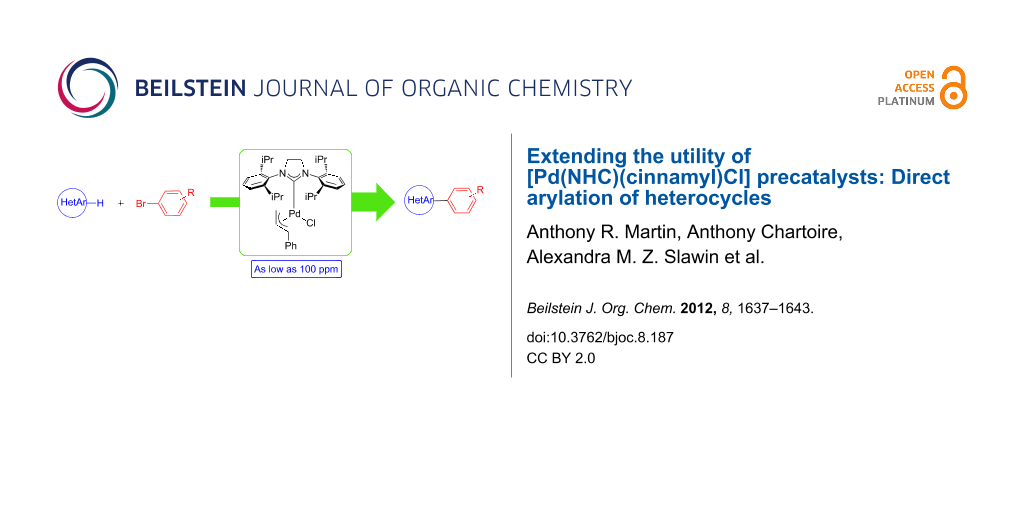
Abstract
The use of [Pd(NHC)(cinnamyl)Cl] precatalysts in the direct arylation of heterocycles has been investigated. Among four different precatalysts, [Pd(SIPr)(cinnamyl)Cl] proved to be the most efficient promoter of the reaction. The C–H functionalization of sulfur- or nitrogen-containing heterocycles has been achieved at low catalyst loadings. These catalyst charges range from 0.1 to 0.01 mol % palladium.

Graphical Abstract
Introduction
As a powerful addition to the classic palladium cross-coupling reactions, C–H bond functionalization has become a growing field of research over the last few years. The ubiquity of C–H bonds makes them a convenient and cost-effective anchoring position within viable substrates, as no derivatisation to form an organometallic reagent is required. Moreover, among the plethora of C–H bonds present on a molecule, it is often possible to target one C–H linkage specifically, taking advantage of directing groups or particular catalyst selectivity [1-5]. Thus, heteroaromatic scaffolds, which are a common feature in biologically relevant compounds and in materials science [6,7] can be selectively arylated as the heteroatom can act as an intrinsic orientating group [8].
Despite the efficiency of well-defined palladium catalysts bearing NHC (N-heterocyclic carbene) ancillary ligands in classical cross-coupling reactions, they have rarely been applied to direct arylation procedures [9-16]. Among the family of [Pd(NHC)] complexes, the [Pd(NHC)(cin)Cl] (cin = cinnamyl) species are known for their ease of activation through the reduction of the metal centre from Pd(II) to Pd(0) [17]. Therefore, we have investigated the use of such precatalysts in the direct arylation of heteroaromatic compounds in order to compare them to ligand-free or phosphine-bearing catalytic systems, and in the end to see whether the reactivity and application scope of these commercially available complexes could be broadened to include C–H bond functionalization transformations.
We now report the activity of the [Pd(NHC)(cin)Cl] complexes 1–4 in the direct arylation of heterocycles with NHC ligands being SIPr (1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene), IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene), IPr* (1,3-bis(2,6-bis(diphenylmethyl)-4-methylphenyl)imidazol-2-ylidene) and IPr*Tol (1,3-bis(2,6-bis(di-p-tolylmethyl)-4-methylphenyl)imidazol-2-ylidene) (Figure 1). Complexes 1 and 2 are commercially available and have proven to be highly efficient in Suzuki–Miyaura coupling and Buchwald–Hartwig amination reactions [17-20]. We have also evaluated the recently reported [Pd(IPr*)(cin)Cl] (3), which has shown potency in Suzuki–Miyaura couplings [21] and Buchwald–Hartwig N-arylations [22] even with challenging substrates. To complete this study and to examine the effect of bulky ligands about the metal centre, we have synthesised a new complex [Pd(IPr*Tol)(cin)Cl] (4), which is a IPr* congener.
![[1860-5397-8-187-1]](/bjoc/content/figures/1860-5397-8-187-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: [Pd(NHC)(cin)Cl] catalysts examined in direct arylation.
Figure 1: [Pd(NHC)(cin)Cl] catalysts examined in direct arylation.
Results and Discussion
The study begins with the preparation of the palladium complex 4. Following the strategy recently reported by Markó [23], we were successful in the synthesis of the IPr*Tol·HCl imidazolium salt 5 in a 53% overall yield (see Supporting Information File 1). Subsequently, 5 was treated with KOt-Bu in dry THF to generate the corresponding free carbene in situ. The expected [Pd(IPr*Tol)(cin)Cl] was then obtained in an excellent yield (97%) by a simple fragmentation of the palladium dimer [{Pd(cin)(µ-Cl)}2] using the free carbene solution (Scheme 1).
![[1860-5397-8-187-i1]](/bjoc/content/inline/1860-5397-8-187-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of [Pd(IPr*Tol)(cin)Cl] (4).
Scheme 1: Synthesis of [Pd(IPr*Tol)(cin)Cl] (4).
The newly synthesized complex 4 was unequivocally characterised by X-ray diffraction [24] (Figure 2, Supporting Information File 2 and Supporting Information File 3) after suitable crystals were grown from slow diffusion of hexane in dichloromethane. Based on this crystal structure, the percentage buried volume (%VBur) of the IPr*Tol ancillary ligand was determined by using the “SambVca” web application [25] and compared to complexes 1–3 (Table 1) [21]. IPr*Tol featured a %VBur in the same range as IPr* (+0.4% difference). SIPr and IPr have been reported as less hindered ligands with %VBur of 37.0 and 36.7, respectively. The length of the Pd–C1 bond in 4 was also examined and is close to the one observed in 3.
![[1860-5397-8-187-2]](/bjoc/content/figures/1860-5397-8-187-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of 4. H atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): Pd1–C1 2.034(0), Pd1–Cl1 2.352(5), Pd1–C85 2.132(8), Pd1–C86 2.119(7), Pd1–C87 2.226(6); C1–Pd1-C85 102.9(5), C85–Pd1–C87 71.2(6), C87–Pd1–Cl1 93.3(8), Cl1–Pd1–C1 91.8(6).
Figure 2: Molecular structure of 4. H atoms were omitted for clarity. Selected bond lengths (Å) and angles (°...
Table 1: Comparison of the %VBur and d(Pd–C1) in the [Pd(NHC)(cin)Cl] family.
| NHC | %VBura | Pd–C1 (Å) |
|---|---|---|
| SIPr | 37.0 | 2.025(7) |
| IPr | 36.7 | 2.041(9) |
| IPr* | 44.6 | 2.038(6) |
| IPr*Tol | 45.0 | 2.034(0) |
a%VBur calculated for a 2.00 Å Pd–C1 length.
With complexes 1–4 in hand, their catalytic activity towards the direct arylation of heteroaromatic compounds was evaluated. For this purpose, the arylation of benzothiophene (6) with 4-bromotoluene (7) was selected as a benchmark reaction (Table 2). This C–H functionalization, initially described by Ohta [26], was then reported by Bhanage and Mori using 2–10 mol % of well-defined palladium catalysts [27,28] (Figure 3). Alternatively, Fagnou and Kappe proposed a Pd/phosphine system involving 1–2 mol % of palladium and 2–4 mol % of phosphine [29,30], but no example of this reaction involving a well-defined [Pd(NHC)] complex has been described. However, it is noteworthy that variously substituted benzothiophene cores have been extensively studied in the direct arylation process [4,31-37].
Table 2: Catalyst screening for the direct arylation of benzothiophene (6).
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-187-i2.svg?max-width=637&scale=1.0)
|
|
| Catalyst | Conversion (%)a |
|---|---|
| [Pd(SIPr)(cin)Cl] (1) | 76 |
| [Pd(IPr)(cin)Cl] (2) | 50 |
| [Pd(IPr*)(cin)Cl] (3) | 8 |
| [Pd(IPr*Tol)(cin)Cl] (4) | 49 |
aConversion of the starting material into C–H arylated product determined by GC, [6] = 0.3 M.
![[1860-5397-8-187-3]](/bjoc/content/figures/1860-5397-8-187-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Previously reported catalytic systems in the direct arylation of benzothiophene (6).
Figure 3: Previously reported catalytic systems in the direct arylation of benzothiophene (6).
Initial screening of precatalysts 1–4 was performed with a 2 mol % loading, by using KOt-Bu as the base, which is known to efficiently activate the [Pd(NHC)(cin)Cl] precatalysts [17]. DMA was selected as the solvent and the reaction was conducted at 140 °C.
This survey showed that 1 is the most efficient precatalyst under these reaction conditions with 76% conversion of the starting material. Precatalysts 2 and 4 exhibited closely related activity, with 50 and 49% conversion, respectively. However, complex 3 gave relatively poor conversion of the benzothiophene (6).
Thus, selecting 1 as the best precatalyst, the use of other solvents, bases and additives was evaluated to optimize the reaction (see the Supporting Information File 1). From this optimization study, it was found that 0.1 mol % of 1 with K2CO3 in DMA as solvent at 140 °C in the presence of a catalytic amount of pivalic acid (30 mol %) generated the best reaction conditions. Under these optimized parameters, a second precatalyst screening was performed. As shown in Table 3, better activity was observed for precatalysts 1 and 2, which have smaller ligands when compared to the NHCs in 3 and 4. This result suggests a strong dependence of the activity on the steric properties of the NHC ligand. Moreover, the small difference between 1 and 2 underlines the fact that the difference in the σ-donation properties of the NHC ligands [38-41] is not likely to play a crucial role in the catalytic activity.
Table 3: Catalyst screening under optimised conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-187-i3.svg?max-width=637&scale=1.0)
|
|
| Catalyst | Conversion (%)a |
|---|---|
| [Pd(SIPr)(cin)Cl] (1) | 80 |
| [Pd(IPr)(cin)Cl] (2) | 75 |
| [Pd(IPr*)(cin)Cl] (3) | 57 |
| [Pd(IPr*Tol)(cin)Cl] (4) | 58 |
aConversion of the starting material into C–H arylated product determined by GC, [6] = 0.3 M.
In comparison with the previously mentioned methodologies to perform this C–H functionalization [27-30], the catalyst loading can be decreased by at least 10-fold without drastically affecting the yield (Table 4, entry 1). Using the optimized reaction conditions, we examined the scope and the limitations of this catalytic system using various aryl bromides and heterocycles (Table 4). It appeared that the sterics of the aryl bromide had almost no impact on the reaction. Indeed, para-, meta- and ortho- substituted aryl bromides could be employed to arylate 6 in good yields. (Table 4, entries 1–3, 77–89%). However, ortho-disubstituted aryl bromide, such as bromomesitylene appeared to be too sterically demanding and led to no conversion (data not shown). Concerning the electronic properties of the aryl bromide, electron-withdrawing (EWG) and electron-donating groups (EDG) were tolerated, although the presence of EWGs resulted in decreased yields (Table 4, entries 4–6, 49–70%). The substrate 4-bromobenzaldehyde was also successfully involved in the direct arylation of 6. Despite its electron-withdrawing nature as well as its high reactivity, the expected biaryl was obtained in moderate yield (Table 4, entry 7). The limits of the scope were determined by switching from benzothiophene (6) to the more sterically demanding 3-methylbenzothiophene (9) (Table 4, entries 8–10). Closely related reactivity was observed for 6 and 9, as these were arylated in comparable yields (Table 4, entry 1 vs 8, 3 vs 9 and 6 vs 10).
Table 4: Palladium-NHC catalysed direct arylation of heterocycles with arylbromides.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-187-i4.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Heterocycles | Products | R | Yieldb |
|---|---|---|---|---|
| 1c |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-187-i5.svg?max-width=637&scale=1.0)
6 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-187-i6.svg?max-width=637&scale=1.0)
8 |
4-Me, 8a | 89% |
| 2c | 3-Me, 8b | 80% | ||
| 3c | 2-Me, 8c | 77% | ||
| 4c | 4-OMe, 8d | 70% | ||
| 5c | 4-Cl, 8e | 49% | ||
| 6c | 4-F, 8f | 53% | ||
| 7c | 4-CHO, 8g | 37% | ||
| 8 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-187-i7.svg?max-width=637&scale=1.0)
9 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-187-i8.svg?max-width=637&scale=1.0)
10 |
4-Me, 10a | 85% |
| 9 | 2-Me, 10b | 83% | ||
| 10 | 4-F, 10c | 52% | ||
| 11 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-187-i9.svg?max-width=637&scale=1.0)
11 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-187-i10.svg?max-width=637&scale=1.0)
12 |
4-Me, 12a | 90% |
| 12 | 4-OMe, 12b | 75% | ||
| 13 | 4-F, 12c | 57% | ||
| 14 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-187-i11.svg?max-width=637&scale=1.0)
13 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-187-i12.svg?max-width=637&scale=1.0)
14 |
4-Me, 14a | 74% |
| 15d | 69% | |||
| 16d | 4-OMe, 14b | 53% | ||
| 17d | 4-F, 14c | 76% | ||
aUnless noted, reactions were performed on 0.6 mmol scale with: Heterocycle (1 equiv), aryl bromide (1 equiv), [Pd(SIPr)(cin)Cl] (0.1 mol %), PivOH (30 mol %), K2CO3 (1.5 equiv), DMA (2 mL), 140 °C. bIsolated yields, average of two independent runs. c6 (1.2 equiv). d[Pd(SIPr)(cin)Cl] (0.01 mol %).
A more challenging heterocycle, 2-methylthiophene (11), was investigated. Simple thiophene rings are known to be less reactive in C–H functionalization reactions [42]; nevertheless, 11 was successfully arylated in moderate to good yields, depending on the electronic properties of the bromobenzene substituents (Table 4, entries 11–13, 57–90%). Electron rich 4-methoxybromobenzene reacted more efficiently than the electron poor 4-fluorobromobenzene. An opposite effect of the electronics was observed by Doucet et al. in their ligandless procedure at low catalyst loadings [43,44]. This is surely due to the nature of the catalyst and thus offers complementary direct arylation methods for thiophene derivatives.
To complete the study, experiments were performed at lower catalyst loading using imidazopyridine (13). This class of substrate has recently been involved, by Doucet et al. [45], in direct arylation with a catalytic charge of Pd(OAc)2 ranging from 0.1 to 0.01 mol %. In our case, comparable yields were obtained when the catalyst loading was decreased from 0.1 to 0.01 mol %, highlighting the high efficiency of the catalytic system (Table 4, entries 14 and 15). Following the same trend as reported by Doucet [45], a better reactivity was observed with bromobenzenes substituted with EWGs compared to with EDGs (Table 4, entries 16 and 17).
Conclusion
In summary, we report here the synthesis and characterization of a new member of the [Pd(NHC)(cin)Cl] family, [Pd(IPr*Tol)(cin)Cl]. The catalytic activity of this family of complexes was surveyed in the direct arylation of heterocycles. The bulkiness of the NHC ligand appears to play a major role in the catalytic efficiency, whereas the σ-donation properties (within the small electronic space examined) have little influence. Among the four complexes, [Pd(SIPr)(cin)Cl] exhibited the highest catalytic efficiency and was investigated for the arylation of various benzothiophenes, thiophene and imidazopyridine. C–H functionalization of such heterocycles was performed in moderate to good yields by using only 0.1–0.01 mol % of precatalyst. This study highlights the fact that [Pd(NHC)(cin)Cl] complexes are multipurpose precatalysts as they may be utilised in various cross-coupling and, now, C–H-bond-functionalization reactions.
Experimental
General procedure for the direct arylation of heterocycles
In a glovebox, a vial containing a stirring bar was charged with K2CO3 (124 mg, 0.9 mmol, 1.5 equiv) and pivalic acid (0.18 mmol, 18 mg, 30 mol %), and sealed with a screw cap fitted with a septum. The heterocycle (0.6 mmol, 1.0 equiv) and/or the arylbromide (0.6 mmol, 1.0 equiv) were added at this point if in solid form, and DMA (1.9 mL) was poured into the vial. Outside of the glovebox, the heterocycle and/or the aryl bromide were added at this point if in liquid form. Finally, [Pd(SIPr)(cin)Cl] (1) was added as a 0.06 M solution in DMA (0.6–6 µmol, 10–100 µL, 0.01–0.1 mol %), and the vial was heated to 140 °C for 16 h. The solution was then cooled down to room temperature, diluted with 40 mL of ethyl acetate, and washed with water (2 × 20 mL) and brine (20 mL). The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude residue was finally purified by either trituration in pentane (if not soluble) or silica-gel column chromatography using pentane as the eluent.
Supporting Information
| Supporting Information File 1: Synthesis and characterization of complex 4; compound characterization data for all the direct arylated products and copies of their 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.9 MB | Download |
| Supporting Information File 2: CIF-Check for compound 4. | ||
| Format: PDF | Size: 124.1 KB | Download |
| Supporting Information File 3: Crystal structure data for compound 4. | ||
| Format: CIF | Size: 85.8 KB | Download |
Acknowledgements
We gratefully acknowledge the EPSRC and the ERC (Advanced Researcher Award-FUNCAT) for funding. We thank the EPSRC National Mass Spectrometry Service Center in Swansea for mass spectrometric analyses. Umicore AG is acknowledged for its generous gifts of material. S.P.N. is a Royal Society Wolfson Research Merit Award holder.
References
-
Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936–946. doi:10.1021/ar300014f
Return to citation in text: [1] -
Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. doi:10.1002/anie.201006368
Return to citation in text: [1] -
Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j
Return to citation in text: [1] -
Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015
Return to citation in text: [1] [2] -
Joucla, L.; Djakovitch, L. Adv. Synth. Catal. 2009, 351, 673–714. doi:10.1002/adsc.200900059
Return to citation in text: [1] -
Eicher, T.; Hauptmann, S.; Speicher, A. The chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] -
Dua, R.; Shrivastava, S.; Sonwane, S. K.; Srivastava, S. K. Adv. Biol. Res. 2011, 5, 120–144.
Return to citation in text: [1] -
Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760
Return to citation in text: [1] -
Kumar, P. V.; Lin, W.-S.; Shen, J.-S.; Nandi, D.; Lee, H. M. Organometallics 2011, 30, 5160–5169. doi:10.1021/om200490k
Return to citation in text: [1] -
Demir, S.; Özdemir, İ.; Arslan, H.; VanDerveer, D. J. Organomet. Chem. 2011, 696, 2589–2593. doi:10.1016/j.jorganchem.2011.03.040
Return to citation in text: [1] -
Chan, K.-T.; Tsai, Y.-H.; Lin, W.-S.; Wu, J.-R.; Chen, S.-J.; Liao, F.-X.; Hu, C.-H.; Lee, H. M. Organometallics 2010, 29, 463–472. doi:10.1021/om9009203
Return to citation in text: [1] -
Lane, B. S.; Brown, M. A.; Sames, D. J. Am. Chem. Soc. 2005, 127, 8050–8057. doi:10.1021/ja043273t
Return to citation in text: [1] -
Touré, B. B.; Lane, B. S.; Sames, D. Org. Lett. 2006, 8, 1979–1982. doi:10.1021/ol053021c
Return to citation in text: [1] -
Normand, A. T.; Cavell, K. J. Eur. J. Inorg. Chem. 2008, 2781–2800. doi:10.1002/ejic.200800323
Return to citation in text: [1] -
Joo, J. M.; Touré, B. B.; Sames, D. J. Org. Chem. 2010, 75, 4911–4920. doi:10.1021/jo100727j
Return to citation in text: [1] -
Ozdemir, İ.; Gök, Y.; Özeroğlu, Ö.; Kaloğlu, M.; Doucet, H.; Bruneau, C. Eur. J. Inorg. Chem. 2010, 1798–1805. doi:10.1002/ejic.200901195
Return to citation in text: [1] -
Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z
Return to citation in text: [1] [2] [3] -
Navarro, O.; Marion, N.; Mei, J.; Nolan, S. P. Chem.–Eur. J. 2006, 12, 5142–5148. doi:10.1002/chem.200600283
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 1440–1449. doi:10.1021/ar800020y
Return to citation in text: [1] -
Egbert, J. D.; Chartoire, A.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2011, 30, 4494–4496. doi:10.1021/om200579h
Return to citation in text: [1] -
Chartoire, A.; Lesieur, M.; Falivene, L.; Slawin, A. M. Z.; Cavallo, L.; Cazin, C. S. J.; Nolan, S. P. Chem.–Eur. J. 2012, 18, 4517–4521. doi:10.1002/chem.201104009
Return to citation in text: [1] [2] -
Chartoire, A.; Frogneux, X.; Nolan, S. P. Adv. Synth. Catal. 2012, 354, 1897–1901. doi:10.1002/adsc.201200207
Return to citation in text: [1] -
Berthon-Gelloz, G.; Siegler, M. A.; Spek, A. L.; Tinant, B.; Reek, J. N. H.; Markó, I. E. Dalton Trans. 2010, 39, 1444–1446. doi:10.1039/b921894g
Return to citation in text: [1] -
CCDC-887349 contains the crystallographic data for 4. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. Eur. J. Inorg. Chem. 2009, 1759–1766. doi:10.1002/ejic.200801160
Return to citation in text: [1] -
Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951–1958. doi:10.3987/COM-90-5467
Return to citation in text: [1] -
Nandurkar, N. S.; Bhanushali, M. J.; Bhor, M. D.; Bhanage, B. M. Tetrahedron Lett. 2008, 49, 1045–1048. doi:10.1016/j.tetlet.2007.11.209
Return to citation in text: [1] [2] -
Tamba, S.; Okubo, Y.; Tanaka, S.; Monguchi, D.; Mori, A. J. Org. Chem. 2010, 75, 6998–7001. doi:10.1021/jo101433g
Return to citation in text: [1] [2] -
Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. J. Org. Chem. 2009, 74, 1826–1834. doi:10.1021/jo8026565
Return to citation in text: [1] [2] -
Baghbanzadeh, M.; Pilger, C.; Kappe, C. O. J. Org. Chem. 2011, 76, 8138–8142. doi:10.1021/jo201516v
Return to citation in text: [1] [2] -
Martins, A.; Alberico, D.; Lautens, M. Org. Lett. 2006, 8, 4827–4829. doi:10.1021/ol061859+
Return to citation in text: [1] -
Martins, A.; Lautens, M. J. Org. Chem. 2008, 73, 8705–8710. doi:10.1021/jo8020105
Return to citation in text: [1] -
Takeda, D.; Yamashita, M.; Hirano, K.; Satoh, T.; Miura, M. Chem. Lett. 2011, 40, 1015–1017. doi:10.1246/cl.2011.1015
Return to citation in text: [1] -
Ohno, H.; Iuchi, M.; Fujii, N.; Tanaka, T. Org. Lett. 2007, 9, 4813–4815. doi:10.1021/ol702141r
Return to citation in text: [1] -
Fournier Dit Chabert, J.; Gozzi, C.; Lemaire, M. Tetrahedron Lett. 2002, 43, 1829–1833. doi:10.1016/S0040-4039(02)00128-4
Return to citation in text: [1] -
David, E.; Perrin, J.; Pellet-Rostaing, S.; Fournier Dit Chabert, J.; Lemaire, M. J. Org. Chem. 2005, 70, 3569–3573. doi:10.1021/jo0500378
Return to citation in text: [1] -
David, E.; Pellet-Rostaing, S.; Lemaire, M. Tetrahedron 2007, 63, 8999–9006. doi:10.1016/j.tet.2007.05.110
Return to citation in text: [1] -
Kelly, R. A., III; Clavier, H.; Giudice, S.; Scott, N. M.; Stevens, E. D.; Bordner, J.; Samardjiev, I.; Hoff, C. D.; Cavallo, L.; Nolan, S. P. Organometallics 2008, 27, 202–210. doi:10.1021/om701001g
Return to citation in text: [1] -
Dorta, R.; Stevens, E. D.; Scott, N. M.; Costabile, C.; Cavallo, L.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2005, 127, 2485–2495. doi:10.1021/ja0438821
Return to citation in text: [1] -
Fantasia, S.; Petersen, J. L.; Jacobsen, H.; Cavallo, L.; Nolan, S. P. Organometallics 2007, 26, 5880–5889. doi:10.1021/om700857j
Return to citation in text: [1] -
Gusev, D. G. Organometallics 2009, 28, 763–770. doi:10.1021/om800933x
Return to citation in text: [1] -
Lapointe, D.; Markiewicz, T.; Whipp, C. J.; Toderian, A.; Fagnou, K. J. Org. Chem. 2011, 76, 749–759. doi:10.1021/jo102081a
Return to citation in text: [1] -
Požgan, F.; Roger, J.; Doucet, H. ChemSusChem 2008, 1, 404–407. doi:10.1002/cssc.200700166
Return to citation in text: [1] -
Roger, J.; Požgan, F.; Doucet, H. Green Chem. 2009, 11, 425–432. doi:10.1039/b819912d
Return to citation in text: [1] -
Fu, H. Y.; Chen, L.; Doucet, H. J. Org. Chem. 2012, 77, 4473–4478. doi:10.1021/jo300528b
Return to citation in text: [1] [2]
| 27. | Nandurkar, N. S.; Bhanushali, M. J.; Bhor, M. D.; Bhanage, B. M. Tetrahedron Lett. 2008, 49, 1045–1048. doi:10.1016/j.tetlet.2007.11.209 |
| 28. | Tamba, S.; Okubo, Y.; Tanaka, S.; Monguchi, D.; Mori, A. J. Org. Chem. 2010, 75, 6998–7001. doi:10.1021/jo101433g |
| 29. | Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. J. Org. Chem. 2009, 74, 1826–1834. doi:10.1021/jo8026565 |
| 30. | Baghbanzadeh, M.; Pilger, C.; Kappe, C. O. J. Org. Chem. 2011, 76, 8138–8142. doi:10.1021/jo201516v |
| 17. | Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z |
| 38. | Kelly, R. A., III; Clavier, H.; Giudice, S.; Scott, N. M.; Stevens, E. D.; Bordner, J.; Samardjiev, I.; Hoff, C. D.; Cavallo, L.; Nolan, S. P. Organometallics 2008, 27, 202–210. doi:10.1021/om701001g |
| 39. | Dorta, R.; Stevens, E. D.; Scott, N. M.; Costabile, C.; Cavallo, L.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2005, 127, 2485–2495. doi:10.1021/ja0438821 |
| 40. | Fantasia, S.; Petersen, J. L.; Jacobsen, H.; Cavallo, L.; Nolan, S. P. Organometallics 2007, 26, 5880–5889. doi:10.1021/om700857j |
| 41. | Gusev, D. G. Organometallics 2009, 28, 763–770. doi:10.1021/om800933x |
| 1. | Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936–946. doi:10.1021/ar300014f |
| 2. | Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. doi:10.1002/anie.201006368 |
| 3. | Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j |
| 4. | Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015 |
| 5. | Joucla, L.; Djakovitch, L. Adv. Synth. Catal. 2009, 351, 673–714. doi:10.1002/adsc.200900059 |
| 17. | Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z |
| 29. | Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. J. Org. Chem. 2009, 74, 1826–1834. doi:10.1021/jo8026565 |
| 30. | Baghbanzadeh, M.; Pilger, C.; Kappe, C. O. J. Org. Chem. 2011, 76, 8138–8142. doi:10.1021/jo201516v |
| 9. | Kumar, P. V.; Lin, W.-S.; Shen, J.-S.; Nandi, D.; Lee, H. M. Organometallics 2011, 30, 5160–5169. doi:10.1021/om200490k |
| 10. | Demir, S.; Özdemir, İ.; Arslan, H.; VanDerveer, D. J. Organomet. Chem. 2011, 696, 2589–2593. doi:10.1016/j.jorganchem.2011.03.040 |
| 11. | Chan, K.-T.; Tsai, Y.-H.; Lin, W.-S.; Wu, J.-R.; Chen, S.-J.; Liao, F.-X.; Hu, C.-H.; Lee, H. M. Organometallics 2010, 29, 463–472. doi:10.1021/om9009203 |
| 12. | Lane, B. S.; Brown, M. A.; Sames, D. J. Am. Chem. Soc. 2005, 127, 8050–8057. doi:10.1021/ja043273t |
| 13. | Touré, B. B.; Lane, B. S.; Sames, D. Org. Lett. 2006, 8, 1979–1982. doi:10.1021/ol053021c |
| 14. | Normand, A. T.; Cavell, K. J. Eur. J. Inorg. Chem. 2008, 2781–2800. doi:10.1002/ejic.200800323 |
| 15. | Joo, J. M.; Touré, B. B.; Sames, D. J. Org. Chem. 2010, 75, 4911–4920. doi:10.1021/jo100727j |
| 16. | Ozdemir, İ.; Gök, Y.; Özeroğlu, Ö.; Kaloğlu, M.; Doucet, H.; Bruneau, C. Eur. J. Inorg. Chem. 2010, 1798–1805. doi:10.1002/ejic.200901195 |
| 4. | Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015 |
| 31. | Martins, A.; Alberico, D.; Lautens, M. Org. Lett. 2006, 8, 4827–4829. doi:10.1021/ol061859+ |
| 32. | Martins, A.; Lautens, M. J. Org. Chem. 2008, 73, 8705–8710. doi:10.1021/jo8020105 |
| 33. | Takeda, D.; Yamashita, M.; Hirano, K.; Satoh, T.; Miura, M. Chem. Lett. 2011, 40, 1015–1017. doi:10.1246/cl.2011.1015 |
| 34. | Ohno, H.; Iuchi, M.; Fujii, N.; Tanaka, T. Org. Lett. 2007, 9, 4813–4815. doi:10.1021/ol702141r |
| 35. | Fournier Dit Chabert, J.; Gozzi, C.; Lemaire, M. Tetrahedron Lett. 2002, 43, 1829–1833. doi:10.1016/S0040-4039(02)00128-4 |
| 36. | David, E.; Perrin, J.; Pellet-Rostaing, S.; Fournier Dit Chabert, J.; Lemaire, M. J. Org. Chem. 2005, 70, 3569–3573. doi:10.1021/jo0500378 |
| 37. | David, E.; Pellet-Rostaing, S.; Lemaire, M. Tetrahedron 2007, 63, 8999–9006. doi:10.1016/j.tet.2007.05.110 |
| 8. | Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760 |
| 26. | Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951–1958. doi:10.3987/COM-90-5467 |
| 6. | Eicher, T.; Hauptmann, S.; Speicher, A. The chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003. |
| 7. | Dua, R.; Shrivastava, S.; Sonwane, S. K.; Srivastava, S. K. Adv. Biol. Res. 2011, 5, 120–144. |
| 27. | Nandurkar, N. S.; Bhanushali, M. J.; Bhor, M. D.; Bhanage, B. M. Tetrahedron Lett. 2008, 49, 1045–1048. doi:10.1016/j.tetlet.2007.11.209 |
| 28. | Tamba, S.; Okubo, Y.; Tanaka, S.; Monguchi, D.; Mori, A. J. Org. Chem. 2010, 75, 6998–7001. doi:10.1021/jo101433g |
| 23. | Berthon-Gelloz, G.; Siegler, M. A.; Spek, A. L.; Tinant, B.; Reek, J. N. H.; Markó, I. E. Dalton Trans. 2010, 39, 1444–1446. doi:10.1039/b921894g |
| 25. | Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. Eur. J. Inorg. Chem. 2009, 1759–1766. doi:10.1002/ejic.200801160 |
| 45. | Fu, H. Y.; Chen, L.; Doucet, H. J. Org. Chem. 2012, 77, 4473–4478. doi:10.1021/jo300528b |
| 22. | Chartoire, A.; Frogneux, X.; Nolan, S. P. Adv. Synth. Catal. 2012, 354, 1897–1901. doi:10.1002/adsc.201200207 |
| 21. | Chartoire, A.; Lesieur, M.; Falivene, L.; Slawin, A. M. Z.; Cavallo, L.; Cazin, C. S. J.; Nolan, S. P. Chem.–Eur. J. 2012, 18, 4517–4521. doi:10.1002/chem.201104009 |
| 45. | Fu, H. Y.; Chen, L.; Doucet, H. J. Org. Chem. 2012, 77, 4473–4478. doi:10.1021/jo300528b |
| 21. | Chartoire, A.; Lesieur, M.; Falivene, L.; Slawin, A. M. Z.; Cavallo, L.; Cazin, C. S. J.; Nolan, S. P. Chem.–Eur. J. 2012, 18, 4517–4521. doi:10.1002/chem.201104009 |
| 42. | Lapointe, D.; Markiewicz, T.; Whipp, C. J.; Toderian, A.; Fagnou, K. J. Org. Chem. 2011, 76, 749–759. doi:10.1021/jo102081a |
| 17. | Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z |
| 18. | Navarro, O.; Marion, N.; Mei, J.; Nolan, S. P. Chem.–Eur. J. 2006, 12, 5142–5148. doi:10.1002/chem.200600283 |
| 19. | Marion, N.; Nolan, S. P. Acc. Chem. Res. 2008, 41, 1440–1449. doi:10.1021/ar800020y |
| 20. | Egbert, J. D.; Chartoire, A.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2011, 30, 4494–4496. doi:10.1021/om200579h |
| 24. | CCDC-887349 contains the crystallographic data for 4. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 43. | Požgan, F.; Roger, J.; Doucet, H. ChemSusChem 2008, 1, 404–407. doi:10.1002/cssc.200700166 |
| 44. | Roger, J.; Požgan, F.; Doucet, H. Green Chem. 2009, 11, 425–432. doi:10.1039/b819912d |
© 2012 Martin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)