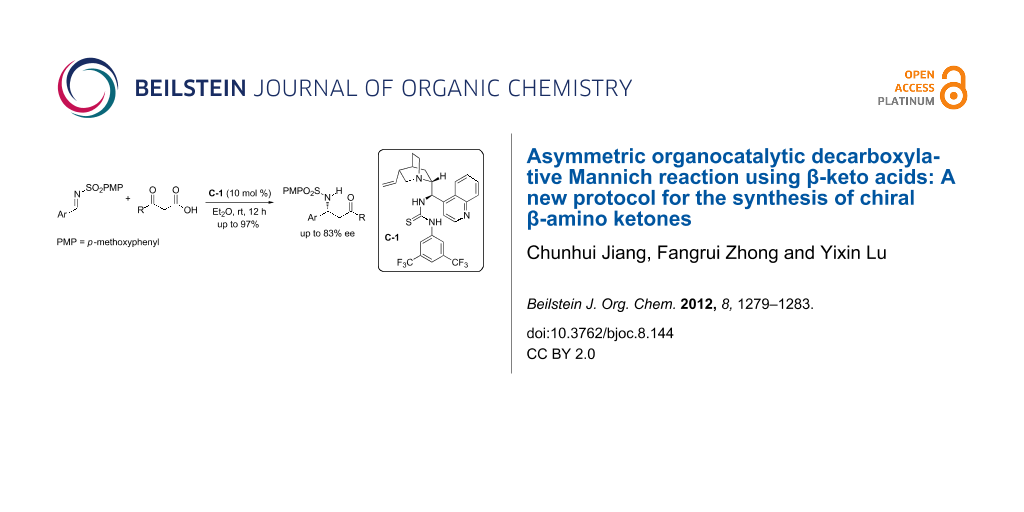
Abstract
The first decarboxylative Mannich reaction employing β-keto acids, catalyzed by cinchonine-derived bifunctional thiourea catalyst has been described. The desired β-amino ketones were obtained in excellent yields and with moderate to good enantioselectivities.

Graphical Abstract
Introduction
Chiral β-amino ketones are an important class of building blocks for the synthesis of 1,3-amino alcohols [1,2], 1,3-amino acids [3] and other bioactive nature products [4-6]. Given their synthetic significance, methods for the asymmetric synthesis of β-amino ketones have been extensively investigated over the past few decades [7]. Among them, the Mukaiyama–Mannich reaction performed with silyl enol ethers and sulfonyl aldimines, catalyzed by a chiral Lewis acid complex, is one of the most important synthetic methods [8-13]. Apparently, direct use of inactivated ketones as a donor would be of great practical value. Indeed, direct approaches such as asymmetric enamine catalysis [14-17] and Brønsted acid catalysis [18] have been reported, through the activation of ketones or aryl imines [19]. However, substrates for the enamine activation are limited to only acetone and cyclic alkyl ketones. Application of aryl methyl ketones in the asymmetric Mannich reaction by enamine catalysis remains elusive. On the other hand, the only chiral Brønsted acid catalytic system based on BINOL-phosphates was reported by Rueping et al. Unfortunately, the yields of the reported reactions were unsatisfactory and the enantioselectivities were modest [20].
In recent years, inspired by the enzymatic synthesis of polyketides and fatty acids in biological systems, the enantioselective decarboxylative reactions of malonic acid half thioesters (MAHTs) have received much attention. In this regard, various electrophiles, including aldehydes, ketones, imines, activated alkenes and azodicarboxylates, have been employed as electrophiles in the presence of metal [21-25] or organocatalysts [26-36].
To provide a practical solution to the low reactivity associated with aryl methyl ketones, we wondered whether β-keto acids could serve as an enolate equivalent to aryl methyl ketones upon decarboxylation (Scheme 1). The proposed addition–decarboxylation sequence is consistent with current mechanistic understanding [31,35,37,38]. However, we cannot exclude an alternative decarboxylation–addition pathway at this stage. In fact, in sharp contrast to the popular use of malonic acid half thioesters (MAHTs) as an ester enolate equivalent in enantioselective decarboxylative additions [39], the employment of β-keto acids as a reaction partner in decarboxylative processes has rarely been explored [37]. Herein, we reported the first decarboxylative Mannich reaction between the β-keto acids and sulfonylimines, affording chiral β-amino ketones in excellent yields and good enantioselectivities.
![[1860-5397-8-144-i1]](/bjoc/content/inline/1860-5397-8-144-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Working hypothesis: Decarboxylative Mannich reaction.
Scheme 1: Working hypothesis: Decarboxylative Mannich reaction.
Findings
In our initial screening, we examined the model reaction between tosylimine 1a and β-keto acid 2a in the presence of a range of bifunctional catalysts (Table 1). We first evaluated the catalytic effects of several cinchona alkaloid derivatives. Commercially available cinchonidine (CD-1) led to the formation of the product with disappointing enantioselectivity (Table 1, entry 1). Quinine-derived sulfonamide [40], β-isocupreidine (β-ICD) [41,42] and biscinchona alkaloid (DHQ)2AQN were all found to be poor catalysts (Table 1, entries 2–4). On the other hand, cinchona alkaloid derived bifunctional thiourea tertiary amine catalysts afforded much improved results (Table 1, entries 5–7). Among them, the cinchonine based thiourea C-1 turned out to be the best catalyst, and the Mannich product was isolated with 58% ee (Table 1, entry 7). In addition, we also examined several other bifunctional catalysts based on amino acids [43,44], including threonine derived Thr-1 [45], and tryptophan based Trp-1 [46], as well as threonine incorporated multifunctional catalyst CD-3 [47]. However, no further improvement could be achieved (Table 1, entries 8–10). The influence of different imines on the reaction was subsequently explored, and it was found that the electronic nature of the sulfonyl protective groups affected the enantioselectivity. While the employment of nosylimine 1b led to decreased enantioselectivity (Table 1, entry 11), replacement of tosylimine 1a with N-(p-methoxybenzenesulfonyl)imine 1c resulted in further improvement, and the product was obtained in 65% ee (Table 1, entry 12). However, when ethoxycarbonylimine 1d was used, nearly racemic products were obtained, suggesting the importance of the sulfonyl group in the asymmetric induction (Table 1, entry 13). Less reactive imines, such as diphenylphosphinoylimine 1e and Cbz-imine 1f, proved to be unsuitable for the reaction (Table 1, entries 14 and 15).
Table 1: Exploration of the decarboxylative addition of β-ketoacids to imines.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-144-i2.svg?max-width=637&scale=1.0)
|
||||
| Entrya | 1 | cat | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 1a | CD-1 | 86 | 17 |
| 2 | 1a | Q-1 | 54 | 13 |
| 3 | 1a | (DHQ)2AQN | 90 | 2 |
| 4 | 1a | β-ICD | 93 | 27 |
| 5 | 1a | CD-2 | 97 | 44 |
| 6 | 1a | QD-1 | 93 | 54 |
| 7 | 1a | C-1 | 95 | 58 |
| 8 | 1a | Thr-1 | 88 | 12 |
| 9 | 1a | Trp-1 | 95 | 21 |
| 10 | 1a | CD-3 | 92 | 13 |
| 11 | 1b | C-1 | 95 | 49 |
| 12 | 1c | C-1 | 96 | 65 |
| 13 | 1d | C-1 | 91 | 5 |
| 14 | 1e | C-1 | trace | – |
| 15 | 1f | C-1 | trace | – |
aReactions were performed with 1 (0.05 mmol), 2a (0.075 mmol) and the catalyst (0.005 mmol) in CHCl3 (0.5 mL). bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase.
A screening of the solvent effect was then followed, and the results are summarized in Table 2. In general, the reaction proceeded very well in common aprotic solvents, and excellent yields were consistently obtained (Table 2, entries 1–9). Enantioselectivity of the reaction varied, and diethyl ether was found to be the best solvent, furnishing the desired product with 72% ee. Employment of other etheric solvents, including methyl tert-butyl ether and dioxane, and lowering reaction temperature did not offer further improvement (Table 2, entries 10–12).
Table 2: Solvent screening.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-144-i3.svg?max-width=637&scale=1.0)
|
|||
| Entrya | Solvent | Yield (%)b | ee (%)c |
|---|---|---|---|
| 1 | CHCl3 | 96 | 65 |
| 2 | THF | 93 | 64 |
| 3 | DCM | 92 | 66 |
| 4 | toluene | 90 | 63 |
| 5 | diethyl ether | 93 | 72 |
| 6 | ethyl acetate | 92 | 66 |
| 7 | benzene | 90 | 67 |
| 8 | DCE | 91 | 66 |
| 9 | acetone | 92 | 52 |
| 10 | methyl tert-butyl ether | 92 | 62 |
| 11 | dioxane | 94 | 65 |
| 12d | diethyl ether | 67 | 65 |
aReactions were performed with 1c (0.05 mmol), 2a (0.075 mmol) and C-1 (0.005 mmol) in the solvent specified (0.5 mL). bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase. dReaction was performed at 0 °C.
To establish the substrate scope, a number of sulfonylimines derived from aromatic aldehydes were employed as acceptors, and the results are summarized in Table 3. In general, the reaction worked well for imines with various substituents at different positions of the phenyl ring, including electron-withdrawing groups, electron-donating groups and halogen atoms, and excellent yields and moderate ee values were obtained (Table 3, entries 1–10). Heterocycles were well-tolerated, and good enantioselectivities were obtained with 2-furyl and thiophen-2-yl containing substrates (Table 3, entries 11 and 12). The aryl groups of β-keto acids could also be varied, and the reaction was applicable to β-keto acids with different aromatic substituents (Table 3, entries 13–17). Furthermore, the reaction was also applicable to alkyl β-keto acids, and comparable chemical yields and enantioselectivities were attainable (Table 3, entries 18–19). The absolute configurations of the products were assigned by comparing the optical rotation of 3a with the value reported in the literature [48] (see the Supporting Information File 1 for details).
Table 3: Substrate scope.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-144-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Ar | R | 3 | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | Ph | Ph | 3c | 93 | 72 |
| 2 | 4-Me-C6H4 | Ph | 3g | 90 | 64 |
| 3 | 4-Br-C6H4 | Ph | 3h | 88 | 61 |
| 4 | 4-CF3-C6H4 | Ph | 3i | 85 | 55 |
| 5 | 4-OMe-C6H4 | Ph | 3j | 97 | 62 |
| 6 | 2-F-C6H4 | Ph | 3k | 89 | 65 |
| 7 | 2-Me-C6H4 | Ph | 3l | 92 | 65 |
| 8 | 2-Br-C6H4 | Ph | 3m | 87 | 59 |
| 9 | 3-Me-C6H4 | Ph | 3n | 97 | 65 |
| 10 | 3-Br-C6H4 | Ph | 3o | 96 | 61 |
| 11 | 2-furyl | Ph | 3p | 94 | 83 |
| 12 | thiophen-2-yl | Ph | 3q | 87 | 77 |
| 13 | Ph | 4-F-C6H4 | 3r | 95 | 64 |
| 14d | Ph | 3-Cl-C6H4 | 3s | 62 | 70 |
| 15 | Ph | 2-naphthyl | 3t | 62 | 69 |
| 16 | Ph | 4-Me-C6H4 | 3u | 93 | 67 |
| 17 | Ph | 2-OMe-C6H4 | 3v | 88 | 60 |
| 18 | Ph | n-Pr | 3w | 92 | 54 |
| 19 | Ph | t-Bu | 3x | 75 | 73 |
aReactions were performed with 1 (0.05 mmol), 2 (0.075 mmol) and C-1 (0.005 mmol) in Et2O (0.5 mL). bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase. dThe catalyst loading was 20 mol %.
In conclusion, we have developed the first organocatalytic decarboxylative Mannich reaction employing β-keto acids as the donor. The reaction was effectively catalyzed by cinchonine-based bifunctional catalyst C-1, and the synthetically useful β-amino ketones were prepared in excellent yields and with moderate to good enantioselectivities. The method reported represents a new protocol for the asymmetric construction of β-amino ketones.
Experimental
General procedure for the decarboxylative Mannich reaction of β-keto acids and aldimines
To a solution of imine 1c (13.8 mg, 0.05 mmol) and C-1 (2.8 mg, 0.005 mmol) in ether (0.5 mL) at room temperature, was added β-keto acid 2a (12.3 mg, 0.075 mmol). The reaction mixture was stirred for 12 h. The solvent was then removed under reduced pressure, and the residue was purified by flash chromatography on silica gel (hexane/ethyl acetate 5:1 to 3:1) to afford 3c as a white solid (18.4 mg, 93% yield).
Supporting Information
| Supporting Information File 1: Characterization data and spectra of synthesized compounds. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Barluenga, J.; Viado, A. L.; Aguilar, E.; Fustero, S.; Olano, B. J. Org. Chem. 1993, 58, 5972–5975. doi:10.1021/jo00074a024
Return to citation in text: [1] -
Enders, D.; Moser, M.; Geibel, G.; Laufer, M. C. Synthesis 2004, 2040–2046. doi:10.1055/s-2004-829142
Return to citation in text: [1] -
Mukhopadhyay, M.; Bhatia, B.; Iqbal, J. Tetrahedron Lett. 1997, 38, 1083–1086. doi:10.1016/S0040-4039(96)02474-4
Return to citation in text: [1] -
Arend, M.; Westerman, B.; Risch, N. Angew. Chem., Int. Ed. 1998, 37, 1044–1070. doi:10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E
Return to citation in text: [1] -
Evans, G. B.; Furneaux, R. H.; Tyler, P. C.; Schramm, V. L. Org. Lett. 2003, 5, 3639–3640. doi:10.1021/ol035293q
Return to citation in text: [1] -
Joshi, N. S.; Whitaker, L. R.; Francis, M. B. J. Am. Chem. Soc. 2004, 126, 15942–15943. doi:10.1021/ja0439017
Return to citation in text: [1] -
Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z
Return to citation in text: [1] -
Ishitani, H.; Ueno, M.; Kobayashi, S. J. Am. Chem. Soc. 1997, 119, 7153–7154. doi:10.1021/ja970498d
Return to citation in text: [1] -
Kobayashi, S.; Ishitani, H.; Ueno, M. J. Am. Chem. Soc. 1998, 120, 431–432. doi:10.1021/ja973527t
Return to citation in text: [1] -
Hagiwara, E.; Fujii, A.; Sodeoka, M. J. Am. Chem. Soc. 1998, 120, 2474–2475. doi:10.1021/ja973962n
Return to citation in text: [1] -
Ishitani, H.; Ueno, M.; Kobayashi, S. J. Am. Chem. Soc. 2000, 122, 8180–8186. doi:10.1021/ja001642p
Return to citation in text: [1] -
Juhl, K.; Gathergood, N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2001, 40, 2995–2997. doi:10.1002/1521-3773(20010817)40:16<2995::AID-ANIE2995>3.0.CO;2-M
Return to citation in text: [1] -
Taggi, A. E.; Hafez, A. E.; Lectka, T. Acc. Chem. Res. 2003, 36, 10–19. doi:10.1021/ar020137p
Return to citation in text: [1] -
Notz, W.; Tanaka, F.; Barbas, C. F., III. Acc. Chem. Res. 2004, 37, 580–591. doi:10.1021/ar0300468
Return to citation in text: [1] -
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016
Return to citation in text: [1] -
Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Angew. Chem., Int. Ed. 2008, 47, 6138–6171. doi:10.1002/anie.200705523
Return to citation in text: [1] -
Kano, T.; Maruoka, K. Chem. Commun. 2008, 5465–5473. doi:10.1039/B809301F
Return to citation in text: [1] -
Akiyama, T. Chem. Rev. 2007, 107, 5744–5758. doi:10.1021/cr068374j
Return to citation in text: [1] -
Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g
Return to citation in text: [1] -
Rueping, M.; Sugiono, E.; Schoepke, F. R. Synlett 2007, 1441–1445. doi:10.1055/s-2007-980369
Return to citation in text: [1] -
Lalic, G.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2003, 125, 2852–2853. doi:10.1021/ja029452x
Return to citation in text: [1] -
Orlandi, S.; Benaglia, M.; Cozzi, F. Tetrahedron Lett. 2004, 45, 1747–1749. doi:10.1016/j.tetlet.2003.12.089
Return to citation in text: [1] -
Magdziak, D.; Lalic, G.; Lee, H. M.; Fortner, K. C.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2005, 127, 7284–7285. doi:10.1021/ja051759j
Return to citation in text: [1] -
Fortner, K. C.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 1032–1033. doi:10.1021/ja0673682
Return to citation in text: [1] -
Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. Chem.–Asian J. 2010, 5, 2351–2354. doi:10.1002/asia.201000540
Return to citation in text: [1] -
Brunner, H.; Müller, J.; Spitzer, J. Monatsh. Chem. 1996, 127, 845–858. doi:10.1007/BF00807023
Return to citation in text: [1] -
Ryu, Y.; Scott, A. I. Tetrahedron Lett. 2003, 44, 7499–7502. doi:10.1016/j.tetlet.2003.08.014
Return to citation in text: [1] -
List, B.; Doehring, A.; Fonseca, M. T. H.; Wobser, K.; van Thienen, H.; Torres, R. R.; Galilea, P. Adv. Synth. Catal. 2005, 347, 1558–1560. doi:10.1002/adsc.200505196
Return to citation in text: [1] -
Blanchet, J.; Baudoux, J.; Amere, M.; Lasne, M.-C.; Rouden, J. Eur. J. Org. Chem. 2008, 5493–5506. doi:10.1002/ejoc.200800759
Return to citation in text: [1] -
Blaquiere, N.; Shore, D. G.; Rousseaux, S.; Fagnou, K. J. Org. Chem. 2009, 74, 6190–6198. doi:10.1021/jo901022j
Return to citation in text: [1] -
Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536
Return to citation in text: [1] [2] -
Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187
Return to citation in text: [1] -
Pan, Y.; Kee, C. W.; Jiang, Z.; Ma, T.; Zhao, Y.; Yang, Y.; Xue, H.; Tan, C.-H. Chem.–Eur. J. 2011, 17, 8363–8370. doi:10.1002/chem.201100687
Return to citation in text: [1] -
Bae, H. Y.; Some, S.; Lee, J. H.; Kim, J.-Y.; Song, M. J.; Lee, S.; Zhang, Y. J.; Song, C. E. Adv. Synth. Catal. 2011, 353, 3196–3202. doi:10.1002/adsc.201100458
Return to citation in text: [1] -
Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.-C.; Rouden, J. Green Chem. 2010, 12, 252–259. doi:10.1039/b915681j
Return to citation in text: [1] [2] -
Hara, N.; Nakamura, S.; Funahashi, Y.; Shibata, N. Adv. Synth. Catal. 2011, 353, 2976–2980. doi:10.1002/adsc.201100410
Return to citation in text: [1] -
Zheng, Y.; Xiong, H.-Y.; Nie, J.; Hua, M.-Q.; Ma, J.-A. Chem. Commun. 2012, 48, 4308–4310. doi:10.1039/c2cc30949a
Return to citation in text: [1] [2] -
Yang, C.-F.; Shen, C.; Wang, J.-Y.; Tian, S.-K. Org. Lett. 2012, 14, 3092–3095. doi:10.1021/ol301180z
Return to citation in text: [1] -
Pan, Y.; Tan, C.-H. Synthesis 2011, 2044–2053. doi:10.1055/s-0030-1260607
Return to citation in text: [1] -
Luo, J.; Xu, L.-W.; Hay, R. A. S.; Lu, Y. Org. Lett. 2009, 11, 437–440. doi:10.1021/ol802486m
Return to citation in text: [1] -
Marcelli, T.; Maarseveen, J. H.; Huiemstra, H. Angew. Chem., Int. Ed. 2006, 45, 7496–7504. doi:10.1002/anie.200602318
Return to citation in text: [1] -
Zhong, F.; Chen, G.-Y.; Lu, Y. Org. Lett. 2011, 13, 82–85. doi:10.1021/ol102597s
Return to citation in text: [1] -
Xu, L.-W.; Lu, Y. Org. Biomol. Chem. 2008, 6, 2047–2053. doi:10.1039/b803116a
Return to citation in text: [1] -
Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807–1821. doi:10.1039/b821070e
Return to citation in text: [1] -
Dou, X.; Han, X.; Lu, Y. Chem.–Eur. J. 2012, 18, 85–89. doi:10.1002/chem.201102796
Return to citation in text: [1] -
Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635
Return to citation in text: [1] -
Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837
Return to citation in text: [1] -
Zhao, C.-H.; Liu, L.; Wang, D.; Chen, Y.-J. Eur. J. Org. Chem. 2006, 2977–2986. doi:10.1002/ejoc.200600147
Return to citation in text: [1]
| 46. | Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635 |
| 43. | Xu, L.-W.; Lu, Y. Org. Biomol. Chem. 2008, 6, 2047–2053. doi:10.1039/b803116a |
| 44. | Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807–1821. doi:10.1039/b821070e |
| 45. | Dou, X.; Han, X.; Lu, Y. Chem.–Eur. J. 2012, 18, 85–89. doi:10.1002/chem.201102796 |
| 1. | Barluenga, J.; Viado, A. L.; Aguilar, E.; Fustero, S.; Olano, B. J. Org. Chem. 1993, 58, 5972–5975. doi:10.1021/jo00074a024 |
| 2. | Enders, D.; Moser, M.; Geibel, G.; Laufer, M. C. Synthesis 2004, 2040–2046. doi:10.1055/s-2004-829142 |
| 8. | Ishitani, H.; Ueno, M.; Kobayashi, S. J. Am. Chem. Soc. 1997, 119, 7153–7154. doi:10.1021/ja970498d |
| 9. | Kobayashi, S.; Ishitani, H.; Ueno, M. J. Am. Chem. Soc. 1998, 120, 431–432. doi:10.1021/ja973527t |
| 10. | Hagiwara, E.; Fujii, A.; Sodeoka, M. J. Am. Chem. Soc. 1998, 120, 2474–2475. doi:10.1021/ja973962n |
| 11. | Ishitani, H.; Ueno, M.; Kobayashi, S. J. Am. Chem. Soc. 2000, 122, 8180–8186. doi:10.1021/ja001642p |
| 12. | Juhl, K.; Gathergood, N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2001, 40, 2995–2997. doi:10.1002/1521-3773(20010817)40:16<2995::AID-ANIE2995>3.0.CO;2-M |
| 13. | Taggi, A. E.; Hafez, A. E.; Lectka, T. Acc. Chem. Res. 2003, 36, 10–19. doi:10.1021/ar020137p |
| 40. | Luo, J.; Xu, L.-W.; Hay, R. A. S.; Lu, Y. Org. Lett. 2009, 11, 437–440. doi:10.1021/ol802486m |
| 7. | Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z |
| 41. | Marcelli, T.; Maarseveen, J. H.; Huiemstra, H. Angew. Chem., Int. Ed. 2006, 45, 7496–7504. doi:10.1002/anie.200602318 |
| 42. | Zhong, F.; Chen, G.-Y.; Lu, Y. Org. Lett. 2011, 13, 82–85. doi:10.1021/ol102597s |
| 4. | Arend, M.; Westerman, B.; Risch, N. Angew. Chem., Int. Ed. 1998, 37, 1044–1070. doi:10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E |
| 5. | Evans, G. B.; Furneaux, R. H.; Tyler, P. C.; Schramm, V. L. Org. Lett. 2003, 5, 3639–3640. doi:10.1021/ol035293q |
| 6. | Joshi, N. S.; Whitaker, L. R.; Francis, M. B. J. Am. Chem. Soc. 2004, 126, 15942–15943. doi:10.1021/ja0439017 |
| 3. | Mukhopadhyay, M.; Bhatia, B.; Iqbal, J. Tetrahedron Lett. 1997, 38, 1083–1086. doi:10.1016/S0040-4039(96)02474-4 |
| 37. | Zheng, Y.; Xiong, H.-Y.; Nie, J.; Hua, M.-Q.; Ma, J.-A. Chem. Commun. 2012, 48, 4308–4310. doi:10.1039/c2cc30949a |
| 20. | Rueping, M.; Sugiono, E.; Schoepke, F. R. Synlett 2007, 1441–1445. doi:10.1055/s-2007-980369 |
| 26. | Brunner, H.; Müller, J.; Spitzer, J. Monatsh. Chem. 1996, 127, 845–858. doi:10.1007/BF00807023 |
| 27. | Ryu, Y.; Scott, A. I. Tetrahedron Lett. 2003, 44, 7499–7502. doi:10.1016/j.tetlet.2003.08.014 |
| 28. | List, B.; Doehring, A.; Fonseca, M. T. H.; Wobser, K.; van Thienen, H.; Torres, R. R.; Galilea, P. Adv. Synth. Catal. 2005, 347, 1558–1560. doi:10.1002/adsc.200505196 |
| 29. | Blanchet, J.; Baudoux, J.; Amere, M.; Lasne, M.-C.; Rouden, J. Eur. J. Org. Chem. 2008, 5493–5506. doi:10.1002/ejoc.200800759 |
| 30. | Blaquiere, N.; Shore, D. G.; Rousseaux, S.; Fagnou, K. J. Org. Chem. 2009, 74, 6190–6198. doi:10.1021/jo901022j |
| 31. | Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536 |
| 32. | Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187 |
| 33. | Pan, Y.; Kee, C. W.; Jiang, Z.; Ma, T.; Zhao, Y.; Yang, Y.; Xue, H.; Tan, C.-H. Chem.–Eur. J. 2011, 17, 8363–8370. doi:10.1002/chem.201100687 |
| 34. | Bae, H. Y.; Some, S.; Lee, J. H.; Kim, J.-Y.; Song, M. J.; Lee, S.; Zhang, Y. J.; Song, C. E. Adv. Synth. Catal. 2011, 353, 3196–3202. doi:10.1002/adsc.201100458 |
| 35. | Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.-C.; Rouden, J. Green Chem. 2010, 12, 252–259. doi:10.1039/b915681j |
| 36. | Hara, N.; Nakamura, S.; Funahashi, Y.; Shibata, N. Adv. Synth. Catal. 2011, 353, 2976–2980. doi:10.1002/adsc.201100410 |
| 19. | Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g |
| 31. | Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536 |
| 35. | Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.-C.; Rouden, J. Green Chem. 2010, 12, 252–259. doi:10.1039/b915681j |
| 37. | Zheng, Y.; Xiong, H.-Y.; Nie, J.; Hua, M.-Q.; Ma, J.-A. Chem. Commun. 2012, 48, 4308–4310. doi:10.1039/c2cc30949a |
| 38. | Yang, C.-F.; Shen, C.; Wang, J.-Y.; Tian, S.-K. Org. Lett. 2012, 14, 3092–3095. doi:10.1021/ol301180z |
| 47. | Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837 |
| 14. | Notz, W.; Tanaka, F.; Barbas, C. F., III. Acc. Chem. Res. 2004, 37, 580–591. doi:10.1021/ar0300468 |
| 15. | Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016 |
| 16. | Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Angew. Chem., Int. Ed. 2008, 47, 6138–6171. doi:10.1002/anie.200705523 |
| 17. | Kano, T.; Maruoka, K. Chem. Commun. 2008, 5465–5473. doi:10.1039/B809301F |
| 21. | Lalic, G.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2003, 125, 2852–2853. doi:10.1021/ja029452x |
| 22. | Orlandi, S.; Benaglia, M.; Cozzi, F. Tetrahedron Lett. 2004, 45, 1747–1749. doi:10.1016/j.tetlet.2003.12.089 |
| 23. | Magdziak, D.; Lalic, G.; Lee, H. M.; Fortner, K. C.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc. 2005, 127, 7284–7285. doi:10.1021/ja051759j |
| 24. | Fortner, K. C.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 1032–1033. doi:10.1021/ja0673682 |
| 25. | Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. Chem.–Asian J. 2010, 5, 2351–2354. doi:10.1002/asia.201000540 |
| 48. | Zhao, C.-H.; Liu, L.; Wang, D.; Chen, Y.-J. Eur. J. Org. Chem. 2006, 2977–2986. doi:10.1002/ejoc.200600147 |
© 2012 Jiang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)