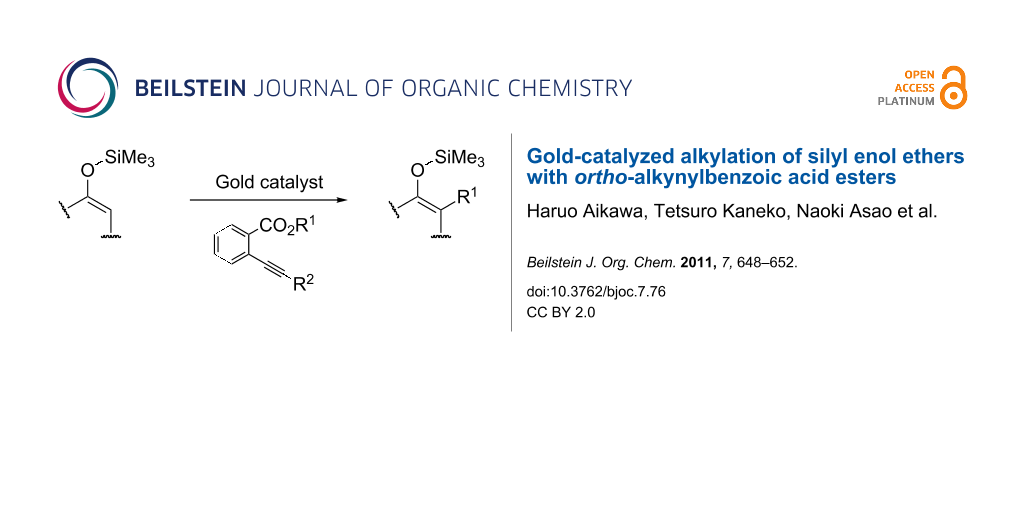
Abstract
Unprecedented alkylation of silyl enol ethers has been developed by the use of ortho-alkynylbenzoic acid alkyl esters as alkylating agents in the presence of a gold catalyst. The reaction probably proceeds through the gold-induced in situ construction of leaving groups and subsequent nucleophilic attack on the silyl enol ethers. The generated leaving compound abstracts a proton to regenerate the silyl enol ether structure.

Graphical Abstract
Findings
Silyl enol ethers have been widely used in organic synthesis as effective carbon nucleophiles for the construction of carbon frameworks [1-4]. Generally, they react with a variety of electrophiles to give carbonyl compounds as products due to cleavage of the silicon–oxygen bond. For example, the Lewis acid-catalyzed reaction of silyl enol ethers with alkyl halides is well known as one of the most efficient preparative methods for regio-defined α-alkylated ketones (path a in Scheme 1) [5-17]. In contrast, in this paper, we report a gold-catalyzed reaction of silyl enol ethers with ortho-alkynylbenzoic acid esters which leads to the formation of α-alkylated silyl enol ethers (path b).
![[1860-5397-7-76-i1]](/bjoc/content/inline/1860-5397-7-76-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Alkylation of silyl enol ethers.
Scheme 1: Alkylation of silyl enol ethers.
We examined the reactions of silyl enol ether 1a with ortho-alkynylbenzoic acid benzyl esters 2 in the presence of gold catalysts under several reaction conditions and the results are summarized in Table 1 [18-21]. With a cationic gold catalyst, derived from Ph3PAuCl and AgClO4, the reaction of 1a with 2a proceeded at 80 °C over 2 h and the benzylated silyl enol ether 3a was obtained in 35% yield, along with the eliminated isocoumarin 4a and recovered 2a in 32% and 65% yields, respectively (entry 1). On the other hand, no products were obtained from the reaction of 1a with benzyl benzoate (having no alkynyl group at the ortho-position) under similar reaction conditions. These results clearly show that the alkynyl moiety of ester 2a is essential for the formation of 3a. It is well known that concerted pericyclic ene-type reaction of silyl enol ethers with electrophiles, such as aldehydes or ketones, gives functionalized silyl enol ethers without desilylation [22-36]. To the best of our knowledge, however, this is the first example of the introduction of simple alkyl groups through a substitution-type reaction [37-40]. The chemical yield was increased to 55% by use of sterically hindered (o-Tol)3PAuCl as the gold catalyst (entry 2). Besides benzene, (CH2Cl)2 and 1,4-dioxane were also suitable solvents (entries 3 and 4). The use of 5 equiv of 1a improved the chemical yield and 3a was obtained in 72% yield (entry 5). The catalyst derived from AgOTf gave a better yield, although a longer reaction time was required (entry 6). Analogously, the reaction with 2b, with a butyl group at the alkynyl terminus, gave 3a in 75% yield (entry 7). In the current catalyst system using AgOTf, TfOH might be produced during the reactions due to the decomposition of AgOTf with a trace amount of water, which could be present in the reaction medium. However, the alkylation of 1a with 2a did not proceed at all with 5 mol % of TfOH. This result clearly indicates that the gold complex functions as a catalyst in the current transformations.
Table 1: Gold-catalyzed alkylation of silyl enol ethera.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-76-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 2 | AgX | Solvent | Conditions | Yield (%)b |
|---|---|---|---|---|---|
| 1c | 2a | AgClO4 | benzene | 80 °C, 2 h | 35 |
| 2 | 2a | AgClO4 | benzene | 80 °C, 2 h | 55 |
| 3 | 2a | AgClO4 | (CH2Cl)2 | 80 °C, 2 h | 44 |
| 4 | 2a | AgClO4 | dioxane | 100 °C, 2 h | 58 |
| 5d | 2a | AgClO4 | dioxane | 100 °C, 1 h | 72 |
| 6d | 2a | AgOTf | dioxane | 100 °C, 10 h | 80 |
| 7d | 2b | AgOTf | dioxane | 80 °C, 5 h | 75 |
aReaction conditions: 0.25 M solution of 2 was treated with 1a (3 equiv) in the presence of the gold catalyst. bNMR yield using CH2Br2 as an internal standard. cPh3PAuCl was used instead of (o-Tol)3PAuCl. d5 equiv of 1a was used.
We next examined the substrate generality with several types of silyl enol ethers 1 and esters 2 (Table 2). Treatment of five-membered silyl enol ether, cyclopentenyloxytrimethylsilane (1b), with 2b in the presence of the gold catalyst gave the corresponding benzylated product 3b in 61% yield (entry 1). It is worth mentioning that benzo-fused silyl enol ether 1c is suitable for this transformation as shown in entries 2 and 3, whereas it cannot be used for ene-reaction due to the lack of a hydrogen atom at the α’-position. Not only cyclic silyl enol ethers but also an acyclic silyl enol ether underwent the reaction. Thus, 1d reacted stereoselectively with 2a to yield E-3e. Interestingly, the formation of the isomeric Z-3e was not detected at all (entry 4) [41]. The reaction of 1a with allyl ester 2d proceeded smoothly and the corresponding allylated product 3f was obtained in 70% yield (entry 5) [42].
Table 2: Gold-catalyzed alkylation of silyl enol ethera.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-76-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | 1 | 2 | R1 | R2 | 3 | Yield (%)b | |
|---|---|---|---|---|---|---|---|
| 1c | 1b |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-76-i7.svg?max-width=637&scale=1.0)
|
2b | Bn | Bu | 3b | 61 |
| 2 | 1c |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-76-i8.svg?max-width=637&scale=1.0)
|
2b | Bn | Bu | 3c | 70 |
| 3d | 1c |
|
2c |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-76-i9.svg?max-width=637&scale=1.0)
|
Ph | 3d | 60e |
| 4c,f | 1d |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-76-i10.svg?max-width=637&scale=1.0)
|
2a | Bn | Ph | 3e | 61 |
| 5 | 1a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-76-i12.svg?max-width=637&scale=1.0)
|
2d |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-76-i11.svg?max-width=637&scale=1.0)
|
Ph | 3f | 70 |
aReaction conditions: 0.25 M solution of 2 was treated with 1 (5 equiv) in the presence of the gold catalyst. bNMR yield using CH2Br2 as an internal standard. c10 mol % of the catalyst was used. d3 equiv of 1 was used. eYield of isolated product. fAgOTf was used instead of AgClO4.
A plausible mechanism for the gold-catalyzed alkylation of silyl enol ethers is shown in Scheme 2. The gold catalyst enhances the electrophilicity of the alkynyl moiety of 2, leading to the formation of a cationic intermediate 6 via the intramolecular nucleophilic attack of the carbonyl oxygen on the alkyne as shown in 5. Due to the high leaving ability of the isocoumarin moiety of 6, the silyl enol ether 1 attacks the R group to give the intermediate 7 together with the gold complex 8 as a leaving compound [43-46]. In the case of ordinary substitution reactions with alkyl halides (path a in Scheme 1), generated halide ions would attack the silyl group, due to their strong affinities with the silicon atom, and cleave the silicon–oxygen bond of 7. However, in the present reaction system, intermediate 8 would prefer to act as a base and abstract a proton, Ha, from the α-position rather than attack the silyl group as a nucleophile, probably due to steric and electronic reasons. For these reasons, deprotonation of 7 occurs to give the product 3 together with 4 as a final leaving compound.
![[1860-5397-7-76-i2]](/bjoc/content/inline/1860-5397-7-76-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanism for the alkylation of silyl enol ether.
Scheme 2: Plausible mechanism for the alkylation of silyl enol ether.
On the other hand, in the case of reactions with silyl enol ethers having a proton, Hb, at the α’-position, compound 9 might be produced through the deprotonation of Hb by 8. However, such products were not obtained in any of the examples studied. These results imply that isomerism from 9 to 3 would occur during the reaction. Thus, compound 1e was prepared according to a known procedure and treated with the gold catalyst at 100 °C for 2 h (Scheme 3). As expected, the isomerization of the double bond occurred and 3a was obtained in 80% yield. This result shows that the indirect pathway from 7 to 3 via deprotonation of Hb is also possible. In addition, it was found that the reaction of 1f, having no hydrogen at the α-position, proceeded smoothly and α,α-dialkyl silyl enol ether 3g was obtained in good yield (Scheme 4). Obviously, this result supports the possibility of the indirect pathway.
![[1860-5397-7-76-i3]](/bjoc/content/inline/1860-5397-7-76-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Gold-catalyzed isomerism of silyl enol ether.
Scheme 3: Gold-catalyzed isomerism of silyl enol ether.
![[1860-5397-7-76-i4]](/bjoc/content/inline/1860-5397-7-76-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gold-catalyzed alkylation of tetra-substituted silyl enol ether.
Scheme 4: Gold-catalyzed alkylation of tetra-substituted silyl enol ether.
In conclusion, we have developed an unprecedented alkylation method for silyl enol ethers, using a gold catalyst and ortho-alkynylbenzoic acid esters as alkylating agents. The reaction probably proceeds through the gold-induced in situ construction of a leaving group and subsequent nucleophilic attack on the silyl enol ether. Unlike ordinary leaving groups, such as halide ions, the generated leaving compound 8 acts as a base and abstracts a proton to regenerate the silyl enol ether structure. The current protocol can also be used with substrates having no hydrogen at the α-position, such as 1f. Further studies to elucidate the mechanism of this reaction and to extend the scope of synthetic utility are underway.
References
-
Brownbridge, P. Synthesis 1983, 1–28. doi:10.1055/s-1983-30204
Return to citation in text: [1] -
Brownbridge, P. Synthesis 1983, 85–104. doi:10.1055/s-1983-30234
Return to citation in text: [1] -
Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u
Return to citation in text: [1] -
Gawronski, J.; Wascinska, N.; Gajewy, J. Chem. Rev. 2008, 108, 5227–5252. doi:10.1021/cr800421c
Return to citation in text: [1] -
Chan, T. H.; Paterson, I.; Pinsonnault, J. Tetrahedron Lett. 1977, 18, 4183–4186. doi:10.1016/S0040-4039(01)83460-2
Return to citation in text: [1] -
Reetz, M. T.; Maier, W. F. Angew. Chem., Int. Ed. Engl. 1978, 17, 48–49. doi:10.1002/anie.197800481
Return to citation in text: [1] -
Paterson, I.; Fleming, I. Tetrahedron Lett. 1979, 20, 995–998. doi:10.1016/S0040-4039(01)86072-X
Return to citation in text: [1] -
Paterson, I. Tetrahedron Lett. 1979, 20, 1519–1520. doi:10.1016/S0040-4039(01)86195-5
Return to citation in text: [1] -
Takagaki, H.; Yasuda, N.; Asaoka, M.; Takei, H. Bull. Chem. Soc. Jpn. 1979, 52, 1241–1242. doi:10.1246/bcsj.52.1241
Return to citation in text: [1] -
Paterson, I.; Fleming, I. Tetrahedron Lett. 1979, 20, 2179–2182. doi:10.1016/S0040-4039(01)86295-X
Return to citation in text: [1] -
Reetz, M. T.; Hüttenhain, S.; Walz, P.; Löwe, U. Tetrahedron Lett. 1979, 20, 4971–4974. doi:10.1016/S0040-4039(01)86764-2
Return to citation in text: [1] -
Jefford, C. W.; Sledeski, A. W.; Lelandais, P.; Boukouvalas, J. Tetrahedron Lett. 1992, 33, 1855–1858. doi:10.1016/S0040-4039(00)74160-8
Return to citation in text: [1] -
Angers, P.; Canonne, P. Tetrahedron Lett. 1994, 35, 367–370. doi:10.1016/0040-4039(94)85055-0
Return to citation in text: [1] -
Nishibayashi, Y.; Wakiji, I.; Ishii, Y.; Uemura, S.; Hidai, M. J. Am. Chem. Soc. 2001, 123, 3393–3394. doi:10.1021/ja015670z
Return to citation in text: [1] -
Matsuda, I.; Wakamatsu, S.; Komori, K.-i.; Makino, T.; Itoh, K. Tetrahedron Lett. 2002, 43, 1043–1046. doi:10.1016/S0040-4039(01)02297-3
Return to citation in text: [1] -
Zhan, Z.-p.; Cai, X.-b.; Wang, S.-p.; Yu, J.-l.; Liu, H.-j.; Cui, Y.-y. J. Org. Chem. 2007, 72, 9838–9841. doi:10.1021/jo701782g
Return to citation in text: [1] -
Rubenbauer, P.; Bach, T. Tetrahedron Lett. 2008, 49, 1305–1309. doi:10.1016/j.tetlet.2007.12.092
Return to citation in text: [1] -
Asao, N.; Aikawa, H.; Tago, S.; Umetsu, K. Org. Lett. 2007, 9, 4299–4302. doi:10.1021/ol701861d
Return to citation in text: [1] -
Umetsu, K.; Asao, N. Tetrahedron Lett. 2008, 49, 7046–7049. doi:10.1016/j.tetlet.2008.09.146
Return to citation in text: [1] -
Aikawa, H.; Tago, S.; Umetsu, K.; Haginiwa, N.; Asao, N. Tetrahedron 2009, 65, 1774–1784. doi:10.1016/j.tet.2008.12.033
Return to citation in text: [1] -
Jean, M.; Renault, J.; van de Weghe, P.; Asao, N. Tetrahedron Lett. 2010, 51, 378–381. doi:10.1016/j.tetlet.2009.11.025
Return to citation in text: [1] -
Wada, M.; Nishihara, Y.; Akiba, K.-y. Tetrahedron Lett. 1984, 25, 5405–5408. doi:10.1016/S0040-4039(01)91296-1
Return to citation in text: [1] -
Magnus, P.; Mugrage, B. J. Am. Chem. Soc. 1990, 112, 462–464. doi:10.1021/ja00157a079
Return to citation in text: [1] -
Maruoka, K.; Concepcion, A. B.; Hirayama, N.; Yamamoto, H. J. Am. Chem. Soc. 1990, 112, 7422–7423. doi:10.1021/ja00176a068
Return to citation in text: [1] -
Magnus, P.; Coldham, I. J. Am. Chem. Soc. 1991, 113, 672–673. doi:10.1021/ja00002a044
Return to citation in text: [1] -
Tanino, K.; Takahashi, M.; Murayama, K.; Kuwajima, I. J. Org. Chem. 1992, 57, 7009–7010. doi:10.1021/jo00052a005
Return to citation in text: [1] -
Mikami, K.; Matsukawa, S. J. Am. Chem. Soc. 1993, 115, 7039–7040. doi:10.1021/ja00068a098
Return to citation in text: [1] -
Shoda, H.; Nakamura, T.; Tanino, K.; Kuwajima, I. Tetrahedron Lett. 1993, 34, 6281–6284. doi:10.1016/S0040-4039(00)73732-4
Return to citation in text: [1] -
Magnus, P.; Lacour, J.; Coldham, I.; Mugrage, B.; Bauta, W. B. Tetrahedron 1995, 51, 11087–11110. doi:10.1016/0040-4020(95)00696-6
Return to citation in text: [1] -
Mikami, K.; Matsukawa, S.; Nagashima, M.; Funabashi, H.; Morishima, H. Tetrahedron Lett. 1997, 38, 579–582. doi:10.1016/S0040-4039(96)02376-3
Return to citation in text: [1] -
Ishii, A.; Kojima, J.; Mikami, K. Org. Lett. 1999, 1, 2013–2016. doi:10.1021/ol990330s
Return to citation in text: [1] -
Ruck, R. T.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 2882–2883. doi:10.1021/ja025588j
Return to citation in text: [1] -
Ruck, R. T.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2003, 42, 4771–4774. doi:10.1002/anie.200351591
Return to citation in text: [1] -
Gil, R.; Eternot, M.; Guillerez, M.-G.; Collin, J. Tetrahedron 2004, 60, 3085–3090. doi:10.1016/j.tet.2004.01.082
Return to citation in text: [1] -
Hutson, G. E.; Dave, A. H.; Rawal, V. H. Org. Lett. 2007, 9, 3869–3872. doi:10.1021/ol071342d
Return to citation in text: [1] -
Mikami, K.; Kawakami, Y.; Akiyama, K.; Aikawa, K. J. Am. Chem. Soc. 2007, 129, 12950–12951. doi:10.1021/ja076539f
Return to citation in text: [1] -
Miura, K.; Taniguchi, M.; Nozaki, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1990, 31, 6391–6394. doi:10.1016/S0040-4039(00)97073-4
Return to citation in text: [1] -
Miura, K.; Takeyama, Y.; Oshima, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1991, 64, 1542–1553. doi:10.1246/bcsj.64.1542
Return to citation in text: [1] -
Miura, T.; Kiyota, K.; Kusama, H.; Iwasawa, N. Org. Lett. 2005, 7, 1445–1447. doi:10.1021/ol0473694
Return to citation in text: [1] -
Miura, T.; Kiyota, K.; Kusama, H.; Iwasawa, N. J. Organomet. Chem. 2007, 692, 562–568. doi:10.1016/j.jorganchem.2006.08.037
Return to citation in text: [1] -
Reich, H. J.; Holtan, R. C.; Bolm, C. J. Am. Chem. Soc. 1990, 112, 5609–5617. doi:10.1021/ja00170a026
Return to citation in text: [1] -
Staben, S. T.; Kennedy-Smith, J. J.; Huang, D.; Corkey, B. K.; LaLonde, R. L.; Toste, F. D. Angew. Chem., Int. Ed. 2006, 45, 5991–5994. doi:10.1002/anie.200602035
Return to citation in text: [1] -
Hotha, S.; Kashyap, S. J. Am. Chem. Soc. 2006, 128, 9620–9621. doi:10.1021/ja062425c
Return to citation in text: [1] -
Li, Y.; Yang, Y.; Yu, B. Tetrahedron Lett. 2008, 49, 3604–3608. doi:10.1016/j.tetlet.2008.04.017
Return to citation in text: [1] -
Mamidyala, S. K.; Finn, M. G. J. Org. Chem. 2009, 74, 8417–8420. doi:10.1021/jo901857x
Return to citation in text: [1] -
Shi, Y.; Roth, K. E.; Ramgren, S. D.; Blum, S. A. J. Am. Chem. Soc. 2009, 131, 18022–18023. doi:10.1021/ja9068497
Return to citation in text: [1]
| 1. | Brownbridge, P. Synthesis 1983, 1–28. doi:10.1055/s-1983-30204 |
| 2. | Brownbridge, P. Synthesis 1983, 85–104. doi:10.1055/s-1983-30234 |
| 3. | Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u |
| 4. | Gawronski, J.; Wascinska, N.; Gajewy, J. Chem. Rev. 2008, 108, 5227–5252. doi:10.1021/cr800421c |
| 37. | Miura, K.; Taniguchi, M.; Nozaki, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1990, 31, 6391–6394. doi:10.1016/S0040-4039(00)97073-4 |
| 38. | Miura, K.; Takeyama, Y.; Oshima, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1991, 64, 1542–1553. doi:10.1246/bcsj.64.1542 |
| 39. | Miura, T.; Kiyota, K.; Kusama, H.; Iwasawa, N. Org. Lett. 2005, 7, 1445–1447. doi:10.1021/ol0473694 |
| 40. | Miura, T.; Kiyota, K.; Kusama, H.; Iwasawa, N. J. Organomet. Chem. 2007, 692, 562–568. doi:10.1016/j.jorganchem.2006.08.037 |
| 22. | Wada, M.; Nishihara, Y.; Akiba, K.-y. Tetrahedron Lett. 1984, 25, 5405–5408. doi:10.1016/S0040-4039(01)91296-1 |
| 23. | Magnus, P.; Mugrage, B. J. Am. Chem. Soc. 1990, 112, 462–464. doi:10.1021/ja00157a079 |
| 24. | Maruoka, K.; Concepcion, A. B.; Hirayama, N.; Yamamoto, H. J. Am. Chem. Soc. 1990, 112, 7422–7423. doi:10.1021/ja00176a068 |
| 25. | Magnus, P.; Coldham, I. J. Am. Chem. Soc. 1991, 113, 672–673. doi:10.1021/ja00002a044 |
| 26. | Tanino, K.; Takahashi, M.; Murayama, K.; Kuwajima, I. J. Org. Chem. 1992, 57, 7009–7010. doi:10.1021/jo00052a005 |
| 27. | Mikami, K.; Matsukawa, S. J. Am. Chem. Soc. 1993, 115, 7039–7040. doi:10.1021/ja00068a098 |
| 28. | Shoda, H.; Nakamura, T.; Tanino, K.; Kuwajima, I. Tetrahedron Lett. 1993, 34, 6281–6284. doi:10.1016/S0040-4039(00)73732-4 |
| 29. | Magnus, P.; Lacour, J.; Coldham, I.; Mugrage, B.; Bauta, W. B. Tetrahedron 1995, 51, 11087–11110. doi:10.1016/0040-4020(95)00696-6 |
| 30. | Mikami, K.; Matsukawa, S.; Nagashima, M.; Funabashi, H.; Morishima, H. Tetrahedron Lett. 1997, 38, 579–582. doi:10.1016/S0040-4039(96)02376-3 |
| 31. | Ishii, A.; Kojima, J.; Mikami, K. Org. Lett. 1999, 1, 2013–2016. doi:10.1021/ol990330s |
| 32. | Ruck, R. T.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 2882–2883. doi:10.1021/ja025588j |
| 33. | Ruck, R. T.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2003, 42, 4771–4774. doi:10.1002/anie.200351591 |
| 34. | Gil, R.; Eternot, M.; Guillerez, M.-G.; Collin, J. Tetrahedron 2004, 60, 3085–3090. doi:10.1016/j.tet.2004.01.082 |
| 35. | Hutson, G. E.; Dave, A. H.; Rawal, V. H. Org. Lett. 2007, 9, 3869–3872. doi:10.1021/ol071342d |
| 36. | Mikami, K.; Kawakami, Y.; Akiyama, K.; Aikawa, K. J. Am. Chem. Soc. 2007, 129, 12950–12951. doi:10.1021/ja076539f |
| 18. | Asao, N.; Aikawa, H.; Tago, S.; Umetsu, K. Org. Lett. 2007, 9, 4299–4302. doi:10.1021/ol701861d |
| 19. | Umetsu, K.; Asao, N. Tetrahedron Lett. 2008, 49, 7046–7049. doi:10.1016/j.tetlet.2008.09.146 |
| 20. | Aikawa, H.; Tago, S.; Umetsu, K.; Haginiwa, N.; Asao, N. Tetrahedron 2009, 65, 1774–1784. doi:10.1016/j.tet.2008.12.033 |
| 21. | Jean, M.; Renault, J.; van de Weghe, P.; Asao, N. Tetrahedron Lett. 2010, 51, 378–381. doi:10.1016/j.tetlet.2009.11.025 |
| 5. | Chan, T. H.; Paterson, I.; Pinsonnault, J. Tetrahedron Lett. 1977, 18, 4183–4186. doi:10.1016/S0040-4039(01)83460-2 |
| 6. | Reetz, M. T.; Maier, W. F. Angew. Chem., Int. Ed. Engl. 1978, 17, 48–49. doi:10.1002/anie.197800481 |
| 7. | Paterson, I.; Fleming, I. Tetrahedron Lett. 1979, 20, 995–998. doi:10.1016/S0040-4039(01)86072-X |
| 8. | Paterson, I. Tetrahedron Lett. 1979, 20, 1519–1520. doi:10.1016/S0040-4039(01)86195-5 |
| 9. | Takagaki, H.; Yasuda, N.; Asaoka, M.; Takei, H. Bull. Chem. Soc. Jpn. 1979, 52, 1241–1242. doi:10.1246/bcsj.52.1241 |
| 10. | Paterson, I.; Fleming, I. Tetrahedron Lett. 1979, 20, 2179–2182. doi:10.1016/S0040-4039(01)86295-X |
| 11. | Reetz, M. T.; Hüttenhain, S.; Walz, P.; Löwe, U. Tetrahedron Lett. 1979, 20, 4971–4974. doi:10.1016/S0040-4039(01)86764-2 |
| 12. | Jefford, C. W.; Sledeski, A. W.; Lelandais, P.; Boukouvalas, J. Tetrahedron Lett. 1992, 33, 1855–1858. doi:10.1016/S0040-4039(00)74160-8 |
| 13. | Angers, P.; Canonne, P. Tetrahedron Lett. 1994, 35, 367–370. doi:10.1016/0040-4039(94)85055-0 |
| 14. | Nishibayashi, Y.; Wakiji, I.; Ishii, Y.; Uemura, S.; Hidai, M. J. Am. Chem. Soc. 2001, 123, 3393–3394. doi:10.1021/ja015670z |
| 15. | Matsuda, I.; Wakamatsu, S.; Komori, K.-i.; Makino, T.; Itoh, K. Tetrahedron Lett. 2002, 43, 1043–1046. doi:10.1016/S0040-4039(01)02297-3 |
| 16. | Zhan, Z.-p.; Cai, X.-b.; Wang, S.-p.; Yu, J.-l.; Liu, H.-j.; Cui, Y.-y. J. Org. Chem. 2007, 72, 9838–9841. doi:10.1021/jo701782g |
| 17. | Rubenbauer, P.; Bach, T. Tetrahedron Lett. 2008, 49, 1305–1309. doi:10.1016/j.tetlet.2007.12.092 |
| 43. | Hotha, S.; Kashyap, S. J. Am. Chem. Soc. 2006, 128, 9620–9621. doi:10.1021/ja062425c |
| 44. | Li, Y.; Yang, Y.; Yu, B. Tetrahedron Lett. 2008, 49, 3604–3608. doi:10.1016/j.tetlet.2008.04.017 |
| 45. | Mamidyala, S. K.; Finn, M. G. J. Org. Chem. 2009, 74, 8417–8420. doi:10.1021/jo901857x |
| 46. | Shi, Y.; Roth, K. E.; Ramgren, S. D.; Blum, S. A. J. Am. Chem. Soc. 2009, 131, 18022–18023. doi:10.1021/ja9068497 |
| 42. | Staben, S. T.; Kennedy-Smith, J. J.; Huang, D.; Corkey, B. K.; LaLonde, R. L.; Toste, F. D. Angew. Chem., Int. Ed. 2006, 45, 5991–5994. doi:10.1002/anie.200602035 |
| 41. | Reich, H. J.; Holtan, R. C.; Bolm, C. J. Am. Chem. Soc. 1990, 112, 5609–5617. doi:10.1021/ja00170a026 |
© 2011 Aikawa et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)