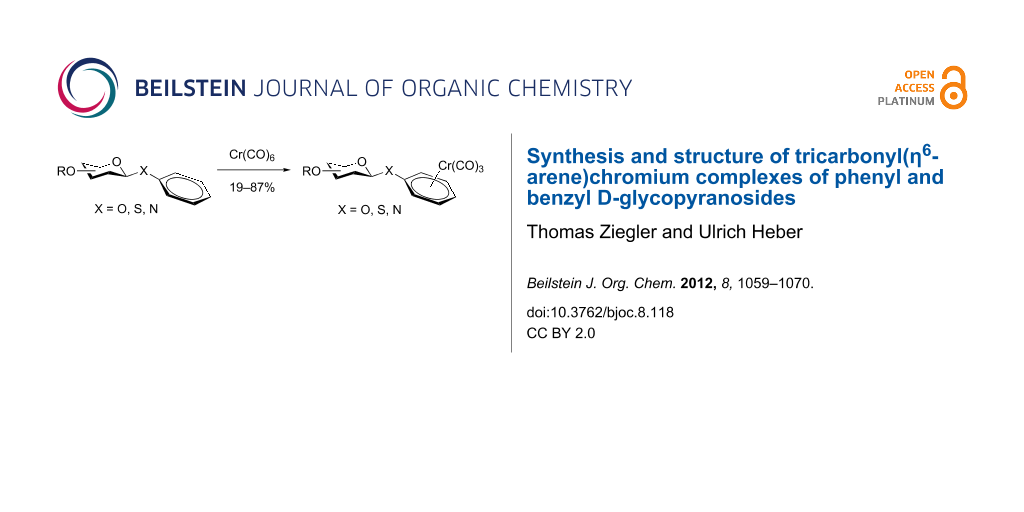
Abstract
A series of 15 glycoside-derived tricarbonyl(η6-arene)chromium complexes were prepared in 19–87% yield by heating fully acetylated or methylated aryl O-, S-, N- and C-glycosides of D-glucopyranose and D-mannopyranose with hexacarbonylchromium. All tricarbonylchromium complexes were fully characterized. The structures of nine crystalline complexes were determined by X-ray diffraction, revealing unusual intra- and intermolecular nonclassical hydrogen bonds.

Graphical Abstract
Introduction
In 1957, Fischer and Öfele published the preparation of tricarbonyl(η6-benzene)chromium, which was the first arene tricarbonylchromium complex [1]. Since then, a plethora of transition-metal complexes of arenes have been prepared, characterized and described in the literature. Among the multitude of transition-metal complexes of aromatic compounds, however, only tricarbonyl(η6-arene)chromium compounds are widely used for organic syntheses [2-4]. This is due to the fact that tricarbonyl(η6-arene)chromium complexes are relatively stable compounds, which can be easily prepared and also easily reconverted into the parent arenes. Furthermore, the tricarbonylchromium group is an electron-withdrawing substituent increasing the acidity of the aromatic protons and the electrophilicity of the aromatic ring and, thus, making the arene more susceptible towards SNAr reactions. Likewise, the benzylic and homo-benzylic positions in tricarbonyl(η6-arene)chromium complexes are more acidic and more prone to solvolysis, nucleophilic substitution and deprotonation than in the parent arenes due to the fact that the tricarbonylchromium ligand stabilizes both benzylic and homo-benzylic carbenium ions and carbanions [5,6]. Asymmetric ortho- or meta-substituted tricarbonyl(η6-arene)chromium compounds display planar chirality, which, in turn, makes these chiral complexes attractive catalysts for enantioselective reactions [6-8].
Despite the broad applications that tricarbonyl(η6-arene)chromium complexes have found in organic synthesis since their discovery in 1957, only a very few tricarbonylchromium complexes of sugar derivatives are known today. Figure 1 shows the types of such carbohydrate-derived chromium complexes that have been described in the literature so far. Complexes of type A and B were obtained from the corresponding glycopyranosides and were studied as substrates for chiral-auxiliary-directed asymmetric ortholithiation and as catalysts for enantioselective Diels–Alder reactions [9-12]. Tricarbonylchromium complexes of type C and D were obtained via benzannulation of glucal-derived pentacarbonylchromium carbenes or by reaction of alkynyl C-glycosides with pentacarbonylchromium carbenes [13].
![[1860-5397-8-118-1]](/bjoc/content/figures/1860-5397-8-118-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Known types of η6-tricarbonylchromium complexes of sugar derivatives [9-13].
Figure 1: Known types of η6-tricarbonylchromium complexes of sugar derivatives [9-13].
Further syntheses and characterizations of more examples of carbohydrate-derived tricarbonyl(η6-arene)chromium complexes are highly desirable in order to allow studies of the rich chemistry of such complexes in greater detail. In this paper we describe the synthesis and structural elucidation of a series of tricarbonyl-(η6-arene)chromium complexes of some simple phenyl and benzyl O-, N-, S- and C-glycosides.
Results and Discussion
Preparation of η6-tricarbonylchromium complexes of glycosides
In general, tricarbonyl(η6-arene)chromium compounds can be prepared in a wide variety of methods [4]. However, the following two methods are the most commonly employed ones: (a) ligand-exchange reaction between an arene and, most conveniently, either naphthalene–Cr(CO)3 complex or (MeCN)3Cr(CO)3 in which the chromium ligand is only weakly bound [14]; (b) simply heating the arene with hexacarbonylchromium in an inert solvent (Mahaffy–Pauson method) [15,16]. Method (a) has the disadvantage that the applied chromium complexes for the ligand exchange reaction are extremely sensitive toward oxidation, due to the weakly bound chromium. For method (b) high-boiling-point solvents, such as di-n-butylether, decalin or dioxane, can be used. The addition of THF to these solvents was shown to prevent the excessive sublimation of Cr(CO)6 during the formation of the arene–chromium complexes [17]. Therefore, we used method (b) for the preparation of tricarbonyl(η6-arene)chromium complexes of glycosides as follows.
One equivalent of phenyl or benzyl glycoside 1 and one equivalent of Cr(CO)6 were dissolved in di-n-butylether containing 10% THF, and the solution was stirred at 140 °C under argon and under the exclusion of light (brown glassware). After the reaction of 1 with Cr(CO)6 was complete (16–96 h), the solvent was evaporated and the crude complexes 2 were purified by chromatography under argon on silica gel with n-hexane/ethyl acetate mixtures as eluent. Crystalline chromium complexes 2 were recrystallized from ethanol, and for suitable crystals X-ray structures were determined. Table 1 summarizes the results for the preparation of the complexes 2 from simple acetylated and methylated phenyl, benzyl and 1-O-benzoyl glycosides 1. All glycosides 1a–k were prepared according to literature procedures (for details see the Supporting Information File 1).
Table 1: Synthesis of η6-tricarbonylchromium complexes 2a–k from glycosides 1a–k and Cr(CO)6 in di-n-butylether/THF 9:1 at 140 °C under Ar and exclusion of light.
| Entry | Glycoside 1 | Time | Complex 2 | Yield |
|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-118-i3.svg?max-width=637&scale=1.0)
1a |
96 h |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-118-i4.svg?max-width=637&scale=1.0)
2a |
29% |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-118-i5.svg?max-width=637&scale=1.0)
1b |
90 h |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-118-i6.svg?max-width=637&scale=1.0)
2b |
87% |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-118-i7.svg?max-width=637&scale=1.0)
1c |
80 h |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-118-i8.svg?max-width=637&scale=1.0)
2c |
29% |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-118-i9.svg?max-width=637&scale=1.0)
1d |
16 h |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-118-i10.svg?max-width=637&scale=1.0)
2d |
19% |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-118-i11.svg?max-width=637&scale=1.0)
1e |
70 h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-118-i12.svg?max-width=637&scale=1.0)
2e |
53% |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-118-i13.svg?max-width=637&scale=1.0)
1f |
42 h |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-118-i14.svg?max-width=637&scale=1.0)
2f |
47% |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-118-i15.svg?max-width=637&scale=1.0)
1g |
42 h |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-118-i16.svg?max-width=637&scale=1.0)
2g |
35% |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-118-i17.svg?max-width=637&scale=1.0)
1h |
67 h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-118-i18.svg?max-width=637&scale=1.0)
2h |
30% |
| 9a |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-118-i19.svg?max-width=637&scale=1.0)
1i |
24 h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-118-i20.svg?max-width=637&scale=1.0)
2i |
46% |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-118-i21.svg?max-width=637&scale=1.0)
1j |
24 h |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-118-i22.svg?max-width=637&scale=1.0)
2j |
49% |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-118-i23.svg?max-width=637&scale=1.0)
1k |
80 h |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-118-i24.svg?max-width=637&scale=1.0)
2k |
81% |
aAnomeric mixture α:β = 1:2.
All reactions of glycosides 1a–k with Cr(CO)6 proceeded smoothly and gave the corresponding tricarbonyl(η6-arene)chromium complexes 2a–k in medium yield. The somewhat lower yields in some cases are due to oxidative decomposition of the products during purification by column chromatography on silica gel.
The reaction times also varied significantly between 16 and 96 h. This was due to the purity of the hexacarbonyl chromium charges we purchased from several companies. In general, however, electron-donating protecting groups in the sugar moiety accelerated the formation of the chromium complexes at the aglycon (Table 1, entries 3 and 4). Treatment of the mannose orthoester 1f (Table 1, entry 6) under the standard conditions applied here resulted in the exclusive formation of tricarbonyl[(2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyloxy)-η6-benzene]chromium (2f) in 47% yield. In the glucose series it is well known that orthoesters similar to 1f rearrange to the corresponding glycosides upon heating [18]. Therefore, it is very likely that 1f isomerized to the corresponding phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside, which was then converted into complex 2f. The direct complexation of orthoester 1f is unlikely because no such chromium complex was detected. The anomeric α-configuration of 2f was proven by decomplexation of an analytical sample with iodine in CHCl3 followed by measurement of the NMR spectrum of the formed glycoside. The latter showed a CH-coupling constant at the anomeric center of 173.9 Hz, which is indicative of an α-anomer [19]. As additional proof for the anomeric configuration of 2f, we also prepared β-anomer 2g (Table 1, entry 7). For the preparation of chromium complex 2i we used a 1:2 anomeric mixture of aminoglucoside 1i, which did not change during the complexation (Table 1, entry 9).
For the anticipated synthesis of a glucose-derived tricarbonyl(η6-pyridine)chromium complex we prepared glycoside 1l by reacting acetobromoglucose with 6-tert-butyl-2-hydroxypyridine [20] under Helferich conditions (Hg(CN)2). Only the corresponding O-glycoside 1l was obtained, and no N-glycoside was formed in this Helferich glycosylation (Scheme 1). Sterically hindered 6-tert-butyl-2-hydroxypyridine was chosen as the aglycon in order to avoid complexation of hexacarbonylchromium with the basic nitrogen atom. However, all attempts to convert 1l into the corresponding tricarbonyl(η6-pyridine)chromium complex failed. Attempts to prepare a chromium complex of 1l by a ligand exchange reaction with naphthalene–Cr(CO)3 or (MeCN)3Cr(CO)3 were also unsuccessful (no further experimental details shown).
![[1860-5397-8-118-i1]](/bjoc/content/inline/1860-5397-8-118-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, we also prepared some sugar-derived tricarbonylchromium complexes of glycosides having a prochiral aglycon, i.e., an ortho-substituted phenyl or benzyl aglycon. Glycosides 1m [21] and 1q [22] were prepared according to the respective literature procedures. Glycosides 1n–p were not known in the literature and were prepared as follows: o-Tolyl 2,3,4-6-tetra-O-acetyl-β-D-glucopyranoside (1m) was first deacetylated (cat. NaOMe in MeOH) followed by treatment with pivaloyl chloride in pyridine to give 1n in 86% yield. Treatment of pentaacetylglucose and 2-tert-butylphenol with BF3-etherate in dichloromethane afforded 1o in 31% yield. Glycoside 1p was prepared by a Helferich glycosylation of o-methylbenzyl alcohol with acetobromoglucose. For further details see Supporting Information File 1. Table 2 summarizes the results of the complexation of glucosides 1m–q affording the diastereomeric tricarbonyl(η6-arene)chromium complexes 2m–q.
Table 2: Synthesis of tricarbonylchromium complexes 2m–q from glycosides 1m–q containing a prochiral aglycon and Cr(CO)6 in di-n-butylether/THF 9:1 at 140 °C under Ar in the dark.
| Entry | Glycoside 1 | Time | Complex 2 | Yield | Ratioa |
|---|---|---|---|---|---|
| 1 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-118-i25.svg?max-width=637&scale=1.0)
1m |
70 h |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-118-i26.svg?max-width=637&scale=1.0)
pR-2m ![[Graphic 25]](/bjoc/content/inline/1860-5397-8-118-i27.svg?max-width=637&scale=1.0)
pS-2m |
76% |
pR-2m:pS-2m
= 7:3b |
| 2 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-118-i28.svg?max-width=637&scale=1.0)
1n R1 = Piv, R2 = Me 1o R1 = Ac, R2 = C(CH3)3 |
no product | – | – | |
| 3 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-118-i29.svg?max-width=637&scale=1.0)
1p |
15 h |
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-118-i30.svg?max-width=637&scale=1.0)
2p |
42% |
pR-2p:pS-2p
= 1:1c |
| 4 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-118-i31.svg?max-width=637&scale=1.0)
1q |
16 h |
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-118-i32.svg?max-width=637&scale=1.0)
2q |
42% |
pR-2q:pS-2q
= 1:1c |
aDetermined by 1H NMR; bdiastereomers can be separated by crystallization; cno separation possible.
Treatment of o-tolyl glucoside 1m with hexacarbonylchromium under the standard conditions for 70 h afforded a 7:3 mixture of the diastereomeric tricarbonylchromium complexes 2m in 76% overall yield (Table 2, entry 1). Upon slow crystallization of the diastereomeric mixture from ethanol, isomer pR-2m could be obtained in pure form. From the mother liquor a small amount of pure isomer pS-2m could be isolated upon repeated recrystallization as well. The absolute configuration of the planar chiral tricarbonyl(η6-o-tolyl)chromium aglycon in both diastereomers 2m could be unambiguously assigned by X-ray crystallography. Since the two diastereomers 2m were not formed in equal amounts during complexation of glucoside 1m, we contemplated that an even higher diastereoselectivity should be obtained when sterically more demanding aglycons or sugar moieties are present during reaction with Cr(CO)6. Therefore, we also reacted glucosides 1n and 1o bearing either bulky pivaloyl groups or a tert-butyl group in the aglycon in the sugar moiety with Cr(CO)6. However, no complexation could be detected even under prolonged reaction time (Table 2, entry 2). Placing the o-tolyl group in a position more distant from the sugar part, as in glucoside 1p (Table 2, entry 3), or using o-tolyl C-glucoside 1q (Table 2, entry 4) gave 1:1 mixtures of the corresponding diastereomers 2p and 2q, respectively. A separation of the diastereomers by crystallization was not possible for complexes 2p and 2q.
The O-acetylated carbohydrate-derived tricarbonyl(η6-arene)chromium complexes prepared here can be deprotected without affecting the chromium complex, as exemplified in Scheme 2. Zemplén deacetylation of 2c afforded tricarbonyl(β-D-glucopyranosyloxy-η6-benzene)chromium (3) in quantitative yield. Glucoside 3 is a good substrate for β-glucosidases, such as almond glucosidase E.C. 3.2.1.21, and is converted into D-glucose and tricarbonyl(η6-phenol)chromium in 98% yield.
![[1860-5397-8-118-i2]](/bjoc/content/inline/1860-5397-8-118-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Deprotection of 2c and enzymatic cleavage of 3.
Scheme 2: Deprotection of 2c and enzymatic cleavage of 3.
Structures of tricarbonyl(η6-arene)chromium complexes of glycosides
In general, complexation of glycosides 1 with Cr(CO)6 to give the corresponding tricarbonyl(η6-arene)chromium compounds 2 does not significantly affect the conformation of the sugar moieties. This is evident from the 1H NMR spectra of compounds 2, which show no significant change of chemical shifts and coupling constants of the carbohydrate protons compared to those of the educts 1. As an example, Table 3 shows the chemical shifts of the protons of compounds 1a and 2a. All NMR signals of the sugar protons remain practically unchanged, whereas the protons of the benzylic methylene group and the benzene moiety appear, as was expected, at higher fields in 2a. Therefore, it can be concluded that the sugar rings remain in the 4C1 conformation. The high-field shifts of the benzylic methylene group and the aromatic moiety are in accordance with other tricarbonyl(η6-arene)chromium complexes. This is further confirmed by the X-ray structures obtained from crystalline chromium complexes.
Table 3: Chemical shifts (δ in ppm) of the protons of compounds 1a and 2a derived from the 1H NMR spectra of the compounds measured in acetone-d6.
| H1 | H2 | H3 | H4 | H5 | H6a | H6b | OCH2 | HAr | |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 4.83 d | 4.98 t | 5.25 t | 5.05 t | 3.97 m | 4.29 dd | 4.15 dd | 4.87 d 4.67 d | 7.28–7.34 m |
| 2a | 4.98 d | 4.98 t | 5.28 t | 5.06 t | 4.01 m | 4.27 dd | 4.16 dd | 4.68 d 4.45 d | 5.56–5.72 m |
For the tricarbonylchromium complexes 2a–e,j,k,m which gave suitable crystals, structures were determined by X-ray diffraction [23]. Crystals were grown for all compounds by slow crystallization of the compounds from ethanol. The conformation of the Cr(CO)3 group in relation to the aromatic ring shows some deviations compared to other tricarbonyl(η6-arene)chromium complexes. In complexes where the benzene ring carries an electron-donating substituent, the Cr(CO)3 group is preferably in an eclipsed orientation, whereas in cases where the benzene carries an electron-withdrawing substituent, a staggered conformation is commonly found [24]. Here, eclipsed conformations of the Cr(CO)3 group were found in compounds 2c and 2e, which both carry an electron-donating glycosyloxy substituent. However, compound 2k, which carries an electron-withdrawing substituent also shows an eclipsed conformation of the Cr(CO)3 group. Likewise, all compounds 2a, 2b, 2d, 2j and 2m show a staggered conformation of the Cr(CO)3 group, although they all carry an electron-donating substituent at the benzene ring. All complexes show unusual intra- and intermolecular nonclassical hydrogen bonds [25], which will be discussed in more details for each individual X-ray structure in the following. For technical details of the X-ray structures see Supporting Information File 1.
Figure 2 shows the asymmetric unit of compound 2a, which contains two slightly differently distorted molecules. Table 4 summarizes some selected atomic distances and angles of the two molecules in the asymmetric unit (2aa refers to the right molecule and 2ab to the left one in Figure 2). Most significantly, the biggest differences are found around the benzylic methylene groups (C7 and C37, respectively). Another significant feature of 2a that was not observed in any other complex 2 is the parallel face-to-face orientation of the two benzene rings. However, the distance of the two ring planes of 3.627 Å and the angle of 16° between the ring planes indicate no π-interaction of the two benzene rings.
![[1860-5397-8-118-2]](/bjoc/content/figures/1860-5397-8-118-2.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP-plot of the asymmetric unit containing two molecules of compound 2a showing 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O1 and H34 and O21 and H4.
Figure 2: ORTEP-plot of the asymmetric unit containing two molecules of compound 2a showing 30% probability e...
The most significant features in the crystal structure of 2a are the two intermolecular nonclassical hydrogen bonds between O1 and H34 and O21 and H4 (Figure 2). That these hydrogen bonds are indeed true bonds is evident from the distances of 2.659 Å for O1/H34 and 2.680 Å for O21/H4 and the angles 101.9° for C1’–O1–H34, 123.7° for C5’–O1–H34, 122.8° for C41–O21–H4 and 105.6° for C41–O21–H4. These distances and angles allow protons H4 and H34 to interact with the lone pairs of the oxygen atoms O1 and O21 of the sugar rings [26]. Most likely, the two nonclassical hydrogen bonds in 2a are the reasons why two molecules crystallize as slightly different pairs; a feature not observed in the other complexes.
Figure 3 shows the structure of compound 2b, which is the anomer of 2a. Once again a nonclassical hydrogen bond is responsible for the interaction of the molecules. Contrary to 2a, in which a nonclassical H-bond was found between the oxygen of a sugar ring and the hydrogen of a benzene ring, the H-bond forms in 2b between the oxygen of a carbonyl group (O13) and the hydrogen of an acetyl group (H26B). However, the length of this H-bond (2.212 Å) and the angles (161.6° for C≡O···H and 159.8° for C–H···O) prove the presence of the H-bond.
![[1860-5397-8-118-3]](/bjoc/content/figures/1860-5397-8-118-3.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP-plot of the asymmetric unit showing two molecules of compound 2b and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O13 and H26B.
Figure 3: ORTEP-plot of the asymmetric unit showing two molecules of compound 2b and 30% probability ellipsoi...
Figure 4 shows the structure of compound 2c. Once again the most significant feature is a nonclassical intermolecular hydrogen bond between the oxygen of a carbonyl group (O12) and the hydrogen (H6’A) at C6 of the neighboring molecule. Distance (2.628 Å) and C≡O···H and C–H···O angles (123 and 101.5°) are indicative of the H-bond. Compound 2c also shows that complexation of an aromatic aglycon with tricarbonylchromium does not have any significant influence on the sugar ring. In Table 5, the distances and angles at the anomeric center of 2c are compared to those for phenyl β-D-glucopyranoside (Ph-Glc) [27] showing that only the anomeric bond is slightly shortened upon complexation.
![[1860-5397-8-118-4]](/bjoc/content/figures/1860-5397-8-118-4.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP-plot of the asymmetrical unit showing two molecules of compound 2c and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O12 and H6’A.
Figure 4: ORTEP-plot of the asymmetrical unit showing two molecules of compound 2c and 30% probability ellips...
![[Graphic 31]](/bjoc/content/inline/1860-5397-8-118-i33.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-8-118-i34.svg?max-width=637&scale=1.0)
Figure 5 shows the structure of compound 2d, which contains a hydrogen bond between carbonyl oxygen O13 of the left molecule and hydrogen H5 of the benzene ring of the right molecule. The length of this hydrogen bond is 2.498 Å and the angles C≡O···H and C–H···O are 166.2 and 158.3°, respectively. All atomic distances and angles of the tricarbonyl(η6-benzene)chromium aglycon are almost identical to those of the acetylated counterpart 2c. However, in 2d, the carbonyl groups are in a staggered conformation (like in compound 2a) whereas in 2c the carbonyl ligands adopt an eclipsed conformation.
![[1860-5397-8-118-5]](/bjoc/content/figures/1860-5397-8-118-5.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP-plot of the asymmetric unit showing two molecules of compound 2d and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O13 and H5.
Figure 5: ORTEP-plot of the asymmetric unit showing two molecules of compound 2d and 30% probability ellipsoi...
All bond distances and angles for both the carbohydrate moiety and the tricarbonyl(η6-benzene)chromium aglycon in compound 2e (Figure 6) are within the values found for all of the other complexes. The carbonyl groups adopt an eclipsed conformation like in 2c. In the crystal, two complexed benzene rings face each other and are aligned parallel, with the tricarbonylchromium groups facing in opposite directions. One carbonyl group of each molecule is placed in between the benzene rings and, thus, allows for the formation of a “network” of three nonclassical H-bonds in the crystal. The lengths and angles of these three hydrogen bonds in 2e are within the typical range for nonclassical H-bonds [28-31] (Table 6).
![[1860-5397-8-118-6]](/bjoc/content/figures/1860-5397-8-118-6.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 6: ORTEP-plot of the asymmetrical unit showing two molecules of compound 2e and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O13 and H3, O11 and H4 and O12 and H5.
Figure 6: ORTEP-plot of the asymmetrical unit showing two molecules of compound 2e and 30% probability ellips...
Figure 7 shows the crystal structure of the S-glycoside 2j in which the molecules are stacked with all benzene rings in an almost symmetric parallel orientation on one side. However, an interaction of the tricarbonyl(η6-benzene)chromium rings can be excluded because the distance between the benzene rings is, with >3.37 Å, too long for such interactions, which only occur at shorter distances [32-34]. A nonclassical hydrogen bond is found between O13 of a carbonyl group and H22a of the acetyl group at O6 of the sugar moiety. The bond lengths and angles of this H-bond are in the expected range (2.709 Å for O···H, 138.2° for angle C–H···O and 93.5° for angle C≡O···H).
![[1860-5397-8-118-7]](/bjoc/content/figures/1860-5397-8-118-7.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 7: ORTEP-plot of the asymmetric unit showing three molecules of compound 2j and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O13 and H22a.
Figure 7: ORTEP-plot of the asymmetric unit showing three molecules of compound 2j and 30% probability ellips...
The structure of the C-glycoside 2k (Figure 8) also shows a “network” or nonclassical hydrogen bond in the crystal. The molecules group similarly to compound 2e; however, the benzene rings between two molecules (Figure 8, molecules on the right side) are not oriented in parallel. Four nonclassical hydrogen bonds form between the oxygen atoms of three carbonyl groups (O12 and O13) and the hydrogen atoms of two methyl groups (H2’ and H22a), one methylene group (H1’) and one aromatic hydrogen (H6). Table 7 summarizes the bond lengths and angles of these nonclassical H-bonds.
![[1860-5397-8-118-8]](/bjoc/content/figures/1860-5397-8-118-8.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 8: ORTEP-plot of the asymmetric unit showing three molecules of compound 2k and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O12 and H2’, O13 and H1’, O11 and H6 and O13 and H22A.
Figure 8: ORTEP-plot of the asymmetric unit showing three molecules of compound 2k and 30% probability ellips...
Figure 9 and Figure 10 show the crystal structures of the two diastereomers 2m. The isomer with (pR)-configuration of the tricarbonyl(η6-benzene)chromium group (Figure 9) shows one intramolecular nonclassical hydrogen bond between carbonyl oxygen O11 and H22B of the 2-O-acetyl group and one intermolecular H-bond between carbonyl oxygen O13 of the right molecule and H24A of the 3-O-acetyl group of the left molecule. All bond lengths and angles of these hydrogen bonds are in the expected range for nonclassical H-bonds.
![[1860-5397-8-118-9]](/bjoc/content/figures/1860-5397-8-118-9.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 9: ORTEP-plot of the asymmetric unit showing two molecules of compound pR-2m and 30% probability ellipsoids; the arrows show the nonclassical intramolecular H-bonds between O11 and H22B and the intermolecular ones between O13 and H24A.
Figure 9: ORTEP-plot of the asymmetric unit showing two molecules of compound pR-2m and 30% probability ellip...
![[1860-5397-8-118-10]](/bjoc/content/figures/1860-5397-8-118-10.png?scale=1.0&max-width=1024&background=FFFFFF)
Figure 10: ORTEP-plot of the asymmetric unit showing three molecules of compound pS-2m and 30% probability ellipsoids; the arrows show the nonclassical H-bonds between O13 and H1’.
Figure 10: ORTEP-plot of the asymmetric unit showing three molecules of compound pS-2m and 30% probability ell...
The structure of the diastereomer 2m with (pS)-configuration of the tricarbonyl(η6-benzene)chromium group (Figure 10) resembles compound 2j (Figure 7) in that the benzene rings are also in a symmetric parallel orientation. However, the distance of the benzene rings is, with 3.178 Å, shorter than in 2j but still too long for an interaction of the aromatic rings. One nonclassical hydrogen bond is found in pS-2m, between the oxygen O13 of a carbonyl group and the anomeric hydrogen H1’ of the sugar moiety. The bond lengths and angles of this H-bond are also in the expected range for nonclassical hydrogen bonds (2.699 Å for O···H, 128.7 degree for angle C–H···O and 167.3 degree for angle C≡O···H).
Conclusion
We have prepared and characterized a series of tricarbonyl(η6-benzene)chromium complexes from phenyl and benzyl O-, N-, S- and C-glycosides, and which were hitherto unknown. The X-ray diffraction of some of these glycoside-derived tricarbonylchromium complexes revealed crystal structures that contain numerous nonclassical hydrogen bonds.
Experimental
General details. All solvents were dried and distilled prior to their use. Reactions were performed under Ar and monitored by TLC on Polygram Sil G/UV silica gel plates from Machery & Nagel. Detection was effected by charring with H2SO4 (5% in EtOH) or by inspection of the TLC plates under UV light. Reactions involving Cr(CO)6 or chromium complexes were performed in brown glassware or in the dark. NMR spectra were recorded on a Bruker ARX 250 spectrometer at 250 MHz for proton spectra and 62.5 MHz for carbon spectra, on a Bruker Avance 400 spectrometer at 400 MHz for proton spectra and 100 MHz for carbon spectra, and on a Bruker AMX 600 spectrometer at 600 MHz for proton spectra and 150 MHz for carbon spectra. Tetramethylsilane was used at the internal standard. Chemical shifts δ are given in parts per million (ppm) and coupling constants in hertz (Hz). All NMR spectra were treated as first-order spectra. HRMS was performed on a Bruker Daltonics APEX 2 FT–ICR spectrometer. FAB–MS was performed on a Finnigan MAT TSQ 70 spectrometer and ionization with Xe. IR spectra were recorded with a Bruker Tensor 27 IR spectrometer. UV spectra were recorded with a Shimadzu UV 2102 PC spectrometer. Elemental analyses were performed on a Hekatech Euro 3000 CHN analyzer. Optical rotations were measured with a Perkin-Elmer Polarimeter 341. Melting points were determined with a Büchi B-540 apparatus and are uncorrected. Preparative chromatography was performed on silica gel (0.032–0.063 mm) from Machery & Nagel with different mixtures of solvents as eluent.
Starting materials. The following glycosides 1 were prepared according to literature procedures: Benzyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (1a) [35], benzyl 2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside (1b) [36], phenyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (1c) [37], phenyl 2,3,4,6-tetra-O-methyl-β-D-glucopyranoside (1d) [38], phenyl 2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside (1e) [39], 3,4,6-tri-O-acetyl-1,2-(1-phenoxy-1-ethylidene)-β-D-mannopyranose (1f) [37], phenyl 2,3,4,6-tetra-O-acetyl-β-D-mannopyranoside (1g) [40], benzoyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (1h) [41], N-phenyl-2,3,4,6-tetra-O-acetyl-D-glucopyranosylamine (1i) [42], phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (1j) [43], 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosylbenzene (1k) [44], 2-methylphenyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (1m) [21], 2-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)methylbenzene (1q) [22].
Chromium complexes: general procedure. A solution of glycoside 1 (1 mol equiv) and Cr(CO)6 (1 mol equiv) in di-n-butylether/THF 9:1 was heated in the dark under Ar at 140 °C until TLC indicated complete consumption of 1 and was then concentrated. Chromatography of the residue under Ar with n-hexane/ethyl acetate 2:1 and immediate concentration of the fractions containing the chromium complex gave 2. Crystalline complexes 2 were slowly recrystallized from ethanol. Suitable crystals were submitted to X-ray crystallographic analysis.
Tricarbonyl[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxymethyl)-η6-benzene]chromium (2a)
Treatment of 1a (3.0 g, 6.8 mmol) and Cr(CO)6 (1.50 g, 6.8 mmol) in di-n-butylether/THF (100 mL) for 96 h according to the general procedure afforded 2a (1.14 g, 29%) as yellow triclinic crystals: Mp 140–141 °C (EtOH); [α]D −11.0 (c 1.0, toluene); IR (KBr): 1952, 1895 cm−1; FAB–MS (m/z): 597 [M + Na]+, 574 [M]+, 490 [M − 3CO]+; 1H NMR (acetone-d6) δ 5.74–5.56 (m, 5H, H-aryl), 5.28 (t, 1H, 3-H), 5.06 (t, 1H, 4-H), 4.98 (t, 1H, 2-H), 4.98 (dd, J1,2 = 7.3 Hz, 1H, 1-H), 4.68, 4.45 (dd, 2H, OCH2Ph), 4.27 (dd, 1H, 6a-H), 4.16 (dd, 1H, 6b-H), 4.01 (m, 1H, 5-H), 2.06–1.94 (m, 12H, OCH3); 13C NMR (acetone-d6) δ 234.1 (Cr-CO), 170.7, 170.3, 170.0, 169.7 (O=CO), 109.3 (C1-aryl), 100.9 (C1), 95.3, 95.2, 94.0, 93.8, 93.8 (C-aryl), 72.8 (C3), 72.6 (C5), 71.9 (C2), 70.0 (OCH2), 69.3 (C4), 62.7 (C6), 20.6 (3C, OCH3), 20.5 (OCH3); Anal. calcd for C24H26CrO13 (574.5): C, 50.18; H, 4.56; found: C, 50.10; H, 4.40.
Supporting Information
| Supporting Information File 1: Experimental data. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Fischer, E. O.; Öfele, K. Chem. Ber. 1957, 90, 2532–2535. doi:10.1002/cber.19570901117
Return to citation in text: [1] -
Davies, S. G. Organotransition Metal Chemistry: Applications to Organic Synthesis; Pergamon Press: Oxford, U.K., 1992.
Return to citation in text: [1] -
Beller, M.; Bolm, C. Transition Metals for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1998.
Return to citation in text: [1] -
Kündig, E. P., Ed. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysts; Topics in Organometallic Chemistry, Vol. 7; Springer, 2004. doi:10.1007/b76615
Return to citation in text: [1] [2] -
Semmelhack, M. F.; Clark, G. R.; Garcia, J. L.; Harrison, J. J.; Thebtaranonth, Y.; Wulff, W.; Yamashita, A. Tetrahedron 1981, 37, 3957–3965. doi:10.1016/S0040-4020(01)93270-3
Return to citation in text: [1] -
Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2007, 36, 1589–1604. doi:10.1039/b606665h
Return to citation in text: [1] [2] -
Bolm, C.; Muñiz, K. Chem. Soc. Rev. 1999, 28, 51–59. doi:10.1039/a801291a
Return to citation in text: [1] -
Pape, A. R.; Kaliappan, K. P.; Kündig, E. P. Chem. Rev. 2000, 100, 2917–2940. doi:10.1021/cr9902852
Return to citation in text: [1] -
Rickard, C. E. F.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1998, 54, 1420–1424. doi:10.1107/S0108270198005976
Return to citation in text: [1] [2] -
Rickard, C. E. F.; Singh, Y.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1999, 55, 1475–1479. doi:10.1107/S0108270199007313
Return to citation in text: [1] [2] -
Han, J. W.; Son, S. U.; Chung, Y. K. J. Org. Chem. 1997, 62, 8264–8267. doi:10.1021/jo9712761
Return to citation in text: [1] [2] -
Shing, T. K. M.; Chow, H.-F.; Chung, I. H. F. Tetrahedron Lett. 1996, 37, 3713–3716. doi:10.1016/0040-4039(96)00648-X
Return to citation in text: [1] [2] -
Dötz, K.-H.; Jäkel, C.; Haase, W.-C. J. Organomet. Chem. 2001, 617–618, 119–132. doi:10.1016/S0022-328X(00)00650-1
Return to citation in text: [1] [2] -
Kündig, E. P.; Perret, C.; Spichiger, S.; Bernardinelli, G. J. Organomet. Chem. 1985, 286, 183–200. doi:10.1016/0022-328X(85)88005-0
Return to citation in text: [1] -
Mahaffy, C. A. L.; Pauson, P. L. Inorg. Synth. 1979, 19, 154–158.
Return to citation in text: [1] -
Silverthorn, W. E. Adv. Organomet. Chem. 1975, 13, 47–137. doi:10.1016/S0065-3055(08)60682-6
Return to citation in text: [1] -
Lee, Y. T.; Choi, S. Y.; Lee, S. I.; Chung, Y. K.; Kang, T. J. Tetrahedron Lett. 2006, 47, 6569–6572. doi:10.1016/j.tetlet.2006.07.011
Return to citation in text: [1] -
Ishido, Y.; Inaba, S.; Matsuno, A.; Yoshino, T.; Umezawa, H. J. Chem. Soc., Perkin Trans. 1 1977, 1382–1390. doi:10.1039/P19770001382
Return to citation in text: [1] -
Nunez, H. A.; Walker, T. E.; Fuentes, R.; O’Conner, J.; Serianni, A.; Barker, R. J. Supramol. Struct. 1977, 6, 535–550. doi:10.1002/jss.400060407
Return to citation in text: [1] -
Baur, J.; Jacobson, H.; Burger, P.; Artus, G.; Berke, H.; Dahlenburg, L. Eur. J. Inorg. Chem. 2000, 1411–1422. doi:10.1002/1099-0682(200007)2000:7<1411::AID-EJIC1411>3.0.CO;2-M
Return to citation in text: [1] -
Helferich, B.; Günther, E.; Winkler, S. Liebigs Ann. Chem. 1934, 508, 192–205. doi:10.1002/jlac.19345080117
Return to citation in text: [1] [2] -
Panigot, M. J.; Curley, R. W., Jr. J. Carbohydr. Chem. 1994, 13, 293–302. doi:10.1080/07328309408009194
Return to citation in text: [1] [2] -
The supplementary crystallographic data for this paper can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif by using the following numbers: CCDC 870827 for 2a, CCDC 870828 for 2b, CCDC 870829 for 2c, CCDC 870830 for 2d, CCDC 870831 for 2e, CCDC 870832 for 2j, CCDC 870833 for 2k, CCDC 870834 for pR-2m and CCDC 870835 for pS-2m.
Return to citation in text: [1] -
Solladié-Cavallo, A. Polyhedron 1985, 4, 901–927. doi:10.1016/S0277-5387(00)84058-9
Return to citation in text: [1] -
Taylor, R.; Kennard, O. J. Am. Chem. Soc. 1982, 104, 5063–5070. doi:10.1021/ja00383a012
Return to citation in text: [1] -
Chesnut, D. B. J. Phys. Chem. A 2000, 104, 11644–11650. doi:10.1021/jp002957u
Return to citation in text: [1] -
Jones, P. G.; Sheldrick, G. M.; Clegg, W.; Kirby, A. J.; Glenn, R. Z. Kristallogr. 1982, 160, 269–274. doi:10.1524/zkri.1982.160.3-4.269
Return to citation in text: [1] -
Brisdou, A. K.; Crossley, I. R.; Pritchard, R. G.; Warren, J. E. Acta Crystallogr., Sect. C 2003, 59, m322–m324. doi:10.1107/S0108270103013787
Return to citation in text: [1] -
Eglin, J. L.; Smith, L. T. Inorg. Chim. Acta 2001, 320, 198–202. doi:10.1016/S0020-1693(01)00475-3
Return to citation in text: [1] -
Bonifaci, C.; Ceccon, A.; Gambaro, A.; Ganis, P.; Santi, S.; Valle, G.; Venzo, A. J. Organomet. Chem. 1995, 492, 35–39. doi:10.1016/0022-328X(94)05280-O
Return to citation in text: [1] -
Braga, D.; Grepioni, F.; Biradha, K.; Pedireddi, V. R.; Desiraju, G. R. J. Am. Chem. Soc. 1995, 117, 3156–3166. doi:10.1021/ja00116a020
Return to citation in text: [1] -
Braga, D.; Grepioni, F. Organometallics 1991, 10, 2563–2569. doi:10.1021/om00054a013
Return to citation in text: [1] -
Braga, D.; Dyson, P. J.; Grepioni, F.; Johnson, B. F. G. Chem. Rev. 1994, 94, 1585–1620. doi:10.1021/cr00030a006
Return to citation in text: [1] -
Gambaro, A.; Ganis, P.; Manoli, F.; Polimeno, A.; Santi, S.; Venzo, A. J. Organomet. Chem. 1999, 583, 126–130. doi:10.1016/S0022-328X(99)00120-5
Return to citation in text: [1] -
Steffan, W.; Vogel, C.; Kristen, H. Carbohydr. Res. 1990, 204, 109–120. doi:10.1016/0008-6215(90)84026-Q
Return to citation in text: [1] -
Utamura, T.; Kuromatsu, K.; Suwa, K.; Koizumi, K.; Shingu, T. Chem. Pharm. Bull. 1986, 34, 2341–2353. doi:10.1248/cpb.34.2341
Return to citation in text: [1] -
Dess, D.; Kleine, H. P.; Weinberg, D. V.; Kaufmann, R. J.; Sidhu, R. S. Synthesis 1981, 883–885. doi:10.1055/s-1981-29631
Return to citation in text: [1] [2] -
Stanssens, D.; De Keukeleire, D.; Vandewalle, M. Tetrahedron: Asymmetry 1990, 1, 547–560. doi:10.1016/S0957-4166(00)80546-7
Return to citation in text: [1] -
Audichya, T. D.; Ingle, T. R.; Bose, J. L. Indian J. Chem. 1973, 11, 704–705.
Return to citation in text: [1] -
Montgomery, E. M.; Richtmyer, N. K.; Hudson, C. S. J. Am. Chem. Soc. 1942, 64, 690–694. doi:10.1021/ja01255a060
Return to citation in text: [1] -
Pfander, H.; Läderach, M. Carbohydr. Res. 1982, 99, 175–179. doi:10.1016/S0008-6215(00)81907-2
Return to citation in text: [1] -
Baker, J. W. J. Chem. Soc. 1928, 1583–1593.
Return to citation in text: [1] -
Dasgupta, F.; Garegg, P. J. Acta Chem. Scand. 1989, 43, 471–475. doi:10.3891/acta.chem.scand.43-0471
Return to citation in text: [1] -
Hur, C. D.; Bonner, W. A. J. Am. Chem. Soc. 1945, 67, 1972–1977. doi:10.1021/ja01227a033
Return to citation in text: [1]
| 39. | Audichya, T. D.; Ingle, T. R.; Bose, J. L. Indian J. Chem. 1973, 11, 704–705. |
| 37. | Dess, D.; Kleine, H. P.; Weinberg, D. V.; Kaufmann, R. J.; Sidhu, R. S. Synthesis 1981, 883–885. doi:10.1055/s-1981-29631 |
| 40. | Montgomery, E. M.; Richtmyer, N. K.; Hudson, C. S. J. Am. Chem. Soc. 1942, 64, 690–694. doi:10.1021/ja01255a060 |
| 1. | Fischer, E. O.; Öfele, K. Chem. Ber. 1957, 90, 2532–2535. doi:10.1002/cber.19570901117 |
| 9. | Rickard, C. E. F.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1998, 54, 1420–1424. doi:10.1107/S0108270198005976 |
| 10. | Rickard, C. E. F.; Singh, Y.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1999, 55, 1475–1479. doi:10.1107/S0108270199007313 |
| 11. | Han, J. W.; Son, S. U.; Chung, Y. K. J. Org. Chem. 1997, 62, 8264–8267. doi:10.1021/jo9712761 |
| 12. | Shing, T. K. M.; Chow, H.-F.; Chung, I. H. F. Tetrahedron Lett. 1996, 37, 3713–3716. doi:10.1016/0040-4039(96)00648-X |
| 21. | Helferich, B.; Günther, E.; Winkler, S. Liebigs Ann. Chem. 1934, 508, 192–205. doi:10.1002/jlac.19345080117 |
| 6. | Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2007, 36, 1589–1604. doi:10.1039/b606665h |
| 7. | Bolm, C.; Muñiz, K. Chem. Soc. Rev. 1999, 28, 51–59. doi:10.1039/a801291a |
| 8. | Pape, A. R.; Kaliappan, K. P.; Kündig, E. P. Chem. Rev. 2000, 100, 2917–2940. doi:10.1021/cr9902852 |
| 22. | Panigot, M. J.; Curley, R. W., Jr. J. Carbohydr. Chem. 1994, 13, 293–302. doi:10.1080/07328309408009194 |
| 5. | Semmelhack, M. F.; Clark, G. R.; Garcia, J. L.; Harrison, J. J.; Thebtaranonth, Y.; Wulff, W.; Yamashita, A. Tetrahedron 1981, 37, 3957–3965. doi:10.1016/S0040-4020(01)93270-3 |
| 6. | Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2007, 36, 1589–1604. doi:10.1039/b606665h |
| 19. | Nunez, H. A.; Walker, T. E.; Fuentes, R.; O’Conner, J.; Serianni, A.; Barker, R. J. Supramol. Struct. 1977, 6, 535–550. doi:10.1002/jss.400060407 |
| 21. | Helferich, B.; Günther, E.; Winkler, S. Liebigs Ann. Chem. 1934, 508, 192–205. doi:10.1002/jlac.19345080117 |
| 2. | Davies, S. G. Organotransition Metal Chemistry: Applications to Organic Synthesis; Pergamon Press: Oxford, U.K., 1992. |
| 3. | Beller, M.; Bolm, C. Transition Metals for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1998. |
| 4. | Kündig, E. P., Ed. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysts; Topics in Organometallic Chemistry, Vol. 7; Springer, 2004. doi:10.1007/b76615 |
| 20. | Baur, J.; Jacobson, H.; Burger, P.; Artus, G.; Berke, H.; Dahlenburg, L. Eur. J. Inorg. Chem. 2000, 1411–1422. doi:10.1002/1099-0682(200007)2000:7<1411::AID-EJIC1411>3.0.CO;2-M |
| 22. | Panigot, M. J.; Curley, R. W., Jr. J. Carbohydr. Chem. 1994, 13, 293–302. doi:10.1080/07328309408009194 |
| 14. | Kündig, E. P.; Perret, C.; Spichiger, S.; Bernardinelli, G. J. Organomet. Chem. 1985, 286, 183–200. doi:10.1016/0022-328X(85)88005-0 |
| 17. | Lee, Y. T.; Choi, S. Y.; Lee, S. I.; Chung, Y. K.; Kang, T. J. Tetrahedron Lett. 2006, 47, 6569–6572. doi:10.1016/j.tetlet.2006.07.011 |
| 43. | Dasgupta, F.; Garegg, P. J. Acta Chem. Scand. 1989, 43, 471–475. doi:10.3891/acta.chem.scand.43-0471 |
| 4. | Kündig, E. P., Ed. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysts; Topics in Organometallic Chemistry, Vol. 7; Springer, 2004. doi:10.1007/b76615 |
| 18. | Ishido, Y.; Inaba, S.; Matsuno, A.; Yoshino, T.; Umezawa, H. J. Chem. Soc., Perkin Trans. 1 1977, 1382–1390. doi:10.1039/P19770001382 |
| 44. | Hur, C. D.; Bonner, W. A. J. Am. Chem. Soc. 1945, 67, 1972–1977. doi:10.1021/ja01227a033 |
| 9. | Rickard, C. E. F.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1998, 54, 1420–1424. doi:10.1107/S0108270198005976 |
| 10. | Rickard, C. E. F.; Singh, Y.; Woodgate, P. D.; Zhao, Z. Acta Crystallogr., Sect. C 1999, 55, 1475–1479. doi:10.1107/S0108270199007313 |
| 11. | Han, J. W.; Son, S. U.; Chung, Y. K. J. Org. Chem. 1997, 62, 8264–8267. doi:10.1021/jo9712761 |
| 12. | Shing, T. K. M.; Chow, H.-F.; Chung, I. H. F. Tetrahedron Lett. 1996, 37, 3713–3716. doi:10.1016/0040-4039(96)00648-X |
| 13. | Dötz, K.-H.; Jäkel, C.; Haase, W.-C. J. Organomet. Chem. 2001, 617–618, 119–132. doi:10.1016/S0022-328X(00)00650-1 |
| 41. | Pfander, H.; Läderach, M. Carbohydr. Res. 1982, 99, 175–179. doi:10.1016/S0008-6215(00)81907-2 |
| 13. | Dötz, K.-H.; Jäkel, C.; Haase, W.-C. J. Organomet. Chem. 2001, 617–618, 119–132. doi:10.1016/S0022-328X(00)00650-1 |
| 15. | Mahaffy, C. A. L.; Pauson, P. L. Inorg. Synth. 1979, 19, 154–158. |
| 16. | Silverthorn, W. E. Adv. Organomet. Chem. 1975, 13, 47–137. doi:10.1016/S0065-3055(08)60682-6 |
| 25. | Taylor, R.; Kennard, O. J. Am. Chem. Soc. 1982, 104, 5063–5070. doi:10.1021/ja00383a012 |
| 23. | The supplementary crystallographic data for this paper can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif by using the following numbers: CCDC 870827 for 2a, CCDC 870828 for 2b, CCDC 870829 for 2c, CCDC 870830 for 2d, CCDC 870831 for 2e, CCDC 870832 for 2j, CCDC 870833 for 2k, CCDC 870834 for pR-2m and CCDC 870835 for pS-2m. |
| 24. | Solladié-Cavallo, A. Polyhedron 1985, 4, 901–927. doi:10.1016/S0277-5387(00)84058-9 |
| 37. | Dess, D.; Kleine, H. P.; Weinberg, D. V.; Kaufmann, R. J.; Sidhu, R. S. Synthesis 1981, 883–885. doi:10.1055/s-1981-29631 |
| 38. | Stanssens, D.; De Keukeleire, D.; Vandewalle, M. Tetrahedron: Asymmetry 1990, 1, 547–560. doi:10.1016/S0957-4166(00)80546-7 |
| 35. | Steffan, W.; Vogel, C.; Kristen, H. Carbohydr. Res. 1990, 204, 109–120. doi:10.1016/0008-6215(90)84026-Q |
| 36. | Utamura, T.; Kuromatsu, K.; Suwa, K.; Koizumi, K.; Shingu, T. Chem. Pharm. Bull. 1986, 34, 2341–2353. doi:10.1248/cpb.34.2341 |
| 28. | Brisdou, A. K.; Crossley, I. R.; Pritchard, R. G.; Warren, J. E. Acta Crystallogr., Sect. C 2003, 59, m322–m324. doi:10.1107/S0108270103013787 |
| 29. | Eglin, J. L.; Smith, L. T. Inorg. Chim. Acta 2001, 320, 198–202. doi:10.1016/S0020-1693(01)00475-3 |
| 30. | Bonifaci, C.; Ceccon, A.; Gambaro, A.; Ganis, P.; Santi, S.; Valle, G.; Venzo, A. J. Organomet. Chem. 1995, 492, 35–39. doi:10.1016/0022-328X(94)05280-O |
| 31. | Braga, D.; Grepioni, F.; Biradha, K.; Pedireddi, V. R.; Desiraju, G. R. J. Am. Chem. Soc. 1995, 117, 3156–3166. doi:10.1021/ja00116a020 |
| 32. | Braga, D.; Grepioni, F. Organometallics 1991, 10, 2563–2569. doi:10.1021/om00054a013 |
| 33. | Braga, D.; Dyson, P. J.; Grepioni, F.; Johnson, B. F. G. Chem. Rev. 1994, 94, 1585–1620. doi:10.1021/cr00030a006 |
| 34. | Gambaro, A.; Ganis, P.; Manoli, F.; Polimeno, A.; Santi, S.; Venzo, A. J. Organomet. Chem. 1999, 583, 126–130. doi:10.1016/S0022-328X(99)00120-5 |
| 26. | Chesnut, D. B. J. Phys. Chem. A 2000, 104, 11644–11650. doi:10.1021/jp002957u |
| 27. | Jones, P. G.; Sheldrick, G. M.; Clegg, W.; Kirby, A. J.; Glenn, R. Z. Kristallogr. 1982, 160, 269–274. doi:10.1524/zkri.1982.160.3-4.269 |
© 2012 Ziegler and Heber; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)