dimeric product but several unidentified side products were detected by TLC and HPLC–MS. Although the full conversion of the isocyanide to the isothiocyanate intermediate was observed by TLC and HPLC–MS, phenol proved to be unreactive under the standard reaction conditions. Consequently, we have turned our attention to different benzylic alcohols that could be utilized to further examine the functional group tolerance of the reaction. Notably, chlorine, bromine and iodine substituents were compatible with the transformation, providing 3m, 3n, 3r and 3s in 81–89% yield. The nitrile derivative 3p was obtained successfully in 62% yield, showing the reactivity difference between the cyano and the isocyano groups. Interestingly, methoxy-substituted thiocarbamates 3o and 3t were obtained in lower 53% and 56% yield, respectively, that decreased further to 34% in the case of the trimethoxy-substituted product 3u. As the methoxy group is inert under the standard reaction conditions, one might assume that the electron-donating ability reduces the stability of the in situ-generated anion, just as in the case of the secondary and tertiary alcohols. The nitro derivative 3q was isolated in 30% yield along with multiple byproducts detected that may be due to possible reductive side-reactions caused by sulfur [91]. To the best of our knowledge, out of the 21 synthesized O-thiocarbamate derivatives (Scheme 2 and Scheme 3), 18 compounds are new, and only 3d, 3e and 3f are known in the literature [92-94]. The new derivatives have been characterized by 1H NMR, 13C NMR, HRMS and melting point. However, the thiocarbamate 3c happened to be unstable and started to decompose after work-up. Therefore, an HPLC–MS spectrum of the reaction mixture after completion of the reaction and an HRMS of the crude product are attached in Supporting Information File 1.
![[1860-5397-15-155-i3]](/bjoc/content/inline/1860-5397-15-155-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of alcohols. Reaction conditions: 1a (1 mmol), S8 (2 mmol), 2 (2mmol), NaH (2 mmol), THF (3 mL), 2 h, under argon atmosphere at 40 °C. Yields refer to isolated yields.
Scheme 3: Substrate scope of alcohols. Reaction conditions: 1a (1 mmol), S8 (2 mmol), 2 (2mmol), NaH (2 mmol)...
Several experiments were performed to discover the reaction conditions that enable the synthesis of O-aryl thiocarbamates. Initially, DBU was used in refluxing dioxane, as this base was shown to provide the appropriate aliphatic O-thiocarbamate. However, in this case only the isothiocyanate intermediate was obtained. Then, trimethylamine was applied in refluxing MeCN [95] or sodium hydroxide in dimethyl sulfoxide (DMSO) at 70 °C. In both cases only the isothiocyanate intermediate and phenol were observed by HPLC–MS but no formation of the desired product.
Then we turned to the synthesis of dithiocarbamate 5a under the standard reaction conditions, but only the isothiocyanate intermediate was obtained (Table 2, entry 1). Thus a new optimization of the reaction conditions became necessary. Similarly to the previous methodology, the base, the solvent, the temperature and the molar excess of the reagents were changed using the model reaction of 2,6-dimethylphenyl isocyanide (1a), sulfur and benzyl mercaptan (4a, Table 2). Initially, NaOH was used in DMSO as shown in entry 2 (Table 2), providing the desired dithiocarbamate 5a in only 22% yield [96]. In order to improve the yield, firstly the temperature was elevated to 70 °C and 100 °C to afford 5a in 38% and 34% yield, respectively (Table 2, entries 3 and 4). Larger excesses of the reagents and the base cut back the yield to 23% (Table 2, entry 5). Using NaH instead of NaOH did not improve the yield of the reaction (Table 2, entry 6), nor did the use of Cs2CO3 (Table 2, entry 7). However, using N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMA) as the solvent provided 5a in 43% and 45% yield, respectively (Table 2, entries 8 and 9), while on the other hand, N-methyl-2-pyrrolidone (NMP) was disadvantageous for the reaction (Table 2, entry 10). It is well-known that at elevated temperatures sulfur may act as an oxidant, which in this case may have compromised the reaction [97-99]. Therefore, the molar excess of sulfur was decreased, providing a positive effect on the reaction affording 5a in 59% yield.
Table 2: Optimization of the reaction conditions for the synthesis of dithiocarbamates.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-155-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Base | Temp. [°C] |
Molar excess
4a/S8/base |
Yield [%]a,b |
| 1 | NaH | THF | 40 | 2:2:2 | 0c |
| 2 | NaOH | DMSO | 40 | 2:2:2 | 22 |
| 3 | NaOH | DMSO | 70 | 2:2:2 | 38 |
| 4 | NaOH | DMSO | 100 | 2:2:2 | 34 |
| 5 | NaOH | DMSO | 70 | 3:3:3 | 23 |
| 6 | NaH | DMSO | 70 | 2:2:2 | 36 |
| 7 | Cs2CO3 | DMSO | 70 | 2:2:2 | 14 |
| 8 | NaOH | DMF | 70 | 2:2:2 | 43 |
| 9 | NaOH | DMA | 70 | 2:2:2 | 45 |
| 10 | NaOH | NMP | 70 | 2:2:2 | 34 |
| 11 | NaOH | DMA | 70 | 2:1.2:2 | 59 |
aReaction conditions: 1a (1 mmol), S8, 4a, base, solvent (3 mL), temperature, 3 h under argon atmosphere. bIsolated yield, unless noted otherwise. cIsothiocyanate intermediate detected by HPLC–MS.
With the optimized reaction conditions in hand, a number of dithiocarbamate derivatives were synthetized (Scheme 4). One might notice the same trend as in the case of thiocarbamates 3g–i, in particular, the primary mercaptans gave the highest yields (5a and 5b), while in the case of secondary (5c and 5d) and tertiary thiols (5e) the products were isolated in lower yields. Although a full conversion of the isocyanide to the isothiocyanate intermediate was observed by TLC and HPLC–MS, thiophenol, likewise to phenol was unreactive under the standard reaction conditions. The generally lower yields, harsher reaction conditions and stronger negative effect of electron-donating groups might be explained with the softer nucleophilicity of the thiols compared to the alcohols [100]. All five dithiocarbamate derivatives synthesized are new and were characterized by 1H NMR, 13C NMR, HRMS and melting point.
![[1860-5397-15-155-i4]](/bjoc/content/inline/1860-5397-15-155-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Substrate scope of thiols. Reaction conditions: 1a (1 mmol), S8 (1.2 mmol), 4 (2 mmol), NaOH (2 mmol), DMAc (3 mL), time, under argon atmosphere at 70 °C. Yields refer to isolated yields.
Scheme 4: Substrate scope of thiols. Reaction conditions: 1a (1 mmol), S8 (1.2 mmol), 4 (2 mmol), NaOH (2 mmo...
After the successful application of various nucleophiles, it was our intention to investigate the scalability of the procedure. Thus, a twenty-fold scale-up of the reaction between the isocyanide 1a, sulfur and methanol (2a) was performed (Scheme 5). In this case, the experimental conditions were necessarily slightly different, as in larger quantities the reaction between the alcohol and NaH needs to be kept under control. Therefore, the mixture of 1a, methanol and THF was added dropwise to a mixture of NaH and sulfur in THF under ice-cooling. After the work-up, no chromatography was necessary and the crude product was purified by recrystallization from hexane/ethyl acetate. The three collected crops of crystals provided the thiocarbamate 3a in a total of 74% yield.
![[1860-5397-15-155-i5]](/bjoc/content/inline/1860-5397-15-155-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
We have envisaged that our multicomponent reaction could be compatible with subsequent one-pot transformations. In order to demonstrate this capability, we performed the multicomponent domino annulation between isocyanide 1a, sulfur and methyl anthranilate (6) in DMSO in the presence of NaOH at 85 °C that provided 3-(2,6-dimethylphenyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (7), a new quinazolinone derivative in 40% yield (Scheme 6). Notably, these heterocycles are known for their use as antitumor [101], anticonvulsant [102] or epidermal growth factor receptor tyrosine kinase inhibitory agents [103], JNK inhibitors [104] or 5-HT3 antagonists [105]. Earlier, a one-pot synthesis of an analogous compound was accomplished by Sayahi et al. starting from isothiocyanates in the presence of CuBr [106].
![[1860-5397-15-155-i6]](/bjoc/content/inline/1860-5397-15-155-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Multicomponent domino synthesis of quinazolinone 7.
Scheme 6: Multicomponent domino synthesis of quinazolinone 7.
As aforementioned, in some cases only isothiocyanate 8 was detected and/or isolated. Thus, in order to gain mechanistic insights on the generation of 3a, we performed a series of experiments (Scheme 7). As shown in Table 1, isocyanide 1a and sulfur did not react in the absence of a base (Table 1, entry 21). Therefore, the reaction was performed in the presence of NaH under the standard reaction conditions, providing 8 in 85% yield (Scheme 7, reaction 1). Notably, the analogous reaction reported by Tan and co-workers using potassium tert-butylate in t-BuOH/dioxane at 55 °C for 6 h resulted in the desired isothiocyanate in only 34% yield [69]. In the next step, we investigated the acylation of the alcohol component by the isothiocyanate (Scheme 7, reaction 2). In the absence of a base, no reaction occurred, and consequenctly the base was necessary for both steps of the thiocarbamate formation. This also explains why two equivalents of base were required. Sodium sulfide as the base provided only traces of 3a suggesting that the activation of sulfur by NaH produces rather a polysulfide anion instead of sodium sulfide [74,107,108] (Scheme 7, reaction 3). It caught our attention that only isothiocyanate was generated in the presence of NaOEt (Table 1, entry 20). We suspected that THF might not be the best solvent for this base, hence the reaction was performed in MeCN providing exclusively 8 both at 40 °C and 70 °C, and the same result was obtained when a solvent mixture of ethanol and THF was used (Scheme 7, reaction 4).
![[1860-5397-15-155-i7]](/bjoc/content/inline/1860-5397-15-155-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on the above experimental results and previous reports [74,107,108], a possible reaction mechanism has been proposed (Scheme 8). Initially, the reaction of elemental sulfur and NaH generates a polysulfide anion that is able to attack the carbenoid carbon atom of isocyanide 1a yielding the isothiocyanate intermediate 8. Then, the present nucleophile (NuH, alcohol or thiol) undergoes a nucleophilic addition on 8 providing thiocarbamate 3a.
![[1860-5397-15-155-i8]](/bjoc/content/inline/1860-5397-15-155-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have developed an efficient, convenient and scalable multicomponent method for the synthesis of O-thiocarbamates and dithiocarbamates under mild reaction conditions. This approach includes an improved catalyst-free synthesis of isothiocyanates from elemental sulfur and isocyanides, and shows good functional group tolerance to halogen, olefin and nitrile groups among others. Moreover, this multicomponent reaction is suitable for a one-pot cascade annulation providing a thioxo dihydroquinazolinone derivative in a metal-free approach. Compared to other reported syntheses of thiocarbamates, this method is highlighted by its simplicity, atom economical nature and green operational method. Out of the 29 synthesized compounds, 18 new O-thiocarbamates, 5 new dithiocarbamates and 1 new thioxodihydroquinazolinone were characterized.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Len, C.; Postel, D.; Ronco, G.; Villa, P.; Goubert, C.; Jeufrault, E.; Mathon, B.; Simon, H. J. Agric. Food Chem. 1997, 45, 3–6. doi:10.1021/jf9607371
Return to citation in text: [1] -
Zhou, Y.; Wang, L.; Han, L.; Meng, F.; Yang, C. Carbohydr. Res. 2009, 344, 1289–1296. doi:10.1016/j.carres.2009.05.008
Return to citation in text: [1] -
Ryder, N. S.; Frank, I.; Dupont, M.-C. Antimicrob. Agents Chemother. 1986, 29, 858–860. doi:10.1128/aac.29.5.858
Return to citation in text: [1] -
Spallarossa, A.; Cesarini, S.; Schenone, S.; Ranise, A. Arch. Pharm. (Weinheim, Ger.) 2009, 342, 344–352. doi:10.1002/ardp.200800212
Return to citation in text: [1] -
Wood, T. F.; Gardner, J. H. J. Am. Chem. Soc. 1941, 63, 2741–2742. doi:10.1021/ja01855a068
Return to citation in text: [1] -
Lin, G.; Shieh, C.-T.; Ho, H.-C.; Chouhwang, J.-Y.; Lin, W.-Y.; Lu, C.-P. Biochemistry 1999, 38, 9971–9981. doi:10.1021/bi982775e
Return to citation in text: [1] -
Cesarini, S.; Spallarossa, A.; Ranise, A.; Fossa, P.; La Colla, P.; Sanna, G.; Collu, G.; Loddo, R. Bioorg. Med. Chem. 2008, 16, 4160–4172. doi:10.1016/j.bmc.2007.12.050
Return to citation in text: [1] -
Cesarini, S.; Spallarossa, A.; Ranise, A.; Bruno, O.; La Colla, P.; Secci, B.; Collu, G.; Loddo, R. Bioorg. Med. Chem. 2008, 16, 4173–4185. doi:10.1016/j.bmc.2007.12.046
Return to citation in text: [1] -
Spallarossa, A.; Cesarini, S.; Ranise, A.; Bruno, O.; Schenone, S.; La Colla, P.; Collu, G.; Sanna, G.; Secci, B.; Loddo, R. Eur. J. Med. Chem. 2009, 44, 1650–1663. doi:10.1016/j.ejmech.2008.09.024
Return to citation in text: [1] -
Cichero, E.; Cesarini, S.; Fossa, P.; Spallarossa, A.; Mosti, L. Eur. J. Med. Chem. 2009, 44, 2059–2070. doi:10.1016/j.ejmech.2008.10.014
Return to citation in text: [1] -
Spallarossa, A.; Cesarini, S.; Ranise, A.; Schenone, S.; Bruno, O.; Borassi, A.; La Colla, P.; Pezzullo, M.; Sanna, G.; Collu, G.; Secci, B.; Loddo, R. Eur. J. Med. Chem. 2009, 44, 2190–2201. doi:10.1016/j.ejmech.2008.10.032
Return to citation in text: [1] -
Zhou, L.; Tan, C. K.; Jiang, X.; Chen, F.; Yeung, Y.-Y. J. Am. Chem. Soc. 2010, 132, 15474–15476. doi:10.1021/ja1048972
Return to citation in text: [1] -
Tan, C. K.; Zhou, L.; Yeung, Y.-Y. Org. Lett. 2011, 13, 2738–2741. doi:10.1021/ol200840e
Return to citation in text: [1] -
Zhou, L.; Chen, J.; Tan, C. K.; Yeung, Y.-Y. J. Am. Chem. Soc. 2011, 133, 9164–9167. doi:10.1021/ja201627h
Return to citation in text: [1] -
Han, W.-Y.; Li, S.-W.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Chem. – Eur. J. 2013, 19, 5551–5556. doi:10.1002/chem.201300206
Return to citation in text: [1] -
Tan, C. K.; Er, J. C.; Yeung, Y.-Y. Tetrahedron Lett. 2014, 55, 1243–1246. doi:10.1016/j.tetlet.2014.01.009
Return to citation in text: [1] -
Tay, D. W.; Leung, G. Y. C.; Yeung, Y.-Y. Angew. Chem., Int. Ed. 2014, 53, 5161–5164. doi:10.1002/anie.201310136
Return to citation in text: [1] -
Steiger, A. K.; Pardue, S.; Kevil, C. G.; Pluth, M. D. J. Am. Chem. Soc. 2016, 138, 7256–7259. doi:10.1021/jacs.6b03780
Return to citation in text: [1] -
Yoshida, S.; Hayashi, Y.; Obitsu, K.; Nakamura, A.; Kikuchi, T.; Sawada, T.; Kimura, T.; Takahashi, T.; Mukuta, T. Org. Process Res. Dev. 2012, 16, 1818–1826. doi:10.1021/op300237b
Return to citation in text: [1] -
Yadav, J. S.; Goreti, R.; Pabbaraja, S.; Sridhar, B. Org. Lett. 2013, 15, 3782–3785. doi:10.1021/ol401760e
Return to citation in text: [1] -
Heyns, A. Y.; Carter, G. A.; Rothwell, K.; Wain, R. L. Ann. Appl. Biol. 1966, 57, 33–51. doi:10.1111/j.1744-7348.1966.tb06864.x
Return to citation in text: [1] -
Rafin, C.; Veignie, E.; Sancholle, M.; Postel, D.; Len, C.; Villa, P.; Ronco, G. J. Agric. Food Chem. 2000, 48, 5283–5287. doi:10.1021/jf0003698
Return to citation in text: [1] -
MacBean, C. The Pesticide Manual, 16th ed.; British Crop Production Council, 2012.
Return to citation in text: [1] -
Gupta, P. K. Herbicides and fungicides. In Biomarkers in Toxicology; Gupta, R. C., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp 409–431. doi:10.1016/b978-0-12-404630-6.00024-5
Return to citation in text: [1] -
Thind, T. S.; Hollomon, D. W. Pest Manage. Sci. 2018, 74, 1547–1551. doi:10.1002/ps.4844
Return to citation in text: [1] -
Carta, F.; Aggarwal, M.; Maresca, A.; Scozzafava, A.; McKenna, R.; Masini, E.; Supuran, C. T. J. Med. Chem. 2012, 55, 1721–1730. doi:10.1021/jm300031j
Return to citation in text: [1] -
Schwimmer, A.; Rubin, D. Method and compositions for inhibiting tumor cell metabolism. U.S. Patent US5,240,914, Aug 31, 1993.
Return to citation in text: [1] -
Boas, U.; Gertz, H.; Christensen, J. B.; Heegaard, P. M. H. Tetrahedron Lett. 2004, 45, 269–272. doi:10.1016/j.tetlet.2003.10.182
Return to citation in text: [1] -
Hatai, J.; Pal, S.; Jose, G. P.; Bandyopadhyay, S. Inorg. Chem. 2012, 51, 10129–10135. doi:10.1021/ic300530f
Return to citation in text: [1] -
Konieczna, D. D.; Blanrue, A.; Wilhelm, R. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 596–604. doi:10.5560/znb.2014-4014
Return to citation in text: [1] -
Seetharamsingh, B.; Ramesh, R.; Dange, S. S.; Khairnar, P. V.; Singhal, S.; Upadhyay, D.; Veeraraghavan, S.; Viswanadha, S.; Vakkalanka, S.; Reddy, D. S. ACS Med. Chem. Lett. 2015, 6, 1105–1110. doi:10.1021/acsmedchemlett.5b00213
Return to citation in text: [1] -
Selvakumar, N.; Kumar, G. S.; Malar Azhagan, A.; Govinda Rajulu, G.; Sharma, S.; Kumar, M. S.; Das, J.; Iqbal, J.; Trehan, S. Eur. J. Med. Chem. 2007, 42, 538–543. doi:10.1016/j.ejmech.2006.10.013
Return to citation in text: [1] -
Sasse, K. Justus Liebigs Ann. Chem. 1970, 735, 158–188. doi:10.1002/jlac.19707350120
Return to citation in text: [1] -
Barma, D. K.; Bandyopadhyay, A.; Capdevila, J. H.; Falck, J. R. Org. Lett. 2003, 5, 4755–4757. doi:10.1021/ol0354573
Return to citation in text: [1] -
Kojima, M.; Nakamura, Y.; Ishikawa, T.; Takeuchi, S. Tetrahedron Lett. 2006, 47, 6309–6314. doi:10.1016/j.tetlet.2006.05.142
Return to citation in text: [1] -
Iwakawa, T.; Tamura, H.; Murabayashi, A.; Hayase, Y. Chem. Pharm. Bull. 1991, 39, 1939–1943. doi:10.1248/cpb.39.1939
Return to citation in text: [1] -
Koketsu, M.; Otsuka, T.; Ishihara, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 443–448. doi:10.1080/10426500490262612
Return to citation in text: [1] -
Gromek, S. M.; deMayo, J. A.; Maxwell, A. T.; West, A. M.; Pavlik, C. M.; Zhao, Z.; Li, J.; Wiemer, A. J.; Zweifach, A.; Balunas, M. J. Bioorg. Med. Chem. 2016, 24, 5183–5196. doi:10.1016/j.bmc.2016.08.040
Return to citation in text: [1] -
Schrader, A. M.; Schroll, A. L.; Barany, G. J. Org. Chem. 2011, 76, 7882–7892. doi:10.1021/jo201329n
Return to citation in text: [1] -
Boeckman, R. K.; Ge, P.; Reed, J. E. Org. Lett. 2001, 3, 3647–3650. doi:10.1021/ol0165645
Return to citation in text: [1] -
Steiger, A. K.; Zhao, Y.; Choi, W. J.; Crammond, A.; Tillotson, M. R.; Pluth, M. D. Biochem. Pharmacol. 2018, 149, 124–130. doi:10.1016/j.bcp.2017.11.004
Return to citation in text: [1] -
Ranu, B. C.; Saha, A.; Banerjee, S. Eur. J. Org. Chem. 2008, 519–523. doi:10.1002/ejoc.200700842
Return to citation in text: [1] -
Safa, K. D.; Tavakkoli Osgoei, S.; Alyari, M. J. Sulfur Chem. 2014, 35, 683–690. doi:10.1080/17415993.2014.953163
Return to citation in text: [1] -
Halimehjani, A. Z.; Hooshmand, S. E.; Shamiri, E. V. Tetrahedron Lett. 2014, 55, 5454–5457. doi:10.1016/j.tetlet.2014.08.017
Return to citation in text: [1] -
Halimehjani, A. Z.; Martens, J.; Schlüter, T. Tetrahedron 2016, 72, 3958–3965. doi:10.1016/j.tet.2016.05.025
Return to citation in text: [1] -
Modarresi-Alam, A. R.; Inaloo, I. D.; Kleinpeter, E. J. Mol. Struct. 2012, 1024, 156–162. doi:10.1016/j.molstruc.2012.05.033
Return to citation in text: [1] -
Tsuji, K.; Nakamura, K.; Konishi, N.; Okumura, H.; Matsuo, M. Chem. Pharm. Bull. 1992, 40, 2399–2409. doi:10.1248/cpb.40.2399
Return to citation in text: [1] -
Hagooly, Y.; Gatenyo, J.; Hagooly, A.; Rozen, S. J. Org. Chem. 2009, 74, 8578–8582. doi:10.1021/jo901560b
Return to citation in text: [1] -
Ranade, S. C.; Hasty, S. J.; Demchenko, A. V. J. Carbohydr. Chem. 2013, 32, 360–379. doi:10.1080/07328303.2013.826670
Return to citation in text: [1] -
Chniti, I.; Sanhoury, M. A. K.; Chehidi, I. J. Fluorine Chem. 2013, 156, 101–105. doi:10.1016/j.jfluchem.2013.09.008
Return to citation in text: [1] -
Chniti, I.; Sanhoury, M. A. K.; Merlet, D.; Chehidi, I. J. Fluorine Chem. 2014, 168, 223–229. doi:10.1016/j.jfluchem.2014.10.015
Return to citation in text: [1] -
Liu, X.; Wang, Z.; Xie, R.; Tang, P.; Yuan, Q. Eur. J. Med. Chem. 2018, 157, 1526–1540. doi:10.1016/j.ejmech.2018.07.038
Return to citation in text: [1] -
Wróblewska, A.; Mlostoń, G. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 509–511. doi:10.1080/10426507.2012.736897
Return to citation in text: [1] -
Zhu, J.-B.; Chen, E. Y.-X. J. Am. Chem. Soc. 2015, 137, 12506–12509. doi:10.1021/jacs.5b08658
Return to citation in text: [1] -
Bassetto, M.; Ferla, S.; Pertusati, F.; Kandil, S.; Westwell, A. D.; Brancale, A.; McGuigan, C. Eur. J. Med. Chem. 2016, 118, 230–243. doi:10.1016/j.ejmech.2016.04.052
Return to citation in text: [1] -
Cai, W.; Wu, J.; Liu, W.; Xie, Y.; Liu, Y.; Zhang, S.; Xu, W.; Tang, L.; Wang, J.; Zhao, G. Molecules 2018, 23, No. 252. doi:10.3390/molecules23020252
Return to citation in text: [1] -
Larsen, C.; Steliou, K.; Harpp, D. N. J. Org. Chem. 1978, 43, 337–339. doi:10.1021/jo00396a035
Return to citation in text: [1] -
Kim, S.; Yi, K. Y. Tetrahedron Lett. 1985, 26, 1661–1664. doi:10.1016/s0040-4039(00)98578-2
Return to citation in text: [1] -
Wong, R.; Dolman, S. J. J. Org. Chem. 2007, 72, 3969–3971. doi:10.1021/jo070246n
Return to citation in text: [1] -
Nath, J.; Ghosh, H.; Yella, R.; Patel, B. K. Eur. J. Org. Chem. 2009, 1849–1851. doi:10.1002/ejoc.200801270
Return to citation in text: [1] -
Zhang, X.; Lee, Y. K.; Kelley, J. A.; Burke, T. R. J. Org. Chem. 2000, 65, 6237–6240. doi:10.1021/jo000139s
Return to citation in text: [1] -
Li, Z.-Y.; Ma, H.-Z.; Han, C.; Xi, H.-T.; Meng, Q.; Chen, X.; Sun, X.-Q. Synthesis 2013, 45, 1667–1674. doi:10.1055/s-0033-1338744
Return to citation in text: [1] -
Jae Nyoung, K.; Ryu, E. K. Tetrahedron Lett. 1993, 34, 8283–8284. doi:10.1016/s0040-4039(00)61411-9
Return to citation in text: [1] -
Kim, J. N.; Jung, K. S.; Lee, H. J.; Son, J. S. Tetrahedron Lett. 1997, 38, 1597–1598. doi:10.1016/s0040-4039(97)00121-4
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 1613–1619. doi:10.3762/bjoc.9.184
Return to citation in text: [1] -
Tanaka, S.; Uemura, S.; Okano, M. Bull. Chem. Soc. Jpn. 1977, 50, 2785–2788. doi:10.1246/bcsj.50.2785
Return to citation in text: [1] -
Bachi, M. D.; Balanov, A.; Bar-Ner, N. J. Org. Chem. 1994, 59, 7752–7758. doi:10.1021/jo00104a035
Return to citation in text: [1] -
Bachi, M. D.; Melman, A. J. Org. Chem. 1995, 60, 6242–6244. doi:10.1021/jo00124a056
Return to citation in text: [1] -
Szabó, T.; Milen, M. Chem. Heterocycl. Compd. 2019, 55, 126–128. doi:10.1007/s10593-019-02427-3
Return to citation in text: [1] [2] -
Tan, W.; Wei, J.; Jiang, X. Org. Lett. 2017, 19, 2166–2169. doi:10.1021/acs.orglett.7b00819
Return to citation in text: [1] -
Reiffen, M.; Hoffmann, R. W. Chem. Ber. 1977, 110, 37–48. doi:10.1002/cber.19771100105
Return to citation in text: [1] -
Cunico, R. F.; Maity, B. C. Org. Lett. 2003, 5, 4947–4949. doi:10.1021/ol030110l
Return to citation in text: [1] -
Mampuys, P.; Zhu, Y.; Vlaar, T.; Ruijter, E.; Orru, R. V. A.; Maes, B. U. W. Angew. Chem., Int. Ed. 2014, 53, 12849–12854. doi:10.1002/anie.201406717
Return to citation in text: [1] -
Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. Synthesis 2014, 46, 3172–3179. doi:10.1055/s-0034-1379327
Return to citation in text: [1] [2] [3] -
Pathare, R. S.; Patil, V.; Kaur, H.; Maurya, A. K.; Agnihotri, V. K.; Khan, S.; Devunuri, N.; Sharon, A.; Sawant, D. M. Org. Biomol. Chem. 2018, 16, 8263–8266. doi:10.1039/c8ob01855c
Return to citation in text: [1] -
Boyer, J. H.; Ramakrishnan, V. T. J. Org. Chem. 1972, 37, 1360–1364. doi:10.1021/jo00974a016
Return to citation in text: [1] -
Zajdlik, A.; Wang, Z.; Hickey, J. L.; Aman, A.; Schimmer, A. D.; Yudin, A. K. Angew. Chem., Int. Ed. 2013, 52, 8411–8415. doi:10.1002/anie.201302818
Return to citation in text: [1] -
Adam, W.; Bargon, R. M.; Bosio, S. G.; Schenk, W. A.; Stalke, D. J. Org. Chem. 2002, 67, 7037–7041. doi:10.1021/jo026042i
Return to citation in text: [1] -
Arisawa, M.; Ashikawa, M.; Suwa, A.; Yamaguchi, M. Tetrahedron Lett. 2005, 46, 1727–1729. doi:10.1016/j.tetlet.2005.01.069
Return to citation in text: [1] -
Farrell, W. S.; Zavalij, P. Y.; Sita, L. R. Organometallics 2016, 35, 2361–2366. doi:10.1021/acs.organomet.6b00302
Return to citation in text: [1] -
Chakrabarty, S.; Choudhary, S.; Doshi, A.; Liu, F.-Q.; Mohan, R.; Ravindra, M. P.; Shah, D.; Yang, X.; Fleming, F. F. Adv. Synth. Catal. 2014, 356, 2135–2196. doi:10.1002/adsc.201400017
Return to citation in text: [1] -
Boyarskiy, V. P.; Bokach, N. A.; Luzyanin, K. V.; Kukushkin, V. Y. Chem. Rev. 2015, 115, 2698–2779. doi:10.1021/cr500380d
Return to citation in text: [1] -
Fujiwara, S.-i.; Shin-Ike, T.; Okada, K.; Aoki, M.; Kambe, N.; Sonoda, N. Tetrahedron Lett. 1992, 33, 7021–7024. doi:10.1016/s0040-4039(00)60922-x
Return to citation in text: [1] -
Feng, W.; Zhang, X.-G. Chem. Commun. 2019, 55, 1144–1147. doi:10.1039/c8cc09190k
Return to citation in text: [1] -
Milen, M.; Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Pongó, L.; Keglevich, G. Tetrahedron Lett. 2013, 54, 5430–5433. doi:10.1016/j.tetlet.2013.07.145
Return to citation in text: [1] -
Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Volk, B.; Keglevich, G.; Milen, M. Tetrahedron 2014, 70, 5711–5719. doi:10.1016/j.tet.2014.06.073
Return to citation in text: [1] -
Varga, V.; Milen, M.; Ábrányi-Balogh, P. Tetrahedron Lett. 2018, 59, 3683–3689. doi:10.1016/j.tetlet.2018.08.063
Return to citation in text: [1] -
Milen, M.; Slégel, P.; Keglevich, P.; Keglevich, G.; Simig, G.; Volk, B. Tetrahedron Lett. 2015, 56, 5697–5700. doi:10.1016/j.tetlet.2015.09.007
Return to citation in text: [1] -
Henderson, R. K.; Jiménez-González, C.; Constable, D. J. C.; Alston, S. R.; Inglis, G. G. A.; Fisher, G.; Sherwood, J.; Binks, S. P.; Curzons, A. D. Green Chem. 2011, 13, 854–862. doi:10.1039/c0gc00918k
Return to citation in text: [1] -
McConvey, I. F.; Woods, D.; Lewis, M.; Gan, Q.; Nancarrow, P. Org. Process Res. Dev. 2012, 16, 612–624. doi:10.1021/op2003503
Return to citation in text: [1] -
Niknam, K.; Kiasat, A. R.; Kazemi, F.; Hossieni, A. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 1385–1389. doi:10.1080/10426500307905
Return to citation in text: [1] -
Burrows, A. A.; Hunter, L. J. Chem. Soc. 1952, 0, 4118–4122. doi:10.1039/jr9520004118
Return to citation in text: [1] -
Degani, I.; Fochi, R.; Magistris, C. Synthesis 2009, 3807–3818. doi:10.1055/s-0029-1218117
Return to citation in text: [1] -
L’Abbé, G.; Sorgeloos, D.; Toppet, S.; King, G. S. D.; Van Meerwelt, L. Bull. Soc. Chim. Belg. 1981, 90, 63–74. doi:10.1002/bscb.19810900111
Return to citation in text: [1] -
Lijuan, M.; Shubai, L.; Lili, S.; Mengqi, W. Method for preparing multi-functional sulfide-bonded collaborative antioxidative stabilizer. Chin. Patent Appl. CN 104529875 A, April 22, 2015.
Return to citation in text: [1] -
Labib, M. B.; Lamie, P. F. Med. Chem. Res. 2016, 25, 2607–2618. doi:10.1007/s00044-016-1703-y
Return to citation in text: [1] -
Nguyen, T. B.; Ermolenko, L.; Retailleau, P.; Al-Mourabit, A. Angew. Chem., Int. Ed. 2014, 53, 13808–13812. doi:10.1002/anie.201408397
Return to citation in text: [1] -
Nguyen, L. A.; Ngo, Q. A.; Retailleau, P.; Nguyen, T. B. Green Chem. 2017, 19, 4289–4293. doi:10.1039/c7gc01825h
Return to citation in text: [1] -
Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 3887–3890. doi:10.1021/acs.orglett.7b01775
Return to citation in text: [1] -
Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Conjugate addition. Organic Chemistry; Oxford University Press: Oxford, U.K., 2001; pp 227–242.
Return to citation in text: [1] -
Al-Suwaidan, I. A.; Alanazi, A. M.; Abdel-Aziz, A. A.-M.; Mohamed, M. A.; El-Azab, A. S. Bioorg. Med. Chem. Lett. 2013, 23, 3935–3941. doi:10.1016/j.bmcl.2013.04.056
Return to citation in text: [1] -
Jatav, V.; Mishra, P.; Kashaw, S.; Stables, J. P. Eur. J. Med. Chem. 2008, 43, 1945–1954. doi:10.1016/j.ejmech.2007.12.003
Return to citation in text: [1] -
Smaill, J. B.; Gonzales, A. J.; Spicer, J. A.; Lee, H.; Reed, J. E.; Sexton, K.; Althaus, I. W.; Zhu, T.; Black, S. L.; Blaser, A.; Denny, W. A.; Ellis, P. A.; Fakhoury, S.; Harvey, P. J.; Hook, K.; McCarthy, F. O. J.; Palmer, B. D.; Rivault, F.; Schlosser, K.; Ellis, T.; Thompson, A. M.; Trachet, E.; Winters, R. T.; Tecle, H.; Bridges, A. J. Med. Chem. 2016, 59, 8103–8124. doi:10.1021/acs.jmedchem.6b00883
Return to citation in text: [1] -
Noble, M. E.; Endicott, J. A.; Johnson, L. N. Science 2004, 303, 1800–1805. doi:10.1126/science.1095920
Return to citation in text: [1] -
Asagarasu, A.; Matsui, T.; Hayashi, H.; Tamaoki, S.; Yamauchi, Y.; Minato, K.; Sato, M. J. Med. Chem. 2010, 53, 7549–7563. doi:10.1021/jm1002292
Return to citation in text: [1] -
Sayahi, M. H.; Saghanezhad, S. J.; Bahadorikhalili, S.; Mahdavi, M. Appl. Organomet. Chem. 2019, 33, e4635. doi:10.1002/aoc.4635
Return to citation in text: [1] -
Okamoto, K.; Yamamoto, T.; Kanbara, T. Synlett 2007, 2687–2690. doi:10.1055/s-2007-991073
Return to citation in text: [1] [2] -
Chen, H.-Y.; Peng, W.-T.; Lee, Y.-H.; Chang, Y.-L.; Chen, Y.-J.; Lai, Y.-C.; Jheng, N.-Y.; Chen, H.-Y. Organometallics 2013, 32, 5514–5522. doi:10.1021/om400784w
Return to citation in text: [1] [2]
| 76. | Boyer, J. H.; Ramakrishnan, V. T. J. Org. Chem. 1972, 37, 1360–1364. doi:10.1021/jo00974a016 |
| 77. | Zajdlik, A.; Wang, Z.; Hickey, J. L.; Aman, A.; Schimmer, A. D.; Yudin, A. K. Angew. Chem., Int. Ed. 2013, 52, 8411–8415. doi:10.1002/anie.201302818 |
| 78. | Adam, W.; Bargon, R. M.; Bosio, S. G.; Schenk, W. A.; Stalke, D. J. Org. Chem. 2002, 67, 7037–7041. doi:10.1021/jo026042i |
| 79. | Arisawa, M.; Ashikawa, M.; Suwa, A.; Yamaguchi, M. Tetrahedron Lett. 2005, 46, 1727–1729. doi:10.1016/j.tetlet.2005.01.069 |
| 80. | Farrell, W. S.; Zavalij, P. Y.; Sita, L. R. Organometallics 2016, 35, 2361–2366. doi:10.1021/acs.organomet.6b00302 |
| 81. | Chakrabarty, S.; Choudhary, S.; Doshi, A.; Liu, F.-Q.; Mohan, R.; Ravindra, M. P.; Shah, D.; Yang, X.; Fleming, F. F. Adv. Synth. Catal. 2014, 356, 2135–2196. doi:10.1002/adsc.201400017 |
| 82. | Boyarskiy, V. P.; Bokach, N. A.; Luzyanin, K. V.; Kukushkin, V. Y. Chem. Rev. 2015, 115, 2698–2779. doi:10.1021/cr500380d |
| 83. | Fujiwara, S.-i.; Shin-Ike, T.; Okada, K.; Aoki, M.; Kambe, N.; Sonoda, N. Tetrahedron Lett. 1992, 33, 7021–7024. doi:10.1016/s0040-4039(00)60922-x |
| 95. | Lijuan, M.; Shubai, L.; Lili, S.; Mengqi, W. Method for preparing multi-functional sulfide-bonded collaborative antioxidative stabilizer. Chin. Patent Appl. CN 104529875 A, April 22, 2015. |
| 96. | Labib, M. B.; Lamie, P. F. Med. Chem. Res. 2016, 25, 2607–2618. doi:10.1007/s00044-016-1703-y |
| 91. | Niknam, K.; Kiasat, A. R.; Kazemi, F.; Hossieni, A. Phosphorus, Sulfur Silicon Relat. Elem. 2003, 178, 1385–1389. doi:10.1080/10426500307905 |
| 92. | Burrows, A. A.; Hunter, L. J. Chem. Soc. 1952, 0, 4118–4122. doi:10.1039/jr9520004118 |
| 93. | Degani, I.; Fochi, R.; Magistris, C. Synthesis 2009, 3807–3818. doi:10.1055/s-0029-1218117 |
| 94. | L’Abbé, G.; Sorgeloos, D.; Toppet, S.; King, G. S. D.; Van Meerwelt, L. Bull. Soc. Chim. Belg. 1981, 90, 63–74. doi:10.1002/bscb.19810900111 |
| 88. | Milen, M.; Slégel, P.; Keglevich, P.; Keglevich, G.; Simig, G.; Volk, B. Tetrahedron Lett. 2015, 56, 5697–5700. doi:10.1016/j.tetlet.2015.09.007 |
| 89. | Henderson, R. K.; Jiménez-González, C.; Constable, D. J. C.; Alston, S. R.; Inglis, G. G. A.; Fisher, G.; Sherwood, J.; Binks, S. P.; Curzons, A. D. Green Chem. 2011, 13, 854–862. doi:10.1039/c0gc00918k |
| 90. | McConvey, I. F.; Woods, D.; Lewis, M.; Gan, Q.; Nancarrow, P. Org. Process Res. Dev. 2012, 16, 612–624. doi:10.1021/op2003503 |
| 84. | Feng, W.; Zhang, X.-G. Chem. Commun. 2019, 55, 1144–1147. doi:10.1039/c8cc09190k |
| 85. | Milen, M.; Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Pongó, L.; Keglevich, G. Tetrahedron Lett. 2013, 54, 5430–5433. doi:10.1016/j.tetlet.2013.07.145 |
| 86. | Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Volk, B.; Keglevich, G.; Milen, M. Tetrahedron 2014, 70, 5711–5719. doi:10.1016/j.tet.2014.06.073 |
| 87. | Varga, V.; Milen, M.; Ábrányi-Balogh, P. Tetrahedron Lett. 2018, 59, 3683–3689. doi:10.1016/j.tetlet.2018.08.063 |
| 97. | Nguyen, T. B.; Ermolenko, L.; Retailleau, P.; Al-Mourabit, A. Angew. Chem., Int. Ed. 2014, 53, 13808–13812. doi:10.1002/anie.201408397 |
| 98. | Nguyen, L. A.; Ngo, Q. A.; Retailleau, P.; Nguyen, T. B. Green Chem. 2017, 19, 4289–4293. doi:10.1039/c7gc01825h |
| 99. | Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 3887–3890. doi:10.1021/acs.orglett.7b01775 |
| 100. | Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Conjugate addition. Organic Chemistry; Oxford University Press: Oxford, U.K., 2001; pp 227–242. |
| 101. | Al-Suwaidan, I. A.; Alanazi, A. M.; Abdel-Aziz, A. A.-M.; Mohamed, M. A.; El-Azab, A. S. Bioorg. Med. Chem. Lett. 2013, 23, 3935–3941. doi:10.1016/j.bmcl.2013.04.056 |
| 74. | Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. Synthesis 2014, 46, 3172–3179. doi:10.1055/s-0034-1379327 |
| 107. | Okamoto, K.; Yamamoto, T.; Kanbara, T. Synlett 2007, 2687–2690. doi:10.1055/s-2007-991073 |
| 108. | Chen, H.-Y.; Peng, W.-T.; Lee, Y.-H.; Chang, Y.-L.; Chen, Y.-J.; Lai, Y.-C.; Jheng, N.-Y.; Chen, H.-Y. Organometallics 2013, 32, 5514–5522. doi:10.1021/om400784w |
| 74. | Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. Synthesis 2014, 46, 3172–3179. doi:10.1055/s-0034-1379327 |
| 107. | Okamoto, K.; Yamamoto, T.; Kanbara, T. Synlett 2007, 2687–2690. doi:10.1055/s-2007-991073 |
| 108. | Chen, H.-Y.; Peng, W.-T.; Lee, Y.-H.; Chang, Y.-L.; Chen, Y.-J.; Lai, Y.-C.; Jheng, N.-Y.; Chen, H.-Y. Organometallics 2013, 32, 5514–5522. doi:10.1021/om400784w |
| 106. | Sayahi, M. H.; Saghanezhad, S. J.; Bahadorikhalili, S.; Mahdavi, M. Appl. Organomet. Chem. 2019, 33, e4635. doi:10.1002/aoc.4635 |
| 69. | Szabó, T.; Milen, M. Chem. Heterocycl. Compd. 2019, 55, 126–128. doi:10.1007/s10593-019-02427-3 |
| 104. | Noble, M. E.; Endicott, J. A.; Johnson, L. N. Science 2004, 303, 1800–1805. doi:10.1126/science.1095920 |
| 105. | Asagarasu, A.; Matsui, T.; Hayashi, H.; Tamaoki, S.; Yamauchi, Y.; Minato, K.; Sato, M. J. Med. Chem. 2010, 53, 7549–7563. doi:10.1021/jm1002292 |
| 102. | Jatav, V.; Mishra, P.; Kashaw, S.; Stables, J. P. Eur. J. Med. Chem. 2008, 43, 1945–1954. doi:10.1016/j.ejmech.2007.12.003 |
| 103. | Smaill, J. B.; Gonzales, A. J.; Spicer, J. A.; Lee, H.; Reed, J. E.; Sexton, K.; Althaus, I. W.; Zhu, T.; Black, S. L.; Blaser, A.; Denny, W. A.; Ellis, P. A.; Fakhoury, S.; Harvey, P. J.; Hook, K.; McCarthy, F. O. J.; Palmer, B. D.; Rivault, F.; Schlosser, K.; Ellis, T.; Thompson, A. M.; Trachet, E.; Winters, R. T.; Tecle, H.; Bridges, A. J. Med. Chem. 2016, 59, 8103–8124. doi:10.1021/acs.jmedchem.6b00883 |
| 1. | Len, C.; Postel, D.; Ronco, G.; Villa, P.; Goubert, C.; Jeufrault, E.; Mathon, B.; Simon, H. J. Agric. Food Chem. 1997, 45, 3–6. doi:10.1021/jf9607371 |
| 2. | Zhou, Y.; Wang, L.; Han, L.; Meng, F.; Yang, C. Carbohydr. Res. 2009, 344, 1289–1296. doi:10.1016/j.carres.2009.05.008 |
| 3. | Ryder, N. S.; Frank, I.; Dupont, M.-C. Antimicrob. Agents Chemother. 1986, 29, 858–860. doi:10.1128/aac.29.5.858 |
| 12. | Zhou, L.; Tan, C. K.; Jiang, X.; Chen, F.; Yeung, Y.-Y. J. Am. Chem. Soc. 2010, 132, 15474–15476. doi:10.1021/ja1048972 |
| 13. | Tan, C. K.; Zhou, L.; Yeung, Y.-Y. Org. Lett. 2011, 13, 2738–2741. doi:10.1021/ol200840e |
| 14. | Zhou, L.; Chen, J.; Tan, C. K.; Yeung, Y.-Y. J. Am. Chem. Soc. 2011, 133, 9164–9167. doi:10.1021/ja201627h |
| 15. | Han, W.-Y.; Li, S.-W.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Chem. – Eur. J. 2013, 19, 5551–5556. doi:10.1002/chem.201300206 |
| 16. | Tan, C. K.; Er, J. C.; Yeung, Y.-Y. Tetrahedron Lett. 2014, 55, 1243–1246. doi:10.1016/j.tetlet.2014.01.009 |
| 17. | Tay, D. W.; Leung, G. Y. C.; Yeung, Y.-Y. Angew. Chem., Int. Ed. 2014, 53, 5161–5164. doi:10.1002/anie.201310136 |
| 38. | Gromek, S. M.; deMayo, J. A.; Maxwell, A. T.; West, A. M.; Pavlik, C. M.; Zhao, Z.; Li, J.; Wiemer, A. J.; Zweifach, A.; Balunas, M. J. Bioorg. Med. Chem. 2016, 24, 5183–5196. doi:10.1016/j.bmc.2016.08.040 |
| 39. | Schrader, A. M.; Schroll, A. L.; Barany, G. J. Org. Chem. 2011, 76, 7882–7892. doi:10.1021/jo201329n |
| 40. | Boeckman, R. K.; Ge, P.; Reed, J. E. Org. Lett. 2001, 3, 3647–3650. doi:10.1021/ol0165645 |
| 41. | Steiger, A. K.; Zhao, Y.; Choi, W. J.; Crammond, A.; Tillotson, M. R.; Pluth, M. D. Biochem. Pharmacol. 2018, 149, 124–130. doi:10.1016/j.bcp.2017.11.004 |
| 6. | Lin, G.; Shieh, C.-T.; Ho, H.-C.; Chouhwang, J.-Y.; Lin, W.-Y.; Lu, C.-P. Biochemistry 1999, 38, 9971–9981. doi:10.1021/bi982775e |
| 7. | Cesarini, S.; Spallarossa, A.; Ranise, A.; Fossa, P.; La Colla, P.; Sanna, G.; Collu, G.; Loddo, R. Bioorg. Med. Chem. 2008, 16, 4160–4172. doi:10.1016/j.bmc.2007.12.050 |
| 8. | Cesarini, S.; Spallarossa, A.; Ranise, A.; Bruno, O.; La Colla, P.; Secci, B.; Collu, G.; Loddo, R. Bioorg. Med. Chem. 2008, 16, 4173–4185. doi:10.1016/j.bmc.2007.12.046 |
| 9. | Spallarossa, A.; Cesarini, S.; Ranise, A.; Bruno, O.; Schenone, S.; La Colla, P.; Collu, G.; Sanna, G.; Secci, B.; Loddo, R. Eur. J. Med. Chem. 2009, 44, 1650–1663. doi:10.1016/j.ejmech.2008.09.024 |
| 10. | Cichero, E.; Cesarini, S.; Fossa, P.; Spallarossa, A.; Mosti, L. Eur. J. Med. Chem. 2009, 44, 2059–2070. doi:10.1016/j.ejmech.2008.10.014 |
| 11. | Spallarossa, A.; Cesarini, S.; Ranise, A.; Schenone, S.; Bruno, O.; Borassi, A.; La Colla, P.; Pezzullo, M.; Sanna, G.; Collu, G.; Secci, B.; Loddo, R. Eur. J. Med. Chem. 2009, 44, 2190–2201. doi:10.1016/j.ejmech.2008.10.032 |
| 42. | Ranu, B. C.; Saha, A.; Banerjee, S. Eur. J. Org. Chem. 2008, 519–523. doi:10.1002/ejoc.200700842 |
| 43. | Safa, K. D.; Tavakkoli Osgoei, S.; Alyari, M. J. Sulfur Chem. 2014, 35, 683–690. doi:10.1080/17415993.2014.953163 |
| 44. | Halimehjani, A. Z.; Hooshmand, S. E.; Shamiri, E. V. Tetrahedron Lett. 2014, 55, 5454–5457. doi:10.1016/j.tetlet.2014.08.017 |
| 45. | Halimehjani, A. Z.; Martens, J.; Schlüter, T. Tetrahedron 2016, 72, 3958–3965. doi:10.1016/j.tet.2016.05.025 |
| 5. | Wood, T. F.; Gardner, J. H. J. Am. Chem. Soc. 1941, 63, 2741–2742. doi:10.1021/ja01855a068 |
| 31. | Seetharamsingh, B.; Ramesh, R.; Dange, S. S.; Khairnar, P. V.; Singhal, S.; Upadhyay, D.; Veeraraghavan, S.; Viswanadha, S.; Vakkalanka, S.; Reddy, D. S. ACS Med. Chem. Lett. 2015, 6, 1105–1110. doi:10.1021/acsmedchemlett.5b00213 |
| 32. | Selvakumar, N.; Kumar, G. S.; Malar Azhagan, A.; Govinda Rajulu, G.; Sharma, S.; Kumar, M. S.; Das, J.; Iqbal, J.; Trehan, S. Eur. J. Med. Chem. 2007, 42, 538–543. doi:10.1016/j.ejmech.2006.10.013 |
| 33. | Sasse, K. Justus Liebigs Ann. Chem. 1970, 735, 158–188. doi:10.1002/jlac.19707350120 |
| 4. | Spallarossa, A.; Cesarini, S.; Schenone, S.; Ranise, A. Arch. Pharm. (Weinheim, Ger.) 2009, 342, 344–352. doi:10.1002/ardp.200800212 |
| 34. | Barma, D. K.; Bandyopadhyay, A.; Capdevila, J. H.; Falck, J. R. Org. Lett. 2003, 5, 4755–4757. doi:10.1021/ol0354573 |
| 35. | Kojima, M.; Nakamura, Y.; Ishikawa, T.; Takeuchi, S. Tetrahedron Lett. 2006, 47, 6309–6314. doi:10.1016/j.tetlet.2006.05.142 |
| 36. | Iwakawa, T.; Tamura, H.; Murabayashi, A.; Hayase, Y. Chem. Pharm. Bull. 1991, 39, 1939–1943. doi:10.1248/cpb.39.1939 |
| 37. | Koketsu, M.; Otsuka, T.; Ishihara, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 443–448. doi:10.1080/10426500490262612 |
| 26. | Carta, F.; Aggarwal, M.; Maresca, A.; Scozzafava, A.; McKenna, R.; Masini, E.; Supuran, C. T. J. Med. Chem. 2012, 55, 1721–1730. doi:10.1021/jm300031j |
| 28. | Boas, U.; Gertz, H.; Christensen, J. B.; Heegaard, P. M. H. Tetrahedron Lett. 2004, 45, 269–272. doi:10.1016/j.tetlet.2003.10.182 |
| 21. | Heyns, A. Y.; Carter, G. A.; Rothwell, K.; Wain, R. L. Ann. Appl. Biol. 1966, 57, 33–51. doi:10.1111/j.1744-7348.1966.tb06864.x |
| 22. | Rafin, C.; Veignie, E.; Sancholle, M.; Postel, D.; Len, C.; Villa, P.; Ronco, G. J. Agric. Food Chem. 2000, 48, 5283–5287. doi:10.1021/jf0003698 |
| 23. | MacBean, C. The Pesticide Manual, 16th ed.; British Crop Production Council, 2012. |
| 24. | Gupta, P. K. Herbicides and fungicides. In Biomarkers in Toxicology; Gupta, R. C., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp 409–431. doi:10.1016/b978-0-12-404630-6.00024-5 |
| 25. | Thind, T. S.; Hollomon, D. W. Pest Manage. Sci. 2018, 74, 1547–1551. doi:10.1002/ps.4844 |
| 29. | Hatai, J.; Pal, S.; Jose, G. P.; Bandyopadhyay, S. Inorg. Chem. 2012, 51, 10129–10135. doi:10.1021/ic300530f |
| 30. | Konieczna, D. D.; Blanrue, A.; Wilhelm, R. Z. Naturforsch., B: J. Chem. Sci. 2014, 69, 596–604. doi:10.5560/znb.2014-4014 |
| 19. | Yoshida, S.; Hayashi, Y.; Obitsu, K.; Nakamura, A.; Kikuchi, T.; Sawada, T.; Kimura, T.; Takahashi, T.; Mukuta, T. Org. Process Res. Dev. 2012, 16, 1818–1826. doi:10.1021/op300237b |
| 20. | Yadav, J. S.; Goreti, R.; Pabbaraja, S.; Sridhar, B. Org. Lett. 2013, 15, 3782–3785. doi:10.1021/ol401760e |
| 18. | Steiger, A. K.; Pardue, S.; Kevil, C. G.; Pluth, M. D. J. Am. Chem. Soc. 2016, 138, 7256–7259. doi:10.1021/jacs.6b03780 |
| 27. | Schwimmer, A.; Rubin, D. Method and compositions for inhibiting tumor cell metabolism. U.S. Patent US5,240,914, Aug 31, 1993. |
| 53. | Wróblewska, A.; Mlostoń, G. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 509–511. doi:10.1080/10426507.2012.736897 |
| 54. | Zhu, J.-B.; Chen, E. Y.-X. J. Am. Chem. Soc. 2015, 137, 12506–12509. doi:10.1021/jacs.5b08658 |
| 55. | Bassetto, M.; Ferla, S.; Pertusati, F.; Kandil, S.; Westwell, A. D.; Brancale, A.; McGuigan, C. Eur. J. Med. Chem. 2016, 118, 230–243. doi:10.1016/j.ejmech.2016.04.052 |
| 56. | Cai, W.; Wu, J.; Liu, W.; Xie, Y.; Liu, Y.; Zhang, S.; Xu, W.; Tang, L.; Wang, J.; Zhao, G. Molecules 2018, 23, No. 252. doi:10.3390/molecules23020252 |
| 46. | Modarresi-Alam, A. R.; Inaloo, I. D.; Kleinpeter, E. J. Mol. Struct. 2012, 1024, 156–162. doi:10.1016/j.molstruc.2012.05.033 |
| 47. | Tsuji, K.; Nakamura, K.; Konishi, N.; Okumura, H.; Matsuo, M. Chem. Pharm. Bull. 1992, 40, 2399–2409. doi:10.1248/cpb.40.2399 |
| 48. | Hagooly, Y.; Gatenyo, J.; Hagooly, A.; Rozen, S. J. Org. Chem. 2009, 74, 8578–8582. doi:10.1021/jo901560b |
| 49. | Ranade, S. C.; Hasty, S. J.; Demchenko, A. V. J. Carbohydr. Chem. 2013, 32, 360–379. doi:10.1080/07328303.2013.826670 |
| 50. | Chniti, I.; Sanhoury, M. A. K.; Chehidi, I. J. Fluorine Chem. 2013, 156, 101–105. doi:10.1016/j.jfluchem.2013.09.008 |
| 51. | Chniti, I.; Sanhoury, M. A. K.; Merlet, D.; Chehidi, I. J. Fluorine Chem. 2014, 168, 223–229. doi:10.1016/j.jfluchem.2014.10.015 |
| 52. | Liu, X.; Wang, Z.; Xie, R.; Tang, P.; Yuan, Q. Eur. J. Med. Chem. 2018, 157, 1526–1540. doi:10.1016/j.ejmech.2018.07.038 |
| 71. | Reiffen, M.; Hoffmann, R. W. Chem. Ber. 1977, 110, 37–48. doi:10.1002/cber.19771100105 |
| 72. | Cunico, R. F.; Maity, B. C. Org. Lett. 2003, 5, 4947–4949. doi:10.1021/ol030110l |
| 73. | Mampuys, P.; Zhu, Y.; Vlaar, T.; Ruijter, E.; Orru, R. V. A.; Maes, B. U. W. Angew. Chem., Int. Ed. 2014, 53, 12849–12854. doi:10.1002/anie.201406717 |
| 74. | Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. Synthesis 2014, 46, 3172–3179. doi:10.1055/s-0034-1379327 |
| 75. | Pathare, R. S.; Patil, V.; Kaur, H.; Maurya, A. K.; Agnihotri, V. K.; Khan, S.; Devunuri, N.; Sharon, A.; Sawant, D. M. Org. Biomol. Chem. 2018, 16, 8263–8266. doi:10.1039/c8ob01855c |
| 69. | Szabó, T.; Milen, M. Chem. Heterocycl. Compd. 2019, 55, 126–128. doi:10.1007/s10593-019-02427-3 |
| 70. | Tan, W.; Wei, J.; Jiang, X. Org. Lett. 2017, 19, 2166–2169. doi:10.1021/acs.orglett.7b00819 |
| 63. | Jae Nyoung, K.; Ryu, E. K. Tetrahedron Lett. 1993, 34, 8283–8284. doi:10.1016/s0040-4039(00)61411-9 |
| 64. | Kim, J. N.; Jung, K. S.; Lee, H. J.; Son, J. S. Tetrahedron Lett. 1997, 38, 1597–1598. doi:10.1016/s0040-4039(97)00121-4 |
| 65. | Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 1613–1619. doi:10.3762/bjoc.9.184 |
| 66. | Tanaka, S.; Uemura, S.; Okano, M. Bull. Chem. Soc. Jpn. 1977, 50, 2785–2788. doi:10.1246/bcsj.50.2785 |
| 67. | Bachi, M. D.; Balanov, A.; Bar-Ner, N. J. Org. Chem. 1994, 59, 7752–7758. doi:10.1021/jo00104a035 |
| 68. | Bachi, M. D.; Melman, A. J. Org. Chem. 1995, 60, 6242–6244. doi:10.1021/jo00124a056 |
| 57. | Larsen, C.; Steliou, K.; Harpp, D. N. J. Org. Chem. 1978, 43, 337–339. doi:10.1021/jo00396a035 |
| 58. | Kim, S.; Yi, K. Y. Tetrahedron Lett. 1985, 26, 1661–1664. doi:10.1016/s0040-4039(00)98578-2 |
| 59. | Wong, R.; Dolman, S. J. J. Org. Chem. 2007, 72, 3969–3971. doi:10.1021/jo070246n |
| 60. | Nath, J.; Ghosh, H.; Yella, R.; Patel, B. K. Eur. J. Org. Chem. 2009, 1849–1851. doi:10.1002/ejoc.200801270 |
| 61. | Zhang, X.; Lee, Y. K.; Kelley, J. A.; Burke, T. R. J. Org. Chem. 2000, 65, 6237–6240. doi:10.1021/jo000139s |
| 62. | Li, Z.-Y.; Ma, H.-Z.; Han, C.; Xi, H.-T.; Meng, Q.; Chen, X.; Sun, X.-Q. Synthesis 2013, 45, 1667–1674. doi:10.1055/s-0033-1338744 |
© 2019 Németh et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)