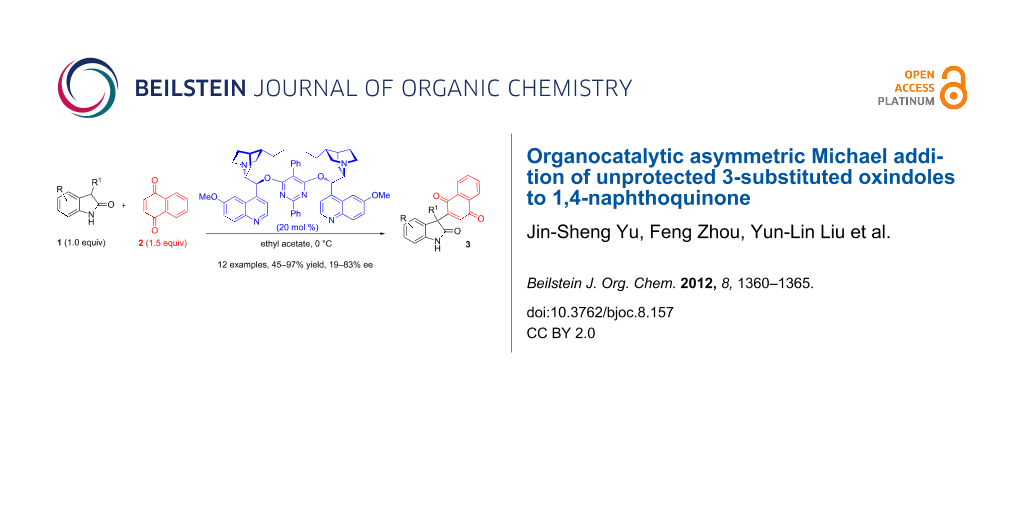
Abstract
We reported the first example of organocatalytic Michael addition of unprotected 3-prochiral oxindoles 1 to 1,4-naphthoquinone. Quinidine derivative (DHQD)2PYR was found to be able to catalyze this reaction in up to 83% ee, with moderate to excellent yields. This method could be used for the synthesis of enantioenriched 3,3-diaryloxindoles, and the catalytic synthesis of which was unprecedented.

Graphical Abstract
Introduction
The catalytic asymmetric synthesis of 3,3-disubstituted oxindoles has recently received great attention because of the wide occurrence of this structural motif in natural products and pharmaceutically active compounds [1-3]. In addition, structure–activity relationship studies have revealed that the absolute configuration and the substituent of the C3 position of oxindole greatly influenced the biological activities [4]. Accordingly, the development of efficient synthetic methods to enable the synthesis of 3,3-disubstituted oxindoles in great structural diversity is of current interest, and much progress had been made in the catalytic enantioselective synthesis of 3-hydroxyoxindoles [5-10], 3-aminooxindoles [11-15] and 3-quaternary oxindoles [16-20]. Despite achievements, the catalytic asymmetric synthesis of 3,3-diaryloxindoles has not been reported. This is possibly due to the challenge in the construction of such congested quaternary stereogenic centers. Only Sammakia tried the SNAr reaction of unprotected 3-phenyloxindole with chiral electron-deficient 5-halooxazoles, promoted by 1.0 equiv of Cs2CO3 [21], with ca. 1:1 diastereoselectivity obtained.
In this context, we are interested in the catalytic economical asymmetric diverse synthesis of 3,3-disubstituted oxindoles, using cheap and easily available starting materials and simple chiral catalysts to facilitate biological evaluation. We have developed the catalytic asymmetric addition of acrolein, allyltrimethylsilane or difluoroenoxysilanes to isatins to furnish differently substituted enantioenriched 3-hydroxyoxindoles [22-24]. For the synthesis of chiral 3-aminooxindoles, we developed the first example of catalytic asymmetric addition of nucleophiles to isatin-derived ketoimines using TMSCN [25] and the amination of unprotected 3-prochiral oxindoles using di-tert-butyl azodicarboxylate [26,27]. To construct the C3 quaternary stereogenic carbon center, we have designed a novel cinchona alkaloid-based phosphoramide bifunctional catalyst to realize a highly enantioselective Michael addition of both unprotected 3-alkyl- and 3-aryloxindoles to nitroolefins [28]. Based on these results, together with our efforts in the synthesis of unsymmetric 3,3-diaryloxindoles [29], we try to develop a catalytic asymmetric method to enantioenriched 3,3-diaryloxindoles.
In 2007, Jørgensen and coworkers pioneered the organocatalytic asymmetric addition reactions to quinones [30,31] which turned out to be a powerful strategy for the α-arylation of β-ketoesters and aldehydes. Inspired by their work, we anticipated that the catalytic asymmetric addition of 3-aryloxindoles to quinones would possibly install a hydroquinone moiety at the C3 position of oxindole to furnish the desired chiral 3,3-diaryloxindoles. It also came to our attention that, while the addition of 3-prochiral oxindole to a variety of Michael acceptors had been studied [32-46], the use of quinones as the Michael acceptor had not been realized. Therefore, in this letter we are going to report our initial results about the catalytic asymmetric Michael addition of unprotected 3-prochiral oxindoles to 1,4-naphthoquinone.
Results and Discussion
We began the reaction development by the evaluation of different chiral catalysts derived from cinchona alkaloids in the reaction of 3-phenyloxindole 1a and 1,4-naphthoquinone (2a), with ethyl acetate (EtOAc) as the solvent at 0 °C (Table 1, Figure 1). A variety of bifunctional cinchona alkaloid-derived catalysts 5–9 were first tried, aiming to facilitate the reaction by the dual activation of both reaction partners, with H-bonding donor moiety of the catalyst to activate quinone 2a and the tertiary amine to deprotonatively activate oxindole 1. The reaction generally proceeded slowly, and only the oxidation product, 1,4-naphthoquinone derivative 3a, was obtained in moderate yield after five days. No hydroquinone product 4 was detected by TLC and NMR analysis of the crude reaction mixture. While the simple quinine and quinidine as catalysts could deliver product 3a in 59% ee (Table 1, entry 2), all other bifunctional catalysts turned out to be much less enantioselective (Table 1, entries 3–5). However, the dinuclear Brønsted base catalysts 10–12 could achieve higher ee for the desired product 3a with comparable yields (Table 1, entries 6–8). When the hydrogenated catalyst 12 was used, 77% ee for product 3a was obtained with 51% yield (Table 1, entry 8). In light of this, we used catalyst 12 for the following screenings.
Table 1: Condition optimization for the reaction of 1a and 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-157-i2.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Cat. | Solvent | Additive | Yield of 3a (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | 5 | EtOAc | – | 52 | 43d |
| 2 | 6 | EtOAc | – | 61 | 59 |
| 3 | 7 | EtOAc | – | 40 | 14 |
| 4 | 8 | EtOAc | – | 34 | 4 |
| 5 | 9 | EtOAc | – | 14 | 15 |
| 6 | 10 | EtOAc | – | 60 | 64d |
| 7 | 11 | EtOAc | – | 64 | 73d |
| 8 | 12 | EtOAc | – | 51 | 77 |
| 9 | 12 | THF | – | 50 | 64 |
| 10 | 12 | Acetone | – | 61 | 64 |
| 11 | 12 | CH3CN | – | 32 | 47 |
| 12 | 12 | DCM | – | 21 | 36 |
| 13 | 12 | Toluene | – | 31 | 73 |
| 14 | 12 | EtOAc | MS 4Å | 43 | 80 |
| 15 | 12 | EtOAc | MS 5Å | 29 | 70 |
| 16 | 12 | EtOAc | H2O (5.0 equiv) | 50 | 78 |
| 17 | 12 | EtOAc | H2O (10.0 equiv) | 50 | 78 |
| 18 | 12 | EtOAc | PhCO2He | 33 | 76 |
| 19 | 12 | EtOAc | (S)-BINOLe | 36 | 40 |
| 20 | 12 | EtOAc | (R)-BINOLe | 43 | 77 |
| 21 | 12 | EtOAc | LiCle | 65 | 4 |
aReactions were run on a 0.10 mmol scale. bIsolated yield. cDetermined by chiral HPLC analysis. dOpposite enantiomer. e10 mol % used.
![[1860-5397-8-157-1]](/bjoc/content/figures/1860-5397-8-157-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cinchona alkaloid-derived catalysts screened for condition optimization (Table 1).
Figure 1: Cinchona alkaloid-derived catalysts screened for condition optimization (Table 1).
We further examined the solvent effects, and found that EtOAc turned out to be the most suitable solvent which afforded product 3a in highest ee (Table 1, entries 8–13). Since the reactivity was unsatisfactory, we further tried the use of some additives to improve the reaction rate. The use of MS 4Å could improve the enantioselectivity to 80%, but decreased the yield from 51% to 43% (Table 1, entry 8 versus 14). The use of MS 5Å had a negative effect on both the reactivity and the enantioselectivity (Table 1, entry 15). Water had no obvious effect on the reaction outcome (Table 1, entries 16 and 17). The addition of acids led to diminished yield and enantioselectivity (Table 1, entries 18–20).
Based on these screenings, we determined to examine the substrate scope by running the reaction at 0 °C in EtOAc, with 20 mol % of (DHQD)2PYR 12 to improve the reactivity. Different substituted 3-prochiral oxindoles were first examined and the results are shown in Table 2. An electron-withdrawing substituent at the C5 position of the oxindole had a positive effect on the reactivity and enantioselectivity of the reaction. The corresponding products 3a–d could be obtained in good to excellent yields with up to 81% ee. Without an electron-withdrawing group, products 3e and 3f were obtained in diminished yields and enantioselectivities. Different aryl substituents at the C3 position were also investigated, the corresponding products 3g–k were obtained in acceptable yields and up to 83% ee. We also tried if this method could be extended to 3-alkyloxindoles but had to find out that product 3l was obtained in only moderate enantioselectivity and yield. The absolute configuration of product 3f was determined to be (S) by chemical transformation to the corresponding known compound [47]; all other products were tentatively assigned in analogy (for details, see Supporting Information File 1).
Table 2: Substrate scope of unprotected 3-prochiral oxindolesa–c.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-157-i3.svg?max-width=637&scale=1.0)
|
|||
| Product, yield, enantioselectivity and reaction time | |||
|---|---|---|---|
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-157-i4.svg?max-width=637&scale=1.0)
3a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-157-i5.svg?max-width=637&scale=1.0)
3b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-157-i6.svg?max-width=637&scale=1.0)
3c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-157-i7.svg?max-width=637&scale=1.0)
3d |
| 70% yield, 79% ee, 5 d | 97% yield, 80% ee, 6 d | 74% yield, 81% ee, 6 d | 71% yield, 79% ee, 8 d |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-157-i8.svg?max-width=637&scale=1.0)
3e |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-157-i9.svg?max-width=637&scale=1.0)
3f |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-157-i10.svg?max-width=637&scale=1.0)
3g |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-157-i11.svg?max-width=637&scale=1.0)
3h |
| 46% yield, 69% ee, 9 d | 51% yield, 70% ee, 9 d | 76% yield, 81% ee, 6 d | 45% yield, 54% ee, 8 d |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-157-i12.svg?max-width=637&scale=1.0)
3i |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-157-i13.svg?max-width=637&scale=1.0)
3j |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-157-i14.svg?max-width=637&scale=1.0)
3k |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-157-i15.svg?max-width=637&scale=1.0)
3l |
| 55% yield, 83% ee, 6 d | 68% yield, 77% ee, 6 d | 53% yield, 65% ee, 6 d | 58% yield, 19% ee, 8 d |
aRun on a 0.25 mmol scale. bIsolated yield. cDetermined by chiral HPLC analysis.
Other quinones such as 2,6-dichloro-1,4-benzoquinone and 1,4-benzoquinone were also examined, however, none of them could react with 3-phenyloxindole 1a to give the desired product.
While the oxidation product 3 was obtained as the only product from the Michael addition, it could be hydrogenated to the corresponding hydroquinone product 4. The free hydroxy groups were protected to prevent re-oxidation. For example, product 3i was reduced and converted to the desired 3,3-diaryloxindole 13 in 57% yield without the loss of ee. We further checked if this protocol could be operated as a “one-pot” sequential reaction. After the reaction of 1i and 2 was run at 0 °C for five days a small amount of oxindole 1i still remained. Then, the reaction was warmed to room temperature, followed by the addition of Pd/C and ammonium formate. When TLC analysis revealed that the hydrogenation of product 3i was completed, acetyl chloride and triethylamine were added. The desired product 13 was obtained in 50% yield with 75% ee (Scheme 1). The diminished enantioselectivity was due to the fact that the remaining oxindole 1i continued to react with 2 at room temperature during the following steps. Even if there is much potential for further improvement in the ee, this sequential reaction represented the first example of catalytic asymmetric synthesis of 3,3-diaryl oxindoles.
![[1860-5397-8-157-i1]](/bjoc/content/inline/1860-5397-8-157-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A one-pot synthesis of enantioenriched 3,3-diaryloxindoles.
Scheme 1: A one-pot synthesis of enantioenriched 3,3-diaryloxindoles.
Conclusion
In summary, we have developed the first example of organocatalytic Michael addition of unprotected 3-prochiral oxindoles to 1,4-naphthoquinone [48] with good to high yields and enantioselectivities. This method could be used for the synthesis of enantioenriched 3,3-diaryloxindoles and the catalytic synthesis of which was unprecedented. The development of new chiral catalysts to improve both the reactivity and enantioselectivity of this reaction is now in progress in our lab.
Supporting Information
| Supporting Information File 1: General experimental procedures and compound characterization. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
The financial support from the NSFC (21172075), the 973 program (2011CB808600), Shanghai Pujiang Program (10PJ1403100), Specialized Research Fund for the Doctoral Program of Higher Education (20090076120007), Innovation Program of SMEC (12ZZ046), and the Fundamental Research Funds for the Central Universities (East China Normal University 11043) are highly appreciated.
References
-
Shen, K.; Liu, X.; Lin, L.; Feng, X. Chem. Sci. 2012, 3, 327. doi:10.1039/c1sc00544h
Return to citation in text: [1] -
Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381. doi:10.1002/adsc.201000161
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Badillo, J. J.; Hanhan, N. V.; Franz, A. K. Curr. Opin. Drug Discovery Dev. 2010, 13, 758.
Return to citation in text: [1] -
Shintani, R.; Inoue, M.; Hayashi, T. Angew. Chem., Int. Ed. 2006, 45, 3353. doi:10.1002/anie.200600392
Return to citation in text: [1] -
Ishimaru, T.; Shibata, N.; Nagai, J.; Nakamura, S.; Toru, T.; Kanemasa, S. J. Am. Chem. Soc. 2006, 128, 16488. doi:10.1021/ja0668825
Return to citation in text: [1] -
Itoh, J.; Han, S. B.; Krische, M. J. Angew. Chem., Int. Ed. 2009, 48, 6313. doi:10.1002/anie.200902328
Return to citation in text: [1] -
Tomita, D.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 6946. doi:10.1021/ja901995a
Return to citation in text: [1] -
Hanhan, N. V.; Sahin, A. H.; Chang, T. W.; Fettinger, J. C.; Franz, A. K. Angew. Chem., Int. Ed. 2010, 49, 744. doi:10.1002/anie.200904393
Return to citation in text: [1] -
Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.; Ma, S.; Duan, W.; Wang, W. Chem.–Eur. J. 2011, 17, 7791. doi:10.1002/chem.201101025
Return to citation in text: [1] -
Zheng, K.; Yin, C.; Liu, X.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 2573. doi:10.1002/anie.201007145
Return to citation in text: [1] -
Jia, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040. doi:10.1039/b810858g
Return to citation in text: [1] -
Cheng, L.; Liu, L.; Wang, D.; Chen, Y.-J. Org. Lett. 2009, 11, 3874. doi:10.1021/ol901405r
Return to citation in text: [1] -
Shen, K.; Liu, X.; Wang, G.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 4684. doi:10.1002/anie.201100758
Return to citation in text: [1] -
Yan, W.; Wang, D.; Feng, J.; Li, P.; Zhao, D.; Wang, R. Org. Lett. 2012, 14, 2512–2515. doi:10.1021/ol3007953
Return to citation in text: [1] -
Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2010, 132, 15534. doi:10.1021/ja1079755
Return to citation in text: [1] -
Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735. doi:10.1038/nchem.730
Return to citation in text: [1] -
Jiang, K.; Jia, Z.-J.; Chen, S.; Wu, L.; Chen, Y.-C. Chem.–Eur. J. 2010, 16, 2852. doi:10.1002/chem.200903009
Return to citation in text: [1] -
Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2007, 46, 8666. doi:10.1002/anie.200703317
Return to citation in text: [1] -
He, R.; Ding, C.; Maruoka, K. Angew. Chem., Int. Ed. 2009, 48, 4559. doi:10.1002/anie.200901277
Return to citation in text: [1] -
Mai, C.-K.; Sammons, M. F.; Sammakia, T. Org. Lett. 2010, 12, 2306. doi:10.1021/ol100666v
Return to citation in text: [1] -
Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176. doi:10.1021/ja107858z
Return to citation in text: [1] -
Cao, Z.-Y.; Zhang, Y.; Ji, C.-B.; Zhou, J. Org. Lett. 2011, 13, 6398. doi:10.1021/ol202705g
Return to citation in text: [1] -
Liu, Y.-L.; Zhou, J. Chem. Commun. 2012, 48, 1919. doi:10.1039/C2CC17140F
Return to citation in text: [1] -
Liu, Y.-L.; Zhou, F.; Cao, J.-J.; Ji, C.-B.; Ding, M.; Zhou, J. Org. Biomol. Chem. 2010, 8, 3847. doi:10.1039/c0ob00174k
Return to citation in text: [1] -
Qian, Z.-Q.; Zhou, F.; Du, T.-P.; Wang, B.-L.; Ding, M.; Zhao, X.-L.; Zhou, J. Chem. Commun. 2009, 6753. doi:10.1039/b915257a
Return to citation in text: [1] -
Zhou, F.; Ding, M.; Liu, Y.-L.; Wang, C.-H.; Ji, C.-B.; Zhang, J.; Zhou, J. Adv. Synth. Catal. 2011, 353, 2945. doi:10.1002/adsc.201100379
Return to citation in text: [1] -
Ding, M.; Zhou, F.; Liu, Y.-L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Chem. Sci. 2011, 2, 2035. doi:10.1039/c1sc00390a
Return to citation in text: [1] -
Zhou, F.; Cao, Z.-Y.; Zhang, J.; Yang, H.-B.; Zhou, J. Chem.–Asian J. 2012, 7, 233. doi:10.1002/asia.201100773
Return to citation in text: [1] -
Alemán, J.; Richter, B.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 5515. doi:10.1002/anie.200701009
Return to citation in text: [1] -
Alemán, J.; Carbrera, S.; Maerten, E.; Overgaad, J.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 5520. doi:10.1002/anie.200701207
Return to citation in text: [1] -
Galzerano, P.; Bencivenni, G.; Pesciaioli, F.; Mazzanti, A.; Giannichi, B.; Sambri, L.; Bartoli, G.; Melchiorre, P. Chem.–Eur. J. 2009, 15, 7846. doi:10.1002/chem.200802466
Return to citation in text: [1] -
Bui, T.; Syed, S.; Barbas, C. F., III. J. Am. Chem. Soc. 2009, 131, 8758. doi:10.1021/ja903520c
Return to citation in text: [1] -
Kato, Y.; Furutachi, M.; Chen, Z.; Mitsunuma, H.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 9168. doi:10.1021/ja903566u
Return to citation in text: [1] -
Bravo, N.; Mon, I.; Companyó, X.; Alba, A.-N.; Moyano, A.; Rios, R. Tetrahedron Lett. 2009, 50, 6624. doi:10.1016/j.tetlet.2009.09.038
Return to citation in text: [1] -
Li, X.; Zhang, B.; Xi, Z.-G.; Luo, S.; Cheng, J.-P. Adv. Synth. Catal. 2010, 352, 416. doi:10.1002/adsc.200900630
Return to citation in text: [1] -
Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2011, 76, 4008. doi:10.1021/jo2004378
Return to citation in text: [1] -
Duan, S.-W.; An, J.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2011, 13, 2290. doi:10.1021/ol200550y
Return to citation in text: [1] -
Zheng, W.; Zhang, Z.; Kaplan, M. J.; Antilla, J. C. J. Am. Chem. Soc. 2011, 133, 3339. doi:10.1021/ja109824x
Return to citation in text: [1] -
Freund, M. H.; Tsogoeva, S. B. Synlett 2011, 503. doi:10.1055/s-0030-1259527
Return to citation in text: [1] -
Liu, L.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Commun. 2012, 48, 1692. doi:10.1039/c2cc17067a
Return to citation in text: [1] -
Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200. doi:10.1021/ol202624f
Return to citation in text: [1] -
Li, L.; Chen, W.; Yang, W.; Pan, Y.; Liu, H.; Tan, C.-H.; Jiang, Z. Chem. Commun. 2012, 48, 5124. doi:10.1039/c2cc31587d
Return to citation in text: [1] -
Liao, Y.-H.; Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. Chem.–Eur. J. 2012, 18, 6679. doi:10.1002/chem.201103293
Return to citation in text: [1] -
Wang, C.; Yang, X.; Enders, D. Chem.–Eur. J. 2012, 18, 4832. doi:10.1002/chem.201200079
Return to citation in text: [1] -
Bergonzini, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 971. doi:10.1002/anie.201107443
Return to citation in text: [1] -
Siau, W.-Y.; Li, W.; Xue, F.; Ren, Q.; Wu, M.; Sun, S.; Guo, H.; Jiang, X.; Wang, J. Chem.–Eur. J. 2012, 18, 9491. doi:10.1002/chem.201200206
During publication of our manuscript, Wang and coworkers reported the Michael addition of N-protected 3-substituted oxindoles to 1,4-naphthoquinone.
Return to citation in text: [1] -
The unprotected 3-substituted oxindoles are less reactive but easier to access than N-Boc protected 3-substituted oxindoles (for discuss, see [28]). While we focused on the use of unprotected 3-prochiral oxindoles for reaction development [26-28], we tried the reaction of N-Boc 3-phenyloxindole and 1,4-naphthoquinone, as suggested by the referee. At the standard reaction condition, the corresponding product was obtained in 47% yield with 67% ee.
Return to citation in text: [1]
| 26. | Qian, Z.-Q.; Zhou, F.; Du, T.-P.; Wang, B.-L.; Ding, M.; Zhao, X.-L.; Zhou, J. Chem. Commun. 2009, 6753. doi:10.1039/b915257a |
| 27. | Zhou, F.; Ding, M.; Liu, Y.-L.; Wang, C.-H.; Ji, C.-B.; Zhang, J.; Zhou, J. Adv. Synth. Catal. 2011, 353, 2945. doi:10.1002/adsc.201100379 |
| 28. | Ding, M.; Zhou, F.; Liu, Y.-L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Chem. Sci. 2011, 2, 2035. doi:10.1039/c1sc00390a |
| 1. | Shen, K.; Liu, X.; Lin, L.; Feng, X. Chem. Sci. 2012, 3, 327. doi:10.1039/c1sc00544h |
| 2. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381. doi:10.1002/adsc.201000161 |
| 3. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975 |
| 16. | Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2010, 132, 15534. doi:10.1021/ja1079755 |
| 17. | Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735. doi:10.1038/nchem.730 |
| 18. | Jiang, K.; Jia, Z.-J.; Chen, S.; Wu, L.; Chen, Y.-C. Chem.–Eur. J. 2010, 16, 2852. doi:10.1002/chem.200903009 |
| 19. | Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2007, 46, 8666. doi:10.1002/anie.200703317 |
| 20. | He, R.; Ding, C.; Maruoka, K. Angew. Chem., Int. Ed. 2009, 48, 4559. doi:10.1002/anie.200901277 |
| 48. | The unprotected 3-substituted oxindoles are less reactive but easier to access than N-Boc protected 3-substituted oxindoles (for discuss, see [28]). While we focused on the use of unprotected 3-prochiral oxindoles for reaction development [26-28], we tried the reaction of N-Boc 3-phenyloxindole and 1,4-naphthoquinone, as suggested by the referee. At the standard reaction condition, the corresponding product was obtained in 47% yield with 67% ee. |
| 11. | Zheng, K.; Yin, C.; Liu, X.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 2573. doi:10.1002/anie.201007145 |
| 12. | Jia, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040. doi:10.1039/b810858g |
| 13. | Cheng, L.; Liu, L.; Wang, D.; Chen, Y.-J. Org. Lett. 2009, 11, 3874. doi:10.1021/ol901405r |
| 14. | Shen, K.; Liu, X.; Wang, G.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 4684. doi:10.1002/anie.201100758 |
| 15. | Yan, W.; Wang, D.; Feng, J.; Li, P.; Zhao, D.; Wang, R. Org. Lett. 2012, 14, 2512–2515. doi:10.1021/ol3007953 |
| 28. | Ding, M.; Zhou, F.; Liu, Y.-L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Chem. Sci. 2011, 2, 2035. doi:10.1039/c1sc00390a |
| 5. | Shintani, R.; Inoue, M.; Hayashi, T. Angew. Chem., Int. Ed. 2006, 45, 3353. doi:10.1002/anie.200600392 |
| 6. | Ishimaru, T.; Shibata, N.; Nagai, J.; Nakamura, S.; Toru, T.; Kanemasa, S. J. Am. Chem. Soc. 2006, 128, 16488. doi:10.1021/ja0668825 |
| 7. | Itoh, J.; Han, S. B.; Krische, M. J. Angew. Chem., Int. Ed. 2009, 48, 6313. doi:10.1002/anie.200902328 |
| 8. | Tomita, D.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 6946. doi:10.1021/ja901995a |
| 9. | Hanhan, N. V.; Sahin, A. H.; Chang, T. W.; Fettinger, J. C.; Franz, A. K. Angew. Chem., Int. Ed. 2010, 49, 744. doi:10.1002/anie.200904393 |
| 10. | Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.; Ma, S.; Duan, W.; Wang, W. Chem.–Eur. J. 2011, 17, 7791. doi:10.1002/chem.201101025 |
| 32. | Galzerano, P.; Bencivenni, G.; Pesciaioli, F.; Mazzanti, A.; Giannichi, B.; Sambri, L.; Bartoli, G.; Melchiorre, P. Chem.–Eur. J. 2009, 15, 7846. doi:10.1002/chem.200802466 |
| 33. | Bui, T.; Syed, S.; Barbas, C. F., III. J. Am. Chem. Soc. 2009, 131, 8758. doi:10.1021/ja903520c |
| 34. | Kato, Y.; Furutachi, M.; Chen, Z.; Mitsunuma, H.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 9168. doi:10.1021/ja903566u |
| 35. | Bravo, N.; Mon, I.; Companyó, X.; Alba, A.-N.; Moyano, A.; Rios, R. Tetrahedron Lett. 2009, 50, 6624. doi:10.1016/j.tetlet.2009.09.038 |
| 36. | Li, X.; Zhang, B.; Xi, Z.-G.; Luo, S.; Cheng, J.-P. Adv. Synth. Catal. 2010, 352, 416. doi:10.1002/adsc.200900630 |
| 37. | Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2011, 76, 4008. doi:10.1021/jo2004378 |
| 38. | Duan, S.-W.; An, J.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2011, 13, 2290. doi:10.1021/ol200550y |
| 39. | Zheng, W.; Zhang, Z.; Kaplan, M. J.; Antilla, J. C. J. Am. Chem. Soc. 2011, 133, 3339. doi:10.1021/ja109824x |
| 40. | Freund, M. H.; Tsogoeva, S. B. Synlett 2011, 503. doi:10.1055/s-0030-1259527 |
| 41. | Liu, L.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Commun. 2012, 48, 1692. doi:10.1039/c2cc17067a |
| 42. | Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200. doi:10.1021/ol202624f |
| 43. | Li, L.; Chen, W.; Yang, W.; Pan, Y.; Liu, H.; Tan, C.-H.; Jiang, Z. Chem. Commun. 2012, 48, 5124. doi:10.1039/c2cc31587d |
| 44. | Liao, Y.-H.; Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. Chem.–Eur. J. 2012, 18, 6679. doi:10.1002/chem.201103293 |
| 45. | Wang, C.; Yang, X.; Enders, D. Chem.–Eur. J. 2012, 18, 4832. doi:10.1002/chem.201200079 |
| 46. | Bergonzini, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 971. doi:10.1002/anie.201107443 |
| 4. | Badillo, J. J.; Hanhan, N. V.; Franz, A. K. Curr. Opin. Drug Discovery Dev. 2010, 13, 758. |
| 47. |
Siau, W.-Y.; Li, W.; Xue, F.; Ren, Q.; Wu, M.; Sun, S.; Guo, H.; Jiang, X.; Wang, J. Chem.–Eur. J. 2012, 18, 9491. doi:10.1002/chem.201200206
During publication of our manuscript, Wang and coworkers reported the Michael addition of N-protected 3-substituted oxindoles to 1,4-naphthoquinone. |
| 26. | Qian, Z.-Q.; Zhou, F.; Du, T.-P.; Wang, B.-L.; Ding, M.; Zhao, X.-L.; Zhou, J. Chem. Commun. 2009, 6753. doi:10.1039/b915257a |
| 27. | Zhou, F.; Ding, M.; Liu, Y.-L.; Wang, C.-H.; Ji, C.-B.; Zhang, J.; Zhou, J. Adv. Synth. Catal. 2011, 353, 2945. doi:10.1002/adsc.201100379 |
| 29. | Zhou, F.; Cao, Z.-Y.; Zhang, J.; Yang, H.-B.; Zhou, J. Chem.–Asian J. 2012, 7, 233. doi:10.1002/asia.201100773 |
| 25. | Liu, Y.-L.; Zhou, F.; Cao, J.-J.; Ji, C.-B.; Ding, M.; Zhou, J. Org. Biomol. Chem. 2010, 8, 3847. doi:10.1039/c0ob00174k |
| 30. | Alemán, J.; Richter, B.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 5515. doi:10.1002/anie.200701009 |
| 31. | Alemán, J.; Carbrera, S.; Maerten, E.; Overgaad, J.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 5520. doi:10.1002/anie.200701207 |
| 22. | Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176. doi:10.1021/ja107858z |
| 23. | Cao, Z.-Y.; Zhang, Y.; Ji, C.-B.; Zhou, J. Org. Lett. 2011, 13, 6398. doi:10.1021/ol202705g |
| 24. | Liu, Y.-L.; Zhou, J. Chem. Commun. 2012, 48, 1919. doi:10.1039/C2CC17140F |
| 21. | Mai, C.-K.; Sammons, M. F.; Sammakia, T. Org. Lett. 2010, 12, 2306. doi:10.1021/ol100666v |
| 28. | Ding, M.; Zhou, F.; Liu, Y.-L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Chem. Sci. 2011, 2, 2035. doi:10.1039/c1sc00390a |
© 2012 Yu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)