Abstract
Backbone modification is a common chemical tool to control the conformation of linear peptides and to explore potentially useful effects on their biochemical and biophysical properties. The thioamide, ψ[CS-NH], group is a nearly isosteric structural mimic of the amide (peptide) functionality. In this paper, we describe the solution synthesis, chemical characterization, preferred conformation, and membrane and biological activities of three, carefully selected, peptide analogues of the lipopeptaibiotic [Leu11-OMe] trichogin GA IV. In each analogue, a single thioamide replacement was incorporated. Sequence positions near the N-terminus, at the center, and near the C-terminus were investigated. Our results indicate that (i) a thioamide linkage is well tolerated in the overall helical conformation of the [Leu11-OMe] lipopeptide analogue and (ii) this backbone modification is compatible with the preservation of its typical membrane leakage and antibiotic properties, although somewhat attenuated.

Graphical Abstract
Introduction
Since their first incorporation into peptides [1], backbone amide surrogates have attracted remarkable attention from organic and medicinal chemists. Not only may these modifications impart to a peptide an increased resistance to enzymatic hydrolysis as well as higher receptor affinity and specificity, but they may also influence its preferred secondary structure.
A ψ[CS-NH] thioamide group is one of the closest mimics of an amide (peptide) linkage. However, it exhibits significantly different chemical and physical properties, some of which are of great potential interest to peptide chemists [2-44]. Among these properties, we highlight the following: (i) The thioamide NH group is more acidic than that of its oxygenated counterpart and consequently it is a stronger H-bonding donor. (ii) Its cis/trans isomerization can be phototriggered by irradiation at about 260 nm [absorption maximum of the -C(=S)NH- π→π* electronic transition]. (iii) It may act as a minimalist, effective quencher for any type of protein and nonprotein fluorophores.
Recently, based on the aforementioned characteristics of the ψ[CS-NH] thioamide bond, we started a program aimed at exploring how thiopeptide groups may affect folding and the related biophysical/biochemical activities of the membrane-active peptaibiotics [45]. This class of compounds represents a subject of long-standing, relevant, interest to our research group. In this paper, we describe our results on the syntheses by solution methods and characterizations of three thioamide-containing [Leu11-OMe] analogues of the short helical lipopeptaibiotic trichogin GA IV [46-50] (Figure 1). We already found that the replacement of the native C-terminal 1,2-amino alcohol leucinol (Lol) by the corresponding α-amino methyl ester (Leu-OMe) alters only slightly the biophysical and biological properties of trichogin GA IV. Conversely, we showed that the Nα-blocking fatty acyl moiety plays a major role in its membrane permeability and antibiotic activity.
![[1860-5397-8-129-1]](/bjoc/content/figures/1860-5397-8-129-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: List of primary structures and abbreviations for the peptides studied in this work. The [Leu11-OMe] trichogin GA IV is termed tric-OMe and its thionated derivatives at position 2, position 5, and position 9 are termed ψ[CS-NH]2, ψ[CS-NH]5, and ψ[CS-NH]9, respectively.
Figure 1: List of primary structures and abbreviations for the peptides studied in this work. The [Leu11-OMe]...
In each analogue, the single ψ[CS-NH] group was strategically incorporated either at an internal position of the amino-acid sequence, ψ[CS-NH]5, or near each of the two ends, ψ[CS-NH]2 and ψ[CS-NH]9. Moreover, the preferred conformations of these analogues were investigated by circular dichroism (CD), FT-IR absorption, and NMR. Finally, we carried out fluorescence leakage experiments in model membranes and antibacterial assays on a large set of both Gram-positive and Gram-negative strains.
Results and Discussion
Peptide synthesis
The total syntheses of the three monothionated ψ[CS-NH] trichogin GA IV analogues at positions 2, 5, and 9, respectively, were accomplished by using the solution-phase method (Schemes 1–3). Both step-by-step and segment condensation approaches were utilized. The former strategy was used in the preparation of Boc-Gly5-Gly-Leu-Aib-Gly-Ile-Leu11-OMe (segment 5–11, for the synthesis of ψ[CS-NH]2), Boc-Gly9-Ile-Leu11-OMe (segment 9–11), and n-Oct-Aib1-Gly-Leu-Aib4-Ot-Bu (segment 1–4, for the synthesis of ψ[CS-NH]5), and n-Oct-Aib1-Gly-Leu-Aib-Gly-Gly-Leu-Aib8-Ot-Bu (segment 1–8, for the synthesis of ψ[CS-NH]9). The latter strategy, which permits a faster preparation of multiple analogues, afforded (i) ψ[CS-NH]2 by condensation of the thionated 1–4 and the 5–11 segments; (ii) ψ[CS-NH]5 by condensation of the thionated 5–8 and the 9–11 segments followed by condensation of the 1–4 and the resulting, thionated, 5–11 segments; and (iii) ψ[CS-NH]9 by condensation of the 1–8 and the thionated 9–11 segments.
![[1860-5397-8-129-i1]](/bjoc/content/inline/1860-5397-8-129-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of ψ[CS-NH]2. 1: Coupling in the presence of EDC/HOBt. 2: Deprotection by using TFA/DCM. 3: Deprotection by catalytic hydrogenation with Pd/C. 4: Thionation with Lawesson's reagent in THF. 5: Coupling with n-Oct-OH in the presence of EDC/HOBt. 6: Saponification with NaOH/MeOH. 7: Coupling in the presence of EDC/HOAt.
Scheme 1: Synthesis of ψ[CS-NH]2. 1: Coupling in the presence of EDC/HOBt. 2: Deprotection by using TFA/DCM. ...
![[1860-5397-8-129-i2]](/bjoc/content/inline/1860-5397-8-129-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of ψ[CS-NH]5. 1: Coupling in the presence of EDC/HOBt. 2: Deprotection by using TFA/DCM. 3: Deprotection by catalytic hydrogenation with Pd/C. 4: Coupling with Fmoc-OSu in 1,4-dioxane. 5: Thionation with Lawesson's reagent in THF. 6: Coupling with n-Oct-OH in the presence of EDC/HOBt. 7: Coupling in the presence of EDC/HOAt. 8: Deprotection with DEA in DCM.
Scheme 2: Synthesis of ψ[CS-NH]5. 1: Coupling in the presence of EDC/HOBt. 2: Deprotection by using TFA/DCM. ...
![[1860-5397-8-129-i3]](/bjoc/content/inline/1860-5397-8-129-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of ψ[CS-NH]9. 1: Deprotection by catalytic hydrogenation with Pd/C. 2: Coupling with n-Oct-OH in the presence of EDC/HOBt. 3: Coupling in the presence of EDC/HOBt. 4: Deprotection by using TFA/DCM. 5: Thionation with Lawesson's reagent in THF. 6: Coupling in the presence of EDC/HOAt.
Scheme 3: Synthesis of ψ[CS-NH]9. 1: Deprotection by catalytic hydrogenation with Pd/C. 2: Coupling with n-Oc...
For the difficult coupling steps, in particular those involving the segment condensations, the N-[3-(dimethylamino)-propyl]-N'-ethylcarbodiimide (EDC)/7-aza-1-hydroxy-1,2,3-benzotriazole (HOAt) [51] activating method was used, while the EDC/1-hydroxy-1,2,3-benzotriazole (HOBt) [52] method turned out to be appropriate for the formation of the other peptide bonds. The yields of the segment condensation reactions were good (75–83%). An important feature, which makes trichogin GA IV a versatile synthetic platform, is the presence of as many as seven achiral (Aib or Gly) residues spread across its sequence, which allows one to design suitable segments each with an achiral residue at its C-terminus, thus reducing dramatically the usually considerable risk of epimerization during the coupling reactions.
For the best choice of the step involving treatment with the Lawesson's reagent [53], we decided to select the longest segment of our sequences, which, in principle, could permit complete regioselectivity between the peptide bond to be thionated and the other peptide bonds (in addition to the known regioselectivity versus the urethane and ester bonds) [53-56]. In this reaction, regioselectivity is known to be heavily governed by the steric hindrance of the two residues around the peptide bond. Therefore, we synthesized Boc-Gly2ψ[CS-NH]Leu-Aib4-OMe (for the preparation of ψ[CS-NH]2), Fmoc-Gly5ψ[CS-NH]Gly-Leu-Aib8-Ot-Bu (for the preparation of ψ[CS-NH]5), and Boc-Gly9ψ[CS-NH]Ile-Leu11-OMe (for the preparation of ψ[CS-NH]9). Thionation of a peptide as short as a dipeptide is not recommended because a further synthetic step at the C-terminus of this compound would not take place satisfactorily, owing to the easy formation of a poorly reactive 1,3-thiazolidin-5-one. All thionation steps were conducted by use of the Lawesson's reagent under mild conditions (in tetrahydrofuran at room temperature) [53-56]. The only synthetic problem was identified in the preparation of the Fmoc-Gly5ψ[CS-NH]Gly-Leu-Aib8-Ot-Bu, in which case the thionation selectivity on the Gly-Gly peptide bond was incomplete. Indeed, a limited production (about 10%) of the isomeric Fmoc-Gly-Gly6ψ[CS-NH]Leu-Aib-Ot-Bu, with its slightly more hindered, thionated Gly-Leu peptide bond, was observed. In this case, the desired monothionated peptide was separated from its isomer by means of flash chromatography.
For details of the synthetic procedures and characterizations of these analogues (and the synthetic precursor segments as well), see the Experimental section and Supporting Information File 1. Figure 2 gives the RP-HPLC profiles of the three monothionated trichogin GA IV analogues.
![[1860-5397-8-129-2]](/bjoc/content/figures/1860-5397-8-129-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: RP-HPLC profiles obtained for [Leu11-OMe] trichogin GA IV (tric-OMe) and its ψ[CS-NH]2, ψ[CS-NH]5, and ψ[CS-NH]9 analogues.
Figure 2: RP-HPLC profiles obtained for [Leu11-OMe] trichogin GA IV (tric-OMe) and its ψ[CS-NH]2, ψ[CS-NH]5, ...
Conformational analysis
A detailed analysis of the spectroscopic properties and conformational preferences of the three monothionated trichogin GA IV analogues synthesized in this work was performed by using CD, FT-IR absorption, and 2D NMR investigations in different solvents.
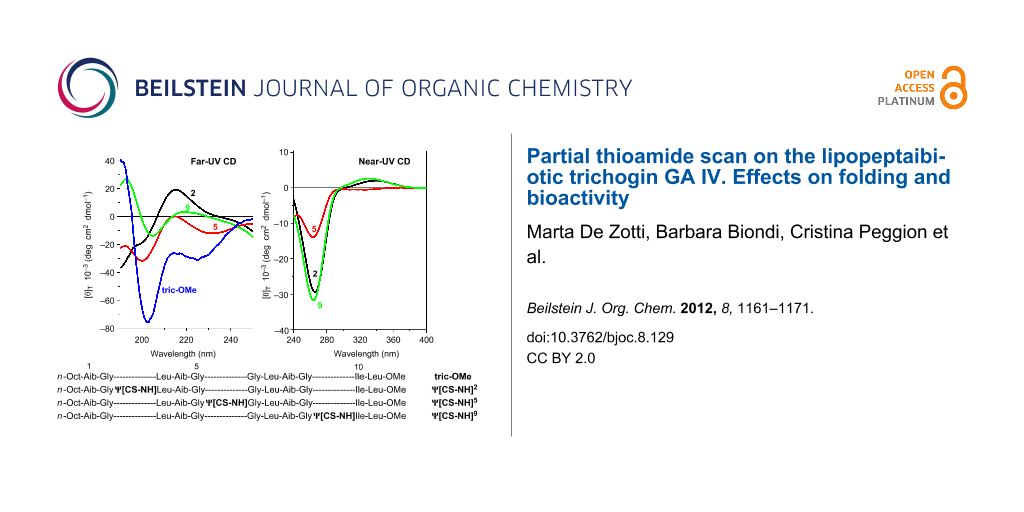
The far- (250–195 nm) and near-UV (400–250 nm) CD spectra of the three peptides, recorded in 2,2,2-trifluoroethanol (TFE) solution, are illustrated in Figure 3. In the near-UV region, in which the peptide chromophore is known not to absorb, both ψ[CS-NH]2 and ψ[CS-NH]9 display two well-separated Cotton effects at about 335 and 265 nm. The longer wavelength band is positive and weak, while the shorter wavelength band is negative and much more intense. Interestingly, the positive band is absent and the negative band is much weaker in ψ[CS-NH]5. The spectral positions of these Cotton effects correspond to a very intense maximum (265 nm) and an extremely weak maximum (320 nm) found in the corresponding region of the UV absorption spectra (not shown) of our peptides in the same solvent. We assign the two bands to the π→π* and n→π* transitions, respectively, of thioamide chromophore [29,30,32-36,40,41,43]. We attribute the overall much less intense CD spectrum of ψ[CS-NH]5 in this region primarily to a thioamide chromophore positioned in this peptide between the two achiral Gly5 and Gly6 residues, i.e., it is significantly far apart from the nearest chiral center (the Leu7 α-carbon).
![[1860-5397-8-129-3]](/bjoc/content/figures/1860-5397-8-129-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Far-UV (panel I) and near-UV (panel II) CD spectra of [Leu11-OMe] trichogin GA IV (tric-OMe) and its ψ[CS-NH]2 (2), ψ[CS-NH]5 (5), and ψ[CS-NH]9 (9) analogues in TFE solution. Peptide concentration: 1 mM.
Figure 3: Far-UV (panel I) and near-UV (panel II) CD spectra of [Leu11-OMe] trichogin GA IV (tric-OMe) and it...
In the far-UV region the CD spectra of all three analogues show multiple (negative/positive) Cotton effects of weak and medium intensities. Here, the bands (shoulders) of ψ[CS-NH]9 are the weakest. In our view, it is risky to attempt to extract any conclusive conformational information from the CD properties of these three isomeric peptides between 195 and 250 nm because, in addition to the expected Cotton effects of the peptide chromophores [57,58], the thioamide chromophore [34,35] is also known to contribute heavily in this spectral region. However, we can safely state that the shape of the CD spectrum of the [Leu11-OMe] trichogin GA IV prototypical peptide [50,59] is most closely paralleled by that of its ψ[CS-NH]5 analogue, although the former is significantly more intense. This finding is not surprising in view of the above-mentioned low influence of the thioamide chromophore on the spectrum of this monothionated compound. It may also suggest that the overall conformation of the lipopeptaibiotic is disturbed by this specific oxygen-to-sulfur exchange only to a limited extent.
The preferred conformations of the three monothionated [Leu11-OMe] trichogin GA IV analogues were more safely analyzed by means of FT-IR absorption and 2D NMR. In this study, we used CDCl3, a solvent of low polarity. In the 3D-structurally informative 3550–3200 cm−1 region at 1.0 mM concentration, the FT-IR spectra of [Leu11-OMe] trichogin GA IV and its analogues are similar and dominated by a broad and intense absorption at 3316–3328 cm−1 (Figure 4), assigned to the N–H stretching mode of the largely prevailing H-bonded peptide groups [60,61]. Additional, very weak bands are visible in the 3450–3400 cm−1 region, mostly attributed to free (solvated) peptide NH groups.
![[1860-5397-8-129-4]](/bjoc/content/figures/1860-5397-8-129-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: FT-IR absorption spectra (3550–3200 cm−1 region) in CDCl3 solution of [Leu11-OMe] trichogin GA IV (tric-OMe) and its ψ[CS-NH]2 (2), ψ[CS-NH]5 (5), and ψ[CS-NH]9 (9) analogues. Peptide concentration: 1 mM.
Figure 4: FT-IR absorption spectra (3550–3200 cm−1 region) in CDCl3 solution of [Leu11-OMe] trichogin GA IV (...
A nonnegligible dilution effect is observed in the spectra of the four peptides between 1.0 and 0.1 mM concentration (not shown). We ascribe this difference to the well-known tendency of trichogin GA IV and its analogues to self-associate above 1.0 mM concentration in CDCl3 solution. We interpret the spectra at 0.1 mM concentration as arising almost exclusively from intramolecular C=O(S)···H–N interactions. It is clear that under these experimental conditions the preferred conformation of [Leu11-OMe] trichogin GA IV [46-50] and its three analogues, all rich in the helix-supporting Aib residues, is highly folded and largely stabilized by intramolecular H-bonds. In the amide I (C=O stretching) region, the maximum of the absorption band of the four peptides is located between 1662 and 1656 cm−1 (not shown), close to the canonical positions of this band [61,62] in α- and 310-helical peptides [63].
A 2D NMR investigation was performed in CD3CN solution for all of the ψ[CS-NH]-containing [Leu11-OMe] trichogin GA IV analogues. The proton resonances were fully assigned following the Wüthrich procedure [64]. Regions of the ROESY spectrum acquired for ψ[CS-NH]9 are shown in Figures 5–7. The patterns of NH(i)–NH(i+1) connectivities (e.g., Figure 5) strongly suggest the onset of an overall helical structure for all analogues. More relevant 3D-structural information was extracted from the fingerprint regions of the ROESY spectra (e.g., Figure 6 and Figure 7), which show evidence for dα(β),N (i, i+2), (i, i+3), and (i, i+4) medium-range connectivities, diagnostic of the presence of mixed 310-/α-helical conformations.
![[1860-5397-8-129-5]](/bjoc/content/figures/1860-5397-8-129-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Region of the amide NH protons in the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solution, 298 K).
Figure 5: Region of the amide NH protons in the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solut...
![[1860-5397-8-129-6]](/bjoc/content/figures/1860-5397-8-129-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fingerprint region of the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solution, 298 K). The CαHi → NHi+2, and CαHi → NHi+3 cross peaks are highlighted in color.
Figure 6: Fingerprint region of the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solution, 298 K)....
![[1860-5397-8-129-7]](/bjoc/content/figures/1860-5397-8-129-7.png?scale=2.3&max-width=1024&background=FFFFFF)
Figure 7: Fingerprint region of the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solution, 298 K). The CβHi → NHi+2, CβHi → NHi+3, and CβHi → NHi+4 cross peaks are highlighted in color.
Figure 7: Fingerprint region of the H/H-ROESY spectrum of ψ[CS-NH]9 (400 MHz, 1 mM in CD3CN solution, 298 K)....
The conformational properties of ψ[CS-NH]9 were further investigated by simulated annealing (SA) and restrained molecular dynamics (MD) calculations. A total of 69 interproton distance restraints, derived from the related ROESY spectrum (Table S-I, Supporting Information File 1), were used in the SA protocol. The structures possessing violations to the NOE restraints lower than 0.5 Å were selected out of the 150 generated structures. The superposition of the 18 structures with a total energy <144 kcal/mol is shown in Figure S-I, Supporting Information File 1. All these structures converge to a well-defined, right-handed, mixed 310-/α-helical conformation throughout the sequence with a backbone average pairwise root-mean-square deviation of 0.49 ± 0.18 Å (deviations from idealized geometry and mean energies for the 18 lowest energy structures are listed in Table S-I, Supporting Information File 1). Due to heavily overlapped ROE signals, the helical structures originated from the MD calculations appear not to superimpose perfectly in correspondence with the first three residues of the sequence. Even with this experimental limit, the residues belonging to the central and C-terminal parts of the sequence showed values of torsion angles corresponding to those characteristic of right-handed helices (Table S-II, Supporting Information File 1). The helical structures are stabilized by intramolecular H-bonds throughout the sequence. H-bonds both of the α- (i←i+4) and 310- (i←i+3) helical types are present, although with a large predominance of the latter type (mean distance: 1.81 ± 0.17 Å; mean angle: 27.4 ± 3.7°), at the N-terminus. Conversely, only the former type (mean distance: 1.53 ± 0.02 Å; mean angle: 27.3 ± 2.9°) was detected at the C-terminus.
In conclusion, the MD calculations based on the restraints derived from the ROESY spectrum of ψ[CS-NH]9 reveal the marked preference of this analogue for a mixed α-/310- helical conformation, with a clear α-helical character at the C-terminus on which the thioamide bond is present. The lowest-energy 3D structure, shown in Figure 8, exhibits the slightly amphipathic character typical of the native peptide, with the poorly hydrophilic Gly residues aligned on the same face of the helix.
![[1860-5397-8-129-8]](/bjoc/content/figures/1860-5397-8-129-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Ribbon representation of the lowest energy (138.7 kcal/mol) 3D structure obtained for ψ[CS-NH]9. All amino acid side chains are shown. The yellow atom on Gly9 refers to sulfur.
Figure 8: Ribbon representation of the lowest energy (138.7 kcal/mol) 3D structure obtained for ψ[CS-NH]9. Al...
Membrane and biological activities
The membrane permeability properties of [Leu11-OMe] trichogin GA IV and its monothionated analogues were tested by measuring the induced leakage of 5(6)-carboxyfluorescein (CF) entrapped in small unilamellar vesicles (SUVs) [65]. For this investigation, the overall negatively charged 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)/1,2-dioleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DOPG) model membrane was exploited. The extent of permeation induced by the three analogues is similar, although 30–40% lower than that of the parent peptaibiotic.
The antimicrobial activities were assessed on a large set of Gram-positive and Gram-negative bacterial strains. We found that [Leu11-OMe] trichogin GA IV is active on the Gram-positive bacteria Staphylococcus aureus and Streptococcus pyrogenes, whereas the spectrum of action of its three analogues is more restricted, as they are not active on the latter strain. The activities of the analogues on S. aureus are comparable, but reduced by about 40% with respect to that of the parent peptaibiotic.
Conclusion
In this work, we performed a partial thioamide scan of the short lipopeptaibiotic [Leu11-OMe] trichogin GA IV by synthesizing and studying three thioamide-containing analogues. The syntheses were accomplished in solution by using a combination of the step-by-step and segment condensation methods. Appropriate tri- and tetrapeptide segments were monothionated by treating the all-amide precursors with the Lawesson's reagent. Our CD, FT-IR absorption, and NMR conformational investigations show that all analogues maintain the mixed 310-/α-helical structure and the self-association propensity of the native lipopeptide. They also preserve, at least to a great extent, its well-established capability to interact with model phospholipid membranes and to exhibit activity against Gram-positive bacterial strains.
Experimental
All reagents and solvents, of analytical grade and purchased from commercial sources, were used without further purification. Melting points were measured by means of a capillary tube immersed in an oil bath (Tottoli apparatus, Büchi) and are uncorrected. Optical rotations [α]D20 (given in units of 10−1 deg·cm2·g−1) were measured at 20 °C on a Perkin-Elmer PE241 polarimeter, with a 1 dm path length cell, at the D-wavelength of sodium (589 nm). The concentration of each compound (c) is given in mg/cL. Mass spectroscopy (electrospray ionization, ESIMS) was performed by using a PerSeptive Biosystem Mariner instrument (Framingham, MA). Analytical TLC and preparative column chromatography were performed on Kieselgel F 254 and Kieselgel 60 (0.040–0.063 mm) (Merck), respectively. The retention factor (Rf) values were determined by using three solvent mixtures as eluants: Rf1: chloroform/ethanol 9:1; Rf2: 1-butanol/acetic acid/water 3:1:1; Rf3: toluene/ethanol 7:1.
General procedure for thionation of peptides
To a solution of the peptide (1 mmol) in anhydrous tetrahydrofuran under nitrogen atmosphere, the Lawesson's reagent (0.6 mmol) was added. The reaction mixture was stirred at rt overnight. After removal of the solvent under reduced pressure, the crude product was purified by flash chromatography with a mixture of ethyl acetate and petroleum ether as eluant.
Peptides Boc-Ile-Leu-OMe [66], Z-Leu-Aib-OMe [67], Z-Leu-Aib-Ot-Bu [68], Boc-Gly-Leu-Aib-OMe [69], Boc-Gly-Ile-Leu-OMe [66], Boc-Leu-Aib-Gly-Ile-Leu-OMe [66], Boc-Gly-Gly-Leu-Aib-Gly-Ile-Leu-OMe [66], Z-Gly-Gly-Leu-Aib-Ot-Bu [59], and Z-Aib-Gly-Leu-Aib-Gly-Gly-Leu-Aib-Ot-Bu [59] are known compounds. Chemical characterizations and selected NMR spectra for the other peptides reported in Schemes 1–3 are given in Supporting Information File 1.
Circular dichroism
The CD spectra were measured on a Jasco (Tokyo, Japan) model J-715 spectropolarimeter equipped with a Haake thermostat (Thermo Fisher Scientific, Waltham, MA). Baselines were corrected by subtracting the solvent contribution. Fused quartz cells of 1.0 mm and 10.0 mm path lengths (Hellma, Mühlheim, Germany) were used. The values are expressed in terms of [θ]T, the total molar ellipticity (deg·cm2·dmol−1). Spectrograde TFE and 99.9% MeOH (Acros Organic, Geel, Belgium) were employed as solvents.
FT-IR absorption
The FT-IR absorption spectra were recorded at 293 K by using a Perkin-Elmer model 1720X FT-IR spectrophotometer, nitrogen-flushed, equipped with a sample-shuttle device, at 2 cm−1 nominal resolution, averaging 100 scans. Solvent (baseline) spectra were recorded under the same conditions. For spectral elaboration, the software SPECTRACALC provided by Galactic (Salem, MA) was employed. Cells with path lengths of 1.0 and 10 mm (with CaF2 windows) were used. Spectrograde deuterated chloroform (99.8%, d) was purchased from Merck (Darmstadt, Germany).
Nuclear magnetic resonance
All 1H NMR experiments were performed on a Bruker AVANCE DMX-600, DRX-400 or AC200 spectrometer operating at 600, 400 or 200 MHz, respectively, using the TOPSPIN software package. Splitting patterns are abbreviated as (s) singlet, (d) doublet, (t) triplet, (q) quartet, (m) multiplet. The homonuclear 2D spectra of the three monothionated [Leu11-OMe] trichogin GA IV analogues were recorded at 298 K, with CD3CN as solvent. All spectra were acquired by recording 512 experiments, each one consisting of 64–80 scans and 2000 data points. The spin systems of protein amino acid residues were identified by using standard DQF-COSY [70] and CLEAN-TOCSY [71,72] spectra. In the latter case, the spin-lock pulse sequence was 70 ms long. A ROESY experiment was exploited for sequence-specific assignment. The mixing time of the ROESY experiment acquired for ψ[CS-NH]9 (peptide concentration: 1.00 mM in CD3CN) and used for interproton distance determination was 200 ms. Interproton distances were obtained by integration of the ROESY spectra using SPARKY 3.111. The calibration was based on the average of the integration values of the cross peaks due to the interactions between the sequential amide protons, set to a distance of 2.80 Å. When peaks could not be integrated because of partial overlap, a distance corresponding to the maximum limit of detection of the experiment (4.0 Å) was assigned to the corresponding proton pair.
MD calculations were carried out using the SA protocol of the XPLOR-NIH 2.9.6 program [73]. For distances involving equivalent or non-stereo-assigned protons, an r−6 averaging was used. The MD calculations involved a minimization stage of 100 cycles, followed by SA and refinement stages. The SA consisted of 30 ps of dynamics at 1,500 K (10,000 cycles, in 3 fs steps) and of 30 ps of cooling from 1,500 to 100 K in 50 K decrements (15,000 cycles, in 2 fs steps). The SA procedure, in which the weights of ROE and nonbonded terms were gradually increased, was followed by 200 cycles of energy minimization. In the SA refinement stage, the system was cooled from 1,000 to 100 K in 50 K decrements (20,000 cycles, in 1 fs steps). Finally, the calculations were completed with 200 cycles of energy minimization by using a NOE force constant of 50 kcal/mol. The generated structures were visualized with the MOLMOL [74] (version 2K.2) program.
Small unilamellar vesicles (SUVs) preparation
DOPE and DOPG were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). The preparation of SUVs was performed in a similar way as described in reference [59].
Leakage from lipid vesicles
The peptide-induced leakage from SUVs was measured at 293 K by using the CF-entrapped vesicle technique [65] and a Perkin Elmer model MPF-66 spectrofluorimeter. SUVs were prepared as described above. The phospholipid concentration was kept constant (0.06 mM), and increasing [peptide]/[lipid] molar ratios (R−1) were obtained by adding aliquots of each non-hydrosoluble, monothionated peptide (or of trichogin GA IV, used as reference compound) as a MeOH solution, keeping the final MeOH concentration below 5% by volume. After rapid and vigorous stirring, the time course of fluorescence change corresponding to CF escape was recorded at 520 nm (6 nm band pass) with λexc 488 nm (3 nm band pass). The percentage of released CF at time t was determined as 100 × (Ft−F0)/(FT−F0), with F0 = fluorescence intensity of vesicles in the absence of peptide, Ft = fluorescence intensity of vesicles at time t in the presence of peptide, and FT = total fluorescence intensity determined by disrupting the vesicles by the addition of 50 μL of a Triton X-100 solution. The experiments were stopped at 20 min.
Antibacterial activity
Peptide antibacterial activity was tested against Gram-positive and Gram-negative bacteria by the standardized disk diffusion Bauer–Kirby method [75] using the Müller–Hinton culture medium at pH 7.2–7.4 as recommended by the National Committee for Clinical Laboratory Standards [76]. The antibacterial activity assay was performed in a similar way to that described in reference [77]. The activity of peptides was tested against clinical isolates of bacteria and reference bacterial strains: Staphylococcus aureus American Type Culture Collection strains (ATCC) 25923, Streptococcus pyogenes ATCC 19615, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Klebsiella pneumoniae ATCC 13883, Salmonella entereditis 13076, and Proteus mirabilis ATCC 10975. The well-known antibiotics bacitracin and tetracyclin (10 μg/disk) were used as controls.
Supporting Information
| Supporting Information File 1: Chemical characterization data for the peptides reported in Schemes 1–3. | ||
| Format: PDF | Size: 247.1 KB | Download |
References
-
Spatola, A. F. Peptide backbone modifications: Structure–activity analysis of peptides containing amide bond surrogates. In Chemistry and Biochemistry of Amino Acids, Peptides and Proteins; Weinstein, B., Ed.; Dekker: New York, 1983; pp 267–357.
Return to citation in text: [1] -
La Cour, T. F. M.; Hansen, H. A. S.; Clausen, K.; Lawesson, S.-O. Int. J. Pept. Protein Res. 1983, 22, 509–512. doi:10.1111/j.1399-3011.1983.tb02122.x
Return to citation in text: [1] -
La Cour, T. F. M. Int. J. Pept. Protein Res. 1987, 30, 564–571. doi:10.1111/j.1399-3011.1987.tb03366.x
Return to citation in text: [1] -
Jensen, O. E.; Lawesson, S.-O.; Bardi, R.; Piazzesi, A. M.; Toniolo, C. Tetrahedron 1985, 41, 5595–5606. doi:10.1016/S0040-4020(01)91361-4
Return to citation in text: [1] -
Bardi, R.; Piazzesi, A. M.; Toniolo, C.; Jensen, O. E.; Omar, R. S.; Senning, A. Biopolymers 1988, 27, 747–761. doi:10.1002/bip.360270504
Return to citation in text: [1] -
Bardi, R.; Piazzesi, A. M.; Toniolo, C.; Jensen, O. E.; Andersen, T. P.; Senning, A. Tetrahedron 1988, 44, 761–769. doi:10.1016/S0040-4020(01)86115-9
Return to citation in text: [1] -
Lehmann, J.; Linden, A.; Heimgartner, H. Tetrahedron 1998, 54, 8721–8736. doi:10.1016/S0040-4020(98)00506-7
Return to citation in text: [1] -
Pradeille, N.; Zerbe, O.; Möhle, K.; Linden, A.; Heimgartner, H. Chem. Biodiversity 2005, 2, 1127–1152. doi:10.1002/cbdv.200590084
Return to citation in text: [1] -
Stamm, S.; Heimgartner, H. Helv. Chim. Acta 2006, 89, 1841–1855. doi:10.1002/hlca.200690178
Return to citation in text: [1] -
Budzowski, A.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2008, 91, 1471–1488. doi:10.1002/hlca.200890160
Return to citation in text: [1] -
Mawad, N.; Pour, F. G.-S.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2010, 93, 2326–2346. doi:10.1002/hlca.201000318
Return to citation in text: [1] -
Liu, Z.; Mehta, S. J.; Lee, K.-S.; Grossman, B.; Qu, H.; Gu, X.; Nichol, G. S.; Hruby, V. J. J. Org. Chem. 2012, 77, 1289–1300. doi:10.1021/jo201753q
Return to citation in text: [1] -
Hua, G.; Fuller, A. L.; Bühl, M.; Slawin, A. M. Z.; Woollins, J. D. Eur. J. Org. Chem. 2011, 3067–3073. doi:10.1002/ejoc.201100226
Return to citation in text: [1] -
Xie, J.; Okano, A.; Pierce, J. G.; James, R. C.; Stamm, S.; Crane, C. M.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 1284–1297. doi:10.1021/ja209937s
Return to citation in text: [1] -
Chen, P.; Qu, J. J. Org. Chem. 2011, 76, 2994–3004. doi:10.1021/jo200403g
Return to citation in text: [1] -
Tran, T. T.; Burgess, A. W.; Treutlein, H.; Perich, J. J. Pept. Res. 2001, 58, 67–78. doi:10.1034/j.1399-3011.2001.00898.x
Return to citation in text: [1] -
Wood, P. A.; Pidcock, E.; Allen, F. H. Acta Crystallogr., Sect. B 2008, 64, 491–496. doi:10.1107/S0108768108015437
Return to citation in text: [1] -
Tran, T. T.; Treutlein, H.; Burgess, A. W. Protein Eng., Des. Sel. 2006, 19, 401–408. doi:10.1093/protein/gzl024
Return to citation in text: [1] -
Tran, T. T.; Zeng, J.; Treutlein, H.; Burgess, A. W. J. Am. Chem. Soc. 2002, 124, 5222–5230. doi:10.1021/ja011916o
Return to citation in text: [1] -
Artis, D. R.; Lipton, M. A. J. Am. Chem. Soc. 1998, 120, 12200–12206. doi:10.1021/ja982398t
Return to citation in text: [1] -
Alemán, C. J. Phys. Chem. A 2001, 105, 6717–6723. doi:10.1021/jp010198p
Return to citation in text: [1] -
Choudhary, A.; Gandla, D.; Krow, G. R.; Raines, R. T. J. Am. Chem. Soc. 2009, 131, 7244–7246. doi:10.1021/ja901188y
Return to citation in text: [1] -
Choudhary, A.; Raines, R. T. ChemBioChem 2011, 12, 1801–1807. doi:10.1002/cbic.201100272
Return to citation in text: [1] -
Cervetto, V.; Pfister, R.; Kolano, C.; Bregy, H.; Heimgartner, H.; Helbing, J. Chem.–Eur. J. 2007, 13, 9004–9011. doi:10.1002/chem.200700396
Return to citation in text: [1] -
Cervetto, V.; Pfister, R.; Helbing, J. J. Phys. Chem. B 2008, 112, 3540–3544. doi:10.1021/jp710611n
Return to citation in text: [1] -
Cervetto, V.; Hamm, P.; Helbing, J. J. Phys. Chem. B 2008, 112, 8398–8405. doi:10.1021/jp801166q
Return to citation in text: [1] -
Bregy, H.; Heimgartner, H.; Helbing, J. J. Phys. Chem. B 2009, 113, 1756–1762. doi:10.1021/jp8089402
Return to citation in text: [1] -
Huang, Y.; Cong, Z.; Yang, L.; Dong, S. J. Pept. Sci. 2008, 14, 1062–1068. doi:10.1002/psc.1042
Return to citation in text: [1] -
Wildemann, D.; Schiene-Fischer, C.; Aumüller, T.; Bachmann, A.; Kiefhaber, T.; Lücke, C.; Fischer, G. J. Am. Chem. Soc. 2007, 129, 4910–4918. doi:10.1021/ja069048o
Return to citation in text: [1] [2] -
Zhao, J.; Micheau, J.-C.; Vargas, C.; Schiene-Fischer, C. Chem.–Eur. J. 2004, 10, 6093–6101. doi:10.1002/chem.200400400
Return to citation in text: [1] [2] -
Bailey, P. D.; Boyd, C. A. R.; Collier, I. D.; Kellett, G. L.; Meredith, D.; Morgan, K. M.; Pettecrew, R.; Price, R. A. Org. Biomol. Chem. 2005, 3, 4038–4039. doi:10.1039/B513274F
Return to citation in text: [1] -
Miwa, J. H.; Pallivathucal, L.; Gowda, S.; Lee, K. E. Org. Lett. 2002, 4, 4655–4657. doi:10.1021/ol027056d
Return to citation in text: [1] [2] -
Miwa, J. H.; Patel, A. K.; Vivatrat, N.; Popek, S. M.; Meyer, A. M. Org. Lett. 2001, 3, 3373–3375. doi:10.1021/ol0166092
Return to citation in text: [1] [2] -
Kajtár, M.; Hollósi, M.; Kajtár, J.; Majer, Z.; Kövér, K. E. Tetrahedron 1986, 42, 3931–3942. doi:10.1016/S0040-4020(01)87548-7
Return to citation in text: [1] [2] [3] -
Hollósi, M.; Kollát, E.; Kajtár, J.; Kajtár, M.; Fasman, G. D. Biopolymers 1990, 30, 1061–1072. doi:10.1002/bip.360301107
Return to citation in text: [1] [2] [3] -
Reiner, A.; Wildemann, D.; Fischer, G.; Kiefhaber, T. J. Am. Chem. Soc. 2008, 130, 8079–8084. doi:10.1021/ja8015044
Return to citation in text: [1] [2] -
Goldberg, J. M.; Batjargal, S.; Petersson, E. J. J. Am. Chem. Soc. 2010, 132, 14718–14720. doi:10.1021/ja1044924
Return to citation in text: [1] -
Goldberg, J. M.; Wissner, R. F.; Klein, A. M.; Petersson, E. J. Chem. Commun. 2012, 48, 1550–1552. doi:10.1039/c1cc14708k
Return to citation in text: [1] -
Satzger, H.; Root, C.; Gilch, P.; Zinth, W.; Wildemann, D.; Fischer, G. J. Phys. Chem. B 2005, 109, 4770–4775. doi:10.1021/jp045151t
Return to citation in text: [1] -
Sifferlen, T.; Rueping, M.; Gademann, K.; Jaun, B.; Seebach, D. Helv. Chim. Acta 1999, 82, 2067–2093. doi:10.1002/(SICI)1522-2675(19991215)82:12<2067::AID-HLCA2067>3.0.CO;2-5
Return to citation in text: [1] [2] -
Bachmann, A.; Wildemann, D.; Praetorius, F.; Fischer, G.; Kiefhaber, T. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3952–3957. doi:10.1073/pnas.1012668108
Return to citation in text: [1] [2] -
Mierke, D. F.; Geyer, A.; Kessler, H. Int. J. Pept. Protein Res. 1994, 44, 325–331. doi:10.1111/j.1399-3011.1994.tb01016.x
Return to citation in text: [1] -
Otani, Y.; Hori, T.; Kawahata, M.; Yamagouchi, K.; Ohwada, T. Tetrahedron 2012, 68, 4418–4428. doi:10.1016/j.tet.2012.01.018
Return to citation in text: [1] [2] -
Banala, S.; Süssmuth, R. D. ChemBioChem 2010, 11, 1335–1337. doi:10.1002/cbic.201000266
Return to citation in text: [1] -
Toniolo, C.; Brückner, H. Peptaibiotics: Fungal Peptides Containing α-Dialkyl α-Amino Acids; Wiley-VCH: Weinheim, Germany, 2009.
Return to citation in text: [1] -
Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C.; Epand, R. F.; Epand, R. M. Cell. Mol. Life Sci. 2001, 58, 1179–1188. doi:10.1007/PL00000932
Return to citation in text: [1] [2] -
Peggion, C.; Formaggio, F.; Crisma, M.; Epand, R. F.; Epand, R. M.; Toniolo, C. J. Pept. Sci. 2003, 9, 679–689. doi:10.1002/psc.500
Return to citation in text: [1] [2] -
Venanzi, M.; Gatto, E.; Bocchinfuso, G.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2006, 7, 43–45. doi:10.1002/cbic.200500271
Return to citation in text: [1] [2] -
Venanzi, M.; Bocchinfuso, G.; Gatto, E.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2009, 10, 91–97. doi:10.1002/cbic.200800617
Return to citation in text: [1] [2] -
De Zotti, M.; Biondi, B.; Formaggio, F.; Toniolo, C.; Stella, L.; Park, Y.; Hahm, K.-S. J. Pept. Sci. 2009, 15, 615–619. doi:10.1002/psc.1135
Return to citation in text: [1] [2] [3] -
Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082
Return to citation in text: [1] -
König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319
Return to citation in text: [1] -
Clausen, K.; Thorsen, M.; Lawesson, S.-O. Tetrahedron 1981, 37, 3635–3639. doi:10.1016/S0040-4020(01)98892-1
Return to citation in text: [1] [2] [3] -
Lajoie, G.; Lépine, F.; Maziak, L.; Belleau, B. Tetrahedron Lett. 1983, 24, 3815–3818. doi:10.1016/S0040-4039(00)94282-5
Return to citation in text: [1] [2] -
Cava, M. P.; Levinson, M. I. Tetrahedron 1985, 41, 5061–5087. doi:10.1016/S0040-4020(01)96753-5
Return to citation in text: [1] [2] -
Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210–5278. doi:10.1021/cr040650b
Return to citation in text: [1] [2] -
Goodman, M.; Toniolo, C. Biopolymers 1968, 6, 1673–1689. doi:10.1002/bip.1968.360061202
Return to citation in text: [1] -
Sreerama, N.; Woody, R. W. Circular dichroism of peptides and proteins. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N.; Nakanishi, K.; Woody, R. W., Eds.; Wiley-VCH: New York, 2000; pp 601–620.
Return to citation in text: [1] -
De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.-S.; Toniolo, C. Org. Biomol. Chem. 2012, 10, 1285–1299. doi:10.1039/c1ob06178j
Return to citation in text: [1] [2] [3] [4] -
Palumbo, M.; da Rin, S.; Bonora, G. M.; Toniolo, C. Makromol. Chem. 1976, 177, 1477–1492. doi:10.1002/macp.1976.021770519
Return to citation in text: [1] -
Moretto, V.; Crisma, M.; Bonora, G. M.; Toniolo, C.; Balaram, H.; Balaram, P. Macromolecules 1989, 22, 2939–2944. doi:10.1021/ma00197a010
Return to citation in text: [1] [2] -
Kennedy, D. F.; Crisma, M.; Toniolo, C.; Chapman, D. Biochemistry 1991, 30, 6541–6548. doi:10.1021/bi00240a026
Return to citation in text: [1] -
Toniolo, C.; Benedetti, E. Trends Biochem. Sci. 1991, 16, 350–353. doi:10.1016/0968-0004(91)90142-I
Return to citation in text: [1] -
Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New York, 1986.
Return to citation in text: [1] -
Auvin-Guette, C.; Rebuffat, S.; Prigent, Y.; Bodo, B. J. Am. Chem. Soc. 1992, 114, 2170–2174. doi:10.1021/ja00032a035
Return to citation in text: [1] [2] -
Gurunath, R.; Balaram, P. Biopolymers 1995, 35, 21–29. doi:10.1002/bip.360350104
Return to citation in text: [1] [2] [3] [4] -
Iida, A.; Yoshimatsu, S.; Sanekata, M.; Fujita, T. Chem. Pharm. Bull. 1990, 38, 2997–3003. doi:10.1248/cpb.38.2997
Return to citation in text: [1] -
Brückner, H.; Koza, A. Amino Acids 2003, 24, 311–323. doi:10.1007/s00726-002-0401-x
Return to citation in text: [1] -
Nagaraj, R.; Balaram, P. Tetrahedron 1981, 37, 1263–1270. doi:10.1016/S0040-4020(01)92061-7
Return to citation in text: [1] -
Rance, M.; Sørensen, O. W.; Bodenhausen, G.; Wagner, G.; Ernst, R. R.; Wüthrich, K. Biochem. Biophys. Res. Commun. 1983, 117, 479–485. doi:10.1016/0006-291X(83)91225-1
Return to citation in text: [1] -
Bax, A.; Davis, D. G. J. Magn. Reson. 1985, 65, 355–360. doi:10.1016/0022-2364(85)90018-6
Return to citation in text: [1] -
Griesinger, C.; Otting, G.; Wüthrich, K.; Ernst, R. R. J. Am. Chem. Soc. 1988, 110, 7870–7872. doi:10.1021/ja00231a044
Return to citation in text: [1] -
Schwieters, C. D.; Kuszewski, J. J.; Tjandra, N.; Clore, G. M. J. Magn. Reson. 2003, 160, 65–73. doi:10.1016/S1090-7807(02)00014-9
(based on X-PLOR 3.851 by A. T. Brunger).
Return to citation in text: [1] -
Koradi, R.; Billeter, M.; Wüthrich, K. J. Mol. Graphics 1996, 14, 51–55. doi:10.1016/0263-7855(96)00009-4
Return to citation in text: [1] -
Bauer, A. W.; Kirby, W. M.; Sherris, J. C.; Turck, M. Am. J. Clin. Pathol. 1966, 45, 493–496.
Return to citation in text: [1] -
Performance Standards for antimicrobial Disk Susceptibility Tests; Approved Standard–Sixth Edition; CLSI: Wayne, PA, 1997; CLSI document M02–A06.
Return to citation in text: [1] -
Oancea, S.; Hilma, G.; Peggion, C.; Formaggio, F.; Toniolo, C. Chem. Biodiversity 2008, 5, 681–692. doi:10.1002/cbdv.200890065
Return to citation in text: [1]
| 59. | De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.-S.; Toniolo, C. Org. Biomol. Chem. 2012, 10, 1285–1299. doi:10.1039/c1ob06178j |
| 70. | Rance, M.; Sørensen, O. W.; Bodenhausen, G.; Wagner, G.; Ernst, R. R.; Wüthrich, K. Biochem. Biophys. Res. Commun. 1983, 117, 479–485. doi:10.1016/0006-291X(83)91225-1 |
| 71. | Bax, A.; Davis, D. G. J. Magn. Reson. 1985, 65, 355–360. doi:10.1016/0022-2364(85)90018-6 |
| 72. | Griesinger, C.; Otting, G.; Wüthrich, K.; Ernst, R. R. J. Am. Chem. Soc. 1988, 110, 7870–7872. doi:10.1021/ja00231a044 |
| 1. | Spatola, A. F. Peptide backbone modifications: Structure–activity analysis of peptides containing amide bond surrogates. In Chemistry and Biochemistry of Amino Acids, Peptides and Proteins; Weinstein, B., Ed.; Dekker: New York, 1983; pp 267–357. |
| 51. | Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082 |
| 46. | Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C.; Epand, R. F.; Epand, R. M. Cell. Mol. Life Sci. 2001, 58, 1179–1188. doi:10.1007/PL00000932 |
| 47. | Peggion, C.; Formaggio, F.; Crisma, M.; Epand, R. F.; Epand, R. M.; Toniolo, C. J. Pept. Sci. 2003, 9, 679–689. doi:10.1002/psc.500 |
| 48. | Venanzi, M.; Gatto, E.; Bocchinfuso, G.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2006, 7, 43–45. doi:10.1002/cbic.200500271 |
| 49. | Venanzi, M.; Bocchinfuso, G.; Gatto, E.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2009, 10, 91–97. doi:10.1002/cbic.200800617 |
| 50. | De Zotti, M.; Biondi, B.; Formaggio, F.; Toniolo, C.; Stella, L.; Park, Y.; Hahm, K.-S. J. Pept. Sci. 2009, 15, 615–619. doi:10.1002/psc.1135 |
| 77. | Oancea, S.; Hilma, G.; Peggion, C.; Formaggio, F.; Toniolo, C. Chem. Biodiversity 2008, 5, 681–692. doi:10.1002/cbdv.200890065 |
| 46. | Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C.; Epand, R. F.; Epand, R. M. Cell. Mol. Life Sci. 2001, 58, 1179–1188. doi:10.1007/PL00000932 |
| 47. | Peggion, C.; Formaggio, F.; Crisma, M.; Epand, R. F.; Epand, R. M.; Toniolo, C. J. Pept. Sci. 2003, 9, 679–689. doi:10.1002/psc.500 |
| 48. | Venanzi, M.; Gatto, E.; Bocchinfuso, G.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2006, 7, 43–45. doi:10.1002/cbic.200500271 |
| 49. | Venanzi, M.; Bocchinfuso, G.; Gatto, E.; Palleschi, A.; Stella, L.; Formaggio, F.; Toniolo, C. ChemBioChem 2009, 10, 91–97. doi:10.1002/cbic.200800617 |
| 50. | De Zotti, M.; Biondi, B.; Formaggio, F.; Toniolo, C.; Stella, L.; Park, Y.; Hahm, K.-S. J. Pept. Sci. 2009, 15, 615–619. doi:10.1002/psc.1135 |
| 61. | Moretto, V.; Crisma, M.; Bonora, G. M.; Toniolo, C.; Balaram, H.; Balaram, P. Macromolecules 1989, 22, 2939–2944. doi:10.1021/ma00197a010 |
| 62. | Kennedy, D. F.; Crisma, M.; Toniolo, C.; Chapman, D. Biochemistry 1991, 30, 6541–6548. doi:10.1021/bi00240a026 |
| 45. | Toniolo, C.; Brückner, H. Peptaibiotics: Fungal Peptides Containing α-Dialkyl α-Amino Acids; Wiley-VCH: Weinheim, Germany, 2009. |
| 50. | De Zotti, M.; Biondi, B.; Formaggio, F.; Toniolo, C.; Stella, L.; Park, Y.; Hahm, K.-S. J. Pept. Sci. 2009, 15, 615–619. doi:10.1002/psc.1135 |
| 59. | De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.-S.; Toniolo, C. Org. Biomol. Chem. 2012, 10, 1285–1299. doi:10.1039/c1ob06178j |
| 75. | Bauer, A. W.; Kirby, W. M.; Sherris, J. C.; Turck, M. Am. J. Clin. Pathol. 1966, 45, 493–496. |
| 2. | La Cour, T. F. M.; Hansen, H. A. S.; Clausen, K.; Lawesson, S.-O. Int. J. Pept. Protein Res. 1983, 22, 509–512. doi:10.1111/j.1399-3011.1983.tb02122.x |
| 3. | La Cour, T. F. M. Int. J. Pept. Protein Res. 1987, 30, 564–571. doi:10.1111/j.1399-3011.1987.tb03366.x |
| 4. | Jensen, O. E.; Lawesson, S.-O.; Bardi, R.; Piazzesi, A. M.; Toniolo, C. Tetrahedron 1985, 41, 5595–5606. doi:10.1016/S0040-4020(01)91361-4 |
| 5. | Bardi, R.; Piazzesi, A. M.; Toniolo, C.; Jensen, O. E.; Omar, R. S.; Senning, A. Biopolymers 1988, 27, 747–761. doi:10.1002/bip.360270504 |
| 6. | Bardi, R.; Piazzesi, A. M.; Toniolo, C.; Jensen, O. E.; Andersen, T. P.; Senning, A. Tetrahedron 1988, 44, 761–769. doi:10.1016/S0040-4020(01)86115-9 |
| 7. | Lehmann, J.; Linden, A.; Heimgartner, H. Tetrahedron 1998, 54, 8721–8736. doi:10.1016/S0040-4020(98)00506-7 |
| 8. | Pradeille, N.; Zerbe, O.; Möhle, K.; Linden, A.; Heimgartner, H. Chem. Biodiversity 2005, 2, 1127–1152. doi:10.1002/cbdv.200590084 |
| 9. | Stamm, S.; Heimgartner, H. Helv. Chim. Acta 2006, 89, 1841–1855. doi:10.1002/hlca.200690178 |
| 10. | Budzowski, A.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2008, 91, 1471–1488. doi:10.1002/hlca.200890160 |
| 11. | Mawad, N.; Pour, F. G.-S.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2010, 93, 2326–2346. doi:10.1002/hlca.201000318 |
| 12. | Liu, Z.; Mehta, S. J.; Lee, K.-S.; Grossman, B.; Qu, H.; Gu, X.; Nichol, G. S.; Hruby, V. J. J. Org. Chem. 2012, 77, 1289–1300. doi:10.1021/jo201753q |
| 13. | Hua, G.; Fuller, A. L.; Bühl, M.; Slawin, A. M. Z.; Woollins, J. D. Eur. J. Org. Chem. 2011, 3067–3073. doi:10.1002/ejoc.201100226 |
| 14. | Xie, J.; Okano, A.; Pierce, J. G.; James, R. C.; Stamm, S.; Crane, C. M.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 1284–1297. doi:10.1021/ja209937s |
| 15. | Chen, P.; Qu, J. J. Org. Chem. 2011, 76, 2994–3004. doi:10.1021/jo200403g |
| 16. | Tran, T. T.; Burgess, A. W.; Treutlein, H.; Perich, J. J. Pept. Res. 2001, 58, 67–78. doi:10.1034/j.1399-3011.2001.00898.x |
| 17. | Wood, P. A.; Pidcock, E.; Allen, F. H. Acta Crystallogr., Sect. B 2008, 64, 491–496. doi:10.1107/S0108768108015437 |
| 18. | Tran, T. T.; Treutlein, H.; Burgess, A. W. Protein Eng., Des. Sel. 2006, 19, 401–408. doi:10.1093/protein/gzl024 |
| 19. | Tran, T. T.; Zeng, J.; Treutlein, H.; Burgess, A. W. J. Am. Chem. Soc. 2002, 124, 5222–5230. doi:10.1021/ja011916o |
| 20. | Artis, D. R.; Lipton, M. A. J. Am. Chem. Soc. 1998, 120, 12200–12206. doi:10.1021/ja982398t |
| 21. | Alemán, C. J. Phys. Chem. A 2001, 105, 6717–6723. doi:10.1021/jp010198p |
| 22. | Choudhary, A.; Gandla, D.; Krow, G. R.; Raines, R. T. J. Am. Chem. Soc. 2009, 131, 7244–7246. doi:10.1021/ja901188y |
| 23. | Choudhary, A.; Raines, R. T. ChemBioChem 2011, 12, 1801–1807. doi:10.1002/cbic.201100272 |
| 24. | Cervetto, V.; Pfister, R.; Kolano, C.; Bregy, H.; Heimgartner, H.; Helbing, J. Chem.–Eur. J. 2007, 13, 9004–9011. doi:10.1002/chem.200700396 |
| 25. | Cervetto, V.; Pfister, R.; Helbing, J. J. Phys. Chem. B 2008, 112, 3540–3544. doi:10.1021/jp710611n |
| 26. | Cervetto, V.; Hamm, P.; Helbing, J. J. Phys. Chem. B 2008, 112, 8398–8405. doi:10.1021/jp801166q |
| 27. | Bregy, H.; Heimgartner, H.; Helbing, J. J. Phys. Chem. B 2009, 113, 1756–1762. doi:10.1021/jp8089402 |
| 28. | Huang, Y.; Cong, Z.; Yang, L.; Dong, S. J. Pept. Sci. 2008, 14, 1062–1068. doi:10.1002/psc.1042 |
| 29. | Wildemann, D.; Schiene-Fischer, C.; Aumüller, T.; Bachmann, A.; Kiefhaber, T.; Lücke, C.; Fischer, G. J. Am. Chem. Soc. 2007, 129, 4910–4918. doi:10.1021/ja069048o |
| 30. | Zhao, J.; Micheau, J.-C.; Vargas, C.; Schiene-Fischer, C. Chem.–Eur. J. 2004, 10, 6093–6101. doi:10.1002/chem.200400400 |
| 31. | Bailey, P. D.; Boyd, C. A. R.; Collier, I. D.; Kellett, G. L.; Meredith, D.; Morgan, K. M.; Pettecrew, R.; Price, R. A. Org. Biomol. Chem. 2005, 3, 4038–4039. doi:10.1039/B513274F |
| 32. | Miwa, J. H.; Pallivathucal, L.; Gowda, S.; Lee, K. E. Org. Lett. 2002, 4, 4655–4657. doi:10.1021/ol027056d |
| 33. | Miwa, J. H.; Patel, A. K.; Vivatrat, N.; Popek, S. M.; Meyer, A. M. Org. Lett. 2001, 3, 3373–3375. doi:10.1021/ol0166092 |
| 34. | Kajtár, M.; Hollósi, M.; Kajtár, J.; Majer, Z.; Kövér, K. E. Tetrahedron 1986, 42, 3931–3942. doi:10.1016/S0040-4020(01)87548-7 |
| 35. | Hollósi, M.; Kollát, E.; Kajtár, J.; Kajtár, M.; Fasman, G. D. Biopolymers 1990, 30, 1061–1072. doi:10.1002/bip.360301107 |
| 36. | Reiner, A.; Wildemann, D.; Fischer, G.; Kiefhaber, T. J. Am. Chem. Soc. 2008, 130, 8079–8084. doi:10.1021/ja8015044 |
| 37. | Goldberg, J. M.; Batjargal, S.; Petersson, E. J. J. Am. Chem. Soc. 2010, 132, 14718–14720. doi:10.1021/ja1044924 |
| 38. | Goldberg, J. M.; Wissner, R. F.; Klein, A. M.; Petersson, E. J. Chem. Commun. 2012, 48, 1550–1552. doi:10.1039/c1cc14708k |
| 39. | Satzger, H.; Root, C.; Gilch, P.; Zinth, W.; Wildemann, D.; Fischer, G. J. Phys. Chem. B 2005, 109, 4770–4775. doi:10.1021/jp045151t |
| 40. | Sifferlen, T.; Rueping, M.; Gademann, K.; Jaun, B.; Seebach, D. Helv. Chim. Acta 1999, 82, 2067–2093. doi:10.1002/(SICI)1522-2675(19991215)82:12<2067::AID-HLCA2067>3.0.CO;2-5 |
| 41. | Bachmann, A.; Wildemann, D.; Praetorius, F.; Fischer, G.; Kiefhaber, T. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3952–3957. doi:10.1073/pnas.1012668108 |
| 42. | Mierke, D. F.; Geyer, A.; Kessler, H. Int. J. Pept. Protein Res. 1994, 44, 325–331. doi:10.1111/j.1399-3011.1994.tb01016.x |
| 43. | Otani, Y.; Hori, T.; Kawahata, M.; Yamagouchi, K.; Ohwada, T. Tetrahedron 2012, 68, 4418–4428. doi:10.1016/j.tet.2012.01.018 |
| 44. | Banala, S.; Süssmuth, R. D. ChemBioChem 2010, 11, 1335–1337. doi:10.1002/cbic.201000266 |
| 60. | Palumbo, M.; da Rin, S.; Bonora, G. M.; Toniolo, C. Makromol. Chem. 1976, 177, 1477–1492. doi:10.1002/macp.1976.021770519 |
| 61. | Moretto, V.; Crisma, M.; Bonora, G. M.; Toniolo, C.; Balaram, H.; Balaram, P. Macromolecules 1989, 22, 2939–2944. doi:10.1021/ma00197a010 |
| 76. | Performance Standards for antimicrobial Disk Susceptibility Tests; Approved Standard–Sixth Edition; CLSI: Wayne, PA, 1997; CLSI document M02–A06. |
| 53. | Clausen, K.; Thorsen, M.; Lawesson, S.-O. Tetrahedron 1981, 37, 3635–3639. doi:10.1016/S0040-4020(01)98892-1 |
| 54. | Lajoie, G.; Lépine, F.; Maziak, L.; Belleau, B. Tetrahedron Lett. 1983, 24, 3815–3818. doi:10.1016/S0040-4039(00)94282-5 |
| 55. | Cava, M. P.; Levinson, M. I. Tetrahedron 1985, 41, 5061–5087. doi:10.1016/S0040-4020(01)96753-5 |
| 56. | Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210–5278. doi:10.1021/cr040650b |
| 57. | Goodman, M.; Toniolo, C. Biopolymers 1968, 6, 1673–1689. doi:10.1002/bip.1968.360061202 |
| 58. | Sreerama, N.; Woody, R. W. Circular dichroism of peptides and proteins. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N.; Nakanishi, K.; Woody, R. W., Eds.; Wiley-VCH: New York, 2000; pp 601–620. |
| 59. | De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.-S.; Toniolo, C. Org. Biomol. Chem. 2012, 10, 1285–1299. doi:10.1039/c1ob06178j |
| 53. | Clausen, K.; Thorsen, M.; Lawesson, S.-O. Tetrahedron 1981, 37, 3635–3639. doi:10.1016/S0040-4020(01)98892-1 |
| 54. | Lajoie, G.; Lépine, F.; Maziak, L.; Belleau, B. Tetrahedron Lett. 1983, 24, 3815–3818. doi:10.1016/S0040-4039(00)94282-5 |
| 55. | Cava, M. P.; Levinson, M. I. Tetrahedron 1985, 41, 5061–5087. doi:10.1016/S0040-4020(01)96753-5 |
| 56. | Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210–5278. doi:10.1021/cr040650b |
| 34. | Kajtár, M.; Hollósi, M.; Kajtár, J.; Majer, Z.; Kövér, K. E. Tetrahedron 1986, 42, 3931–3942. doi:10.1016/S0040-4020(01)87548-7 |
| 35. | Hollósi, M.; Kollát, E.; Kajtár, J.; Kajtár, M.; Fasman, G. D. Biopolymers 1990, 30, 1061–1072. doi:10.1002/bip.360301107 |
| 65. | Auvin-Guette, C.; Rebuffat, S.; Prigent, Y.; Bodo, B. J. Am. Chem. Soc. 1992, 114, 2170–2174. doi:10.1021/ja00032a035 |
| 53. | Clausen, K.; Thorsen, M.; Lawesson, S.-O. Tetrahedron 1981, 37, 3635–3639. doi:10.1016/S0040-4020(01)98892-1 |
| 73. |
Schwieters, C. D.; Kuszewski, J. J.; Tjandra, N.; Clore, G. M. J. Magn. Reson. 2003, 160, 65–73. doi:10.1016/S1090-7807(02)00014-9
(based on X-PLOR 3.851 by A. T. Brunger). |
| 52. | König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319 |
| 29. | Wildemann, D.; Schiene-Fischer, C.; Aumüller, T.; Bachmann, A.; Kiefhaber, T.; Lücke, C.; Fischer, G. J. Am. Chem. Soc. 2007, 129, 4910–4918. doi:10.1021/ja069048o |
| 30. | Zhao, J.; Micheau, J.-C.; Vargas, C.; Schiene-Fischer, C. Chem.–Eur. J. 2004, 10, 6093–6101. doi:10.1002/chem.200400400 |
| 32. | Miwa, J. H.; Pallivathucal, L.; Gowda, S.; Lee, K. E. Org. Lett. 2002, 4, 4655–4657. doi:10.1021/ol027056d |
| 33. | Miwa, J. H.; Patel, A. K.; Vivatrat, N.; Popek, S. M.; Meyer, A. M. Org. Lett. 2001, 3, 3373–3375. doi:10.1021/ol0166092 |
| 34. | Kajtár, M.; Hollósi, M.; Kajtár, J.; Majer, Z.; Kövér, K. E. Tetrahedron 1986, 42, 3931–3942. doi:10.1016/S0040-4020(01)87548-7 |
| 35. | Hollósi, M.; Kollát, E.; Kajtár, J.; Kajtár, M.; Fasman, G. D. Biopolymers 1990, 30, 1061–1072. doi:10.1002/bip.360301107 |
| 36. | Reiner, A.; Wildemann, D.; Fischer, G.; Kiefhaber, T. J. Am. Chem. Soc. 2008, 130, 8079–8084. doi:10.1021/ja8015044 |
| 40. | Sifferlen, T.; Rueping, M.; Gademann, K.; Jaun, B.; Seebach, D. Helv. Chim. Acta 1999, 82, 2067–2093. doi:10.1002/(SICI)1522-2675(19991215)82:12<2067::AID-HLCA2067>3.0.CO;2-5 |
| 41. | Bachmann, A.; Wildemann, D.; Praetorius, F.; Fischer, G.; Kiefhaber, T. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3952–3957. doi:10.1073/pnas.1012668108 |
| 43. | Otani, Y.; Hori, T.; Kawahata, M.; Yamagouchi, K.; Ohwada, T. Tetrahedron 2012, 68, 4418–4428. doi:10.1016/j.tet.2012.01.018 |
| 74. | Koradi, R.; Billeter, M.; Wüthrich, K. J. Mol. Graphics 1996, 14, 51–55. doi:10.1016/0263-7855(96)00009-4 |
| 65. | Auvin-Guette, C.; Rebuffat, S.; Prigent, Y.; Bodo, B. J. Am. Chem. Soc. 1992, 114, 2170–2174. doi:10.1021/ja00032a035 |
| 63. | Toniolo, C.; Benedetti, E. Trends Biochem. Sci. 1991, 16, 350–353. doi:10.1016/0968-0004(91)90142-I |
| 66. | Gurunath, R.; Balaram, P. Biopolymers 1995, 35, 21–29. doi:10.1002/bip.360350104 |
| 59. | De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.-S.; Toniolo, C. Org. Biomol. Chem. 2012, 10, 1285–1299. doi:10.1039/c1ob06178j |
| 66. | Gurunath, R.; Balaram, P. Biopolymers 1995, 35, 21–29. doi:10.1002/bip.360350104 |
| 66. | Gurunath, R.; Balaram, P. Biopolymers 1995, 35, 21–29. doi:10.1002/bip.360350104 |
| 68. | Brückner, H.; Koza, A. Amino Acids 2003, 24, 311–323. doi:10.1007/s00726-002-0401-x |
| 69. | Nagaraj, R.; Balaram, P. Tetrahedron 1981, 37, 1263–1270. doi:10.1016/S0040-4020(01)92061-7 |
| 66. | Gurunath, R.; Balaram, P. Biopolymers 1995, 35, 21–29. doi:10.1002/bip.360350104 |
| 67. | Iida, A.; Yoshimatsu, S.; Sanekata, M.; Fujita, T. Chem. Pharm. Bull. 1990, 38, 2997–3003. doi:10.1248/cpb.38.2997 |
© 2012 De Zotti et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)