Abstract
Recently, a novel chiral cubane-based Schiff base ligand was reported to yield modest enantioselectivity in the Henry reaction. To further explore the utility of this ligand in other asymmetric organic transformations, we evaluated its stereoselectivity in cyclopropanation and Michael addition reactions. Although there was no increase in stereocontrol, upon computational evaluation using both M06L and B3LYP calculations, it was revealed that a pseudo six-membered ring exists, through H-bonding of a cubyl hydrogen to the copper core. This decreases the steric bulk above the copper center and limits the asymmetric control with this ligand.

Graphical Abstract
Introduction
Since the initial synthesis of cubane in 1964 by Eaton and Cole [1,2], numerous studies have been undertaken on its derivatives: Nitrated cubanes are remarkably explosive [3,4]; cubylamines possess antiviral activity [5]; and cubylamides have been shown to be P2X7 receptor antagonists [6]; whereas other cubane derivatives were examined as narcotic antagonists against both μ and ĸ receptors [7], as well as being monoamine oxidase (MAO) inactivators [8-10]. Cubanes have also shown a propensity to undergo cage opening. In particular, syn-tricyclooctadienes are formed when rhodium(I) salts are introduced [11], while with silver(I) or palladium(II) catalysts, cuneane is obtained [12]. Spontaneous cage opening has been observed with cubanol yielding vinylcyclobutenylketene [13,14], whereas dicubyl disulfide is remarkably stable [15]. More recently, 4-iodo-1-vinylcubane was shown to undergo cage opening/rearrangement to form 4-vinyl-trans-β-iodostyrene [16,17], whereas 1-iodocubane-4-carboxaldehyde undergoes cage opening/fragmentation to afford benzaldehyde, which further reacts to give benzyl benzoate [18].
Recently, we synthesized the first cubane-based Schiff base ligand (Figure 1) and screened it in the Henry reaction in the synthesis of β-nitroalcohols [19]. The cubyl moiety can be considered a cross between a tert-butyl and a phenyl group. In fact, the C–H bond of cubane has been shown to have ~31% s-character [20], compared to 25% for a simple alkane and 33% for an aromatic hydrogen. Due to the bulk of the cube it was initially envisioned that there would be high stereocontrol; however, the stereoselectivity was modest-at-best with only the highest ee value of 39% being achieved when copper(I) chloride was used at 65 °C. Therefore, we decided to examine this novel ligand further in the hope of increasing the stereoselectivity in other organic transformations.
![[1860-5397-8-207-1]](/bjoc/content/figures/1860-5397-8-207-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of (1R,2R)-N,N’-bis[(4-iodocuban-1-yl)methylene]-trans-1,2-diamino cyclohexane (1).
Figure 1: Structure of (1R,2R)-N,N’-bis[(4-iodocuban-1-yl)methylene]-trans-1,2-diamino cyclohexane (1).
Results and Discussion
We began by initially evaluating this cubane-based ligand with cyclopropanation reactions. When no chiral ligand was added there was a 2.6:1 ratio of trans to cis products with no ee control. We then introduced our cubane-based chiral ligand 1 to our cyclopropanation protocol with four different copper sources (Table 1). The highest ee value of 14% was found with the use of Cu(I) triflate tetrakisacetonitrile as catalyst. For the reaction with Cu(I) chloride, values of only 1% ee were obtained for both cis and trans products. In all reactions, the trans product was favored over the cis. Overall, this reaction was unsuccessful in obtaining high stereoselectivity, and thus we decided to switch to Michael addition with organomagnesium and organozinc reagents.
Table 1: Cyclopropanation with cubane ligand 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-207-i1.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst precursor | Conv (%)a | cis/transb |
eeb
cis (%) |
eeb
trans (%) |
|---|---|---|---|---|---|
| 1 | Cu(OTf) toluene complex | 53 | 45:55 | 9 | 5 |
| 2 | Cu(OTf) tetrakisacetonitrile | 13 | 29:71 | 14 | 9 |
| 3 | CuCl | 77 | 37:63 | 1 | 1 |
| 4 | Cu(OTf)2 | 82 | 42:58 | 8 | 6 |
aThe conversion was determined by analysis of the 1H NMR spectra. bDetermined by GC analysis using a Chirasil-Dextrin CB column.
Since the copper source that produced the highest ee value with the cyclopropanation above was Cu(I) triflate tetrakisacetonitrile, we decided to initially focus on this catalyst precursor. We were pleased to observe an ee value of 16% in the Michael addition of EtMgBr to 2-cyclohexen-1-one at 0 °C in Et2O (Table 2, entry 1). We next screened a variety of solvents in this reaction. With both CH2Cl2 and THF the ee values significantly dropped (Table 2, entry 2 and 3). As previously reported, CuCl gave the best results with the Henry reaction [19]; hence, we decided to evaluate this copper source. This, however, did not yield any encouraging results with only a maximum ee value of 4% when performed in Et2O (Table 2, entry 4). We next evaluated a Cu(OTf) toluene complex, as well as Cu(OTf)2; however, we only obtained a maximum value of 12% ee with Cu(OTf) 2 in Et2O (Table 2, entry 10). The use of Et2Zn has also been used to provide excellent stereoselectivity in Michael addition reactions [21,22]. However, with our cubane-based chiral ligand 1 we did not have this success. Our best result with this anion source was with Cu(OTf)2 in Et2O reaching an ee value of only 12% (Table 2, entry 20).
Table 2: Asymmetric ethyl addition to 2-cyclohexen-1-one under different conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-207-i2.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst precursor | Alkylating reagent | Solvent | Conv (%)a | ee (%)b |
|---|---|---|---|---|---|
| 1 | Cu(OTf) tetrakisacetonitrile | EtMgBr | Et2O | >99 | 16 (R) |
| 2 | Cu(OTf) tetrakisacetonitrile | EtMgBr | CH2Cl2 | 89 | <1 (S) |
| 3 | Cu(OTf) tetrakisacetonitrile | EtMgBr | THF | >99 | 3 (R) |
| 4 | CuCl | EtMgBr | Et2O | >99 | 4 (S) |
| 5 | CuCl | EtMgBr | CH2Cl2 | 92 | 1 (S) |
| 6 | CuCl | EtMgBr | THF | >99 | 2 (R) |
| 7 | Cu(OTf) toluene complex | EtMgBr | Et2O | >99 | 1 (R) |
| 8 | Cu(OTf) toluene complex | EtMgBr | CH2Cl2 | >99 | 15 (S) |
| 9 | Cu(OTf) toluene complex | EtMgBr | THF | 94 | 11 (R) |
| 10 | Cu(OTf)2 | EtMgBr | Et2O | >99 | 12 (S) |
| 11 | Cu(OTf)2 | EtMgBr | CH2Cl2 | >99 | 5 (S) |
| 12 | Cu(OTf)2 | EtMgBr | THF | >99 | 1 (S) |
| 13 | Cu(OTf) tetrakisacetonitrile | Et2Zn | Et2O | 86 | 3 (S) |
| 14 | Cu(OTf) tetrakisacetonitrile | Et2Zn | CH2Cl2 | 87 | 9 (R) |
| 15 | CuCl | Et2Zn | Et2O | >99 | 9 (S) |
| 16 | CuCl | Et2Zn | CH2Cl2 | >99 | 11 (S) |
| 17 | CuCl | Et2Zn | THF | >99 | 4 (S) |
| 18 | Cu(OTf) toluene complex | Et2Zn | Et2O | >99 | 4 (S) |
| 19 | Cu(OTf) toluene complex | Et2Zn | CH2Cl2 | >99 | 1 (S) |
| 20 | Cu(OTf)2 | Et2Zn | Et2O | >99 | 12 (S) |
| 21 | Cu(OTf)2 | Et2Zn | CH2Cl2 | 97 | 6 (S) |
| 22 | Cu(OTf)2 | Et2Zn | THF | >99 | 1 (S) |
aThe conversion was determined by analysis of the 1H NMR spectra. bDetermined by HPLC (OD Column, 0.5 mL/min, 99.7:0.3 hexanes/iPrOH) after derivatization with (R,R)-1,2-diphenylethane-1,2-diol.
At the onset we had anticipated much stronger results as the large bulky nature of the cube should likely block one side of the copper complex, thus hopefully increasing stereoselectivity. Appropriate crystals for X-ray analysis were not obtained, thus we decided to look into computational simulations. On the basis of earlier assessment studies [23,24], we chose the M06L [25] density functional theory method (and B3LYP [26] for comparison) to locate the global minimum-energy structures for the ligands and their Cu(I) complexes. The computations employed the 6-31+G(d) basis set, and for iodine the LAN2DZdp basis set [27] with an effective core potential was used instead of 6-31+G(d). All calculations were performed with Gaussian09 [28].
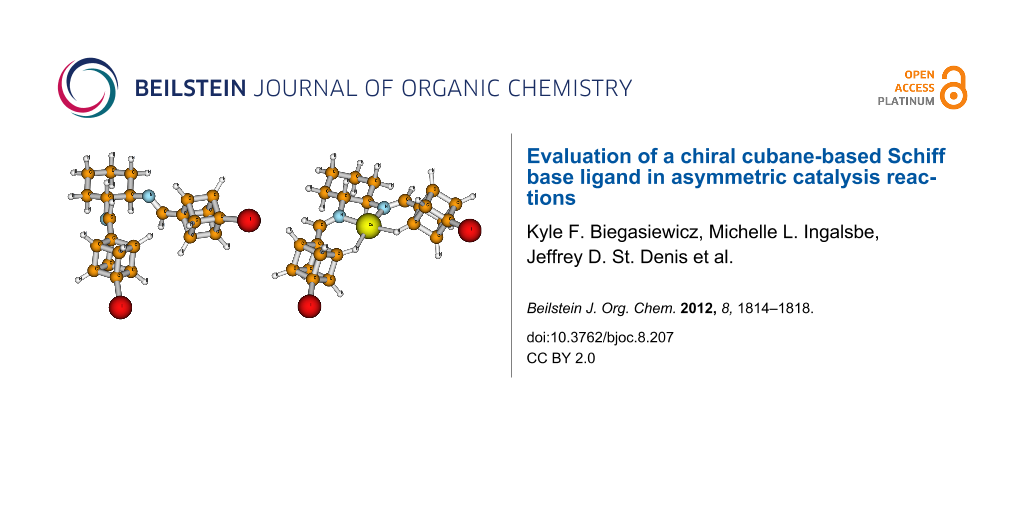
Using both B3LYP and M06L density functional theory methods, we observed N–C–C–N dihedral angles of 65.0 and 62.7°, respectively for the ligand 1 on its own (Figure 2a). When this was complexed with a Cu+1, however, the cyclohexyl moiety retained its chair conformation, but the N–C–C–N dihedral angles dropped to 49.7° and 48.5°, in the B3LYP and M06L calculations, respectively (Figure 2b). In addition, the copper is observed to exhibit either an agostic interaction [29] or a H-bond [30] with the “meta” cubyl hydrogens (with respect to the iodine). The H…Cu bond lengths of 2.06 Å with M06L and 2.29 Å with B3LYP calculations are well within the required H-bonding length of <3.2 Å [31]. In addition, the C…Cu lengths of 2.85 and 3.03 Å with M06L and B3LYP, respectively, were well within the required 4.0 Å limit [31]. This produces pseudo six-membered rings having a C–H…Cu bond angle of 125.1° or 122.9° with M06L and B3LYP calculations, respectively. As Cu(I) is a late transition metal in a low oxidation state [32], the H…Cu bond length is shorter than that of C…Cu [27], and the C–H…Cu bond angle is >100° [33], it can be stated that the H…Cu interaction is a true intermolecular multicenter hetero-acceptor hydrogen bond as opposed to an intermolecular pseudo-agostic bond. Cubyl hydrogens participating in H-bonding have been reported previously with nitrocubanes [34], as well as with dicubyl vic-disulfone [35]. From the computational results it would appear that the cubes play very little role in blocking either side for selective coordination with the copper. In fact, it would appear that the very modest selectivity is due to the axial hydrogens on the cyclohexyl moiety since the copper is best described as being strained square planar with a N–Cu–H bond angle of 93.7° for M06L and 91.0° for B3LYP.
![[1860-5397-8-207-2]](/bjoc/content/figures/1860-5397-8-207-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: M06L DFT global minima for (a) (1R,2R)-N,N’-bis[(4-iodocuban-1-yl)methylene]-trans-1,2-diaminocyclohexane (1) and (b) Cu+1 complex with ligand 1.
Figure 2: M06L DFT global minima for (a) (1R,2R)-N,N’-bis[(4-iodocuban-1-yl)methylene]-trans-1,2-diaminocyclo...
Conclusion
We obtained slight stereoselectivity for both cyclopropanation as well as Michael addition. A maximum ee value of 16% was observed when Michael addition was performed with EtMgBr at 0 °C in Et2O. The poor stereoselectivity is explained computationally by the lack of steric hindrance on either face of the copper complex. In addition, H-bonding with cubyl hydrogens and the copper core yielded a pseudo six-membered ring, which decreased the N–C–C–N dihedral angle.
Supporting Information
| Supporting Information File 1: Gaussian archives of (1R,2R)-N,N’-bis[(4-iodocuban-1-yl)methylene]-trans-1,2-diaminocyclohexane (1) and Cu+1 complex with ligand 1. | ||
| Format: PDF | Size: 283.9 KB | Download |
Acknowledgements
The authors thank the Niagara University Academic Center for Integrated Science and the Rochester Academy of Science for their financial support. MLI would like to thank the Barbara S. Zimmer Memorial Research Award for financial aid. MLC gratefully acknowledges generous allocations of supercomputing time from the Australian National Computational Infrastructure, support from the Australian Research Council under its Centers of Excellence program, and an ARC Future Fellowship. RP would also like to thank Western New England University, College of Pharmacy for generous financial support.
References
-
Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 962. doi:10.1021/ja01059a072
Return to citation in text: [1] -
Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 3157. doi:10.1021/ja01069a041
Return to citation in text: [1] -
Eaton, P. E.; Zhang, M.-X.; Gilardi, R.; Gelber, N.; Iyer, S.; Surapaneni, R. Propellants, Explos., Pyrotech. 2002, 27, 1. doi:10.1002/1521-4087(200203)27:1<1::AID-PREP1>3.0.CO;2-6
Return to citation in text: [1] -
Zhang, M.-X.; Eaton, P. E.; Gilardi, R. Angew. Chem., Int. Ed. 2000, 39, 401. doi:10.1002/(SICI)1521-3773(20000117)39:2<401::AID-ANIE401>3.0.CO;2-P
Return to citation in text: [1] -
Camps, P.; Duque, M. D.; Vázquez, S.; Naesens, L.; De Clercq, E.; Sureda, F. X.; López-Querol, M.; Camins, A.; Pallàs, M.; Prathalingam, S. R.; Kelly, J. M.; Romero, V.; Ivorra, D.; Cortés, D. Bioorg. Med. Chem. 2008, 16, 9925. doi:10.1016/j.bmc.2008.10.028
Return to citation in text: [1] -
Gunosewoyo, H.; Guo, J. L.; Bennett, M. R.; Coster, M. J.; Kassiou, M. Bioorg. Med. Chem. Lett. 2008, 18, 3720. doi:10.1016/j.bmcl.2008.05.062
Return to citation in text: [1] -
Cheng, C.-Y.; Hsin, L.-W.; Lin, Y.-P.; Tao, P.-L.; Jong, T.-T. Bioorg. Med. Chem. 1996, 4, 73. doi:10.1016/0968-0896(95)00165-4
Return to citation in text: [1] -
Zhou, J. J. P.; Li, J.; Upadhyaya, S.; Eaton, P. E.; Silverman, R. B. J. Med. Chem. 1997, 40, 1165. doi:10.1021/jm9606249
Return to citation in text: [1] -
Silverman, R. B.; Zhou, J. J. P.; Eaton, P. E. J. Am. Chem. Soc. 1993, 115, 8841. doi:10.1021/ja00072a048
Return to citation in text: [1] -
Silverman, R. B. Acc. Chem. Res. 1995, 28, 335. doi:10.1021/ar00056a003
Return to citation in text: [1] -
Cassar, L.; Eaton, P. E.; Halpern, J. J. Am. Chem. Soc. 1970, 92, 3515. doi:10.1021/ja00714a075
Return to citation in text: [1] -
Eaton, P. E.; Cassar, L.; Halpern, J. J. Am. Chem. Soc. 1970, 92, 6366. doi:10.1021/ja00724a061
Return to citation in text: [1] -
Cole, T. W., Jr. Ph.D. Thesis, University of Chicago, 1966.
Return to citation in text: [1] -
Hormann, R. E. Ph.D. Thesis, University of Chicago, 1987.
Return to citation in text: [1] -
Priefer, R.; Lee, Y. J.; Barrios, F.; Wosnick, J. H.; Lebuis, A.-M.; Farrell, P. G.; Harpp, D. N.; Sun, A.; Wu, S.; Snyder, J. P. J. Am. Chem. Soc. 2002, 124, 5626. doi:10.1021/ja025823y
Return to citation in text: [1] -
Carroll, V. M.; Harpp, D. N.; Priefer, R. Tetrahedron Lett. 2008, 49, 2677. doi:10.1016/j.tetlet.2008.02.161
Return to citation in text: [1] -
Griffiths, J. R.; Tsanaktsidis, J.; Savage, G. P.; Priefer, R. Thermochim. Acta 2010, 499, 15. doi:10.1016/j.tca.2009.10.015
Return to citation in text: [1] -
Heaphy, P. J.; Griffiths, J. R.; Dietz, C. J.; Savage, G. P.; Priefer, R. Tetrahedron Lett. 2011, 52, 6359. doi:10.1016/j.tetlet.2011.09.037
Return to citation in text: [1] -
Ingalsbe, M. L.; St Denis, J. D.; Gleason, J. L.; Savage, G. P.; Priefer, R. Synthesis 2010, 98. doi:10.1055/s-0029-1217083
Return to citation in text: [1] [2] -
Della, E. W.; Hine, P. T.; Patney, H. K. J. Org. Chem. 1977, 42, 2940. doi:10.1021/jo00437a040
Return to citation in text: [1] -
Shibata, N.; Yoshimura, M.; Yamada, H.; Arakawa, R.; Sakaguchi, S. J. Org. Chem. 2012, 77, 4079. doi:10.1021/jo300472r
Return to citation in text: [1] -
Matsumoto, Y.; Yamada, K.-i.; Tomioka, K. J. Org. Chem. 2008, 73, 4578. doi:10.1021/jo800613h
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157. doi:10.1021/ar700111a
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. doi:10.1063/1.2370993
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi:10.1063/1.464913
Return to citation in text: [1] -
Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284. doi:10.1063/1.448800
Return to citation in text: [1] [2] -
Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] -
Brookhart, M.; Green, M. L. H. J. Organomet. Chem. 1983, 250, 395. doi:10.1016/0022-328X(83)85065-7
Return to citation in text: [1] -
Thakur, T. S.; Desiraju, G. R. Chem. Commun. 2006, 552. doi:10.1039/b514427b
Return to citation in text: [1] -
Jeffrey, G. A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, 1997.
Return to citation in text: [1] [2] -
Braga, D.; Grepioni, F.; Desiraju, G. R. Chem. Rev. 1998, 98, 1375. doi:10.1021/cr960091b
Return to citation in text: [1] -
Braga, D.; Grepioni, F.; Tedesco, E.; Biradha, K.; Desiraju, G. R. Organometallics 1997, 16, 1846. doi:10.1021/om9608364
Return to citation in text: [1] -
Zhou, G.; Zhang, J.-L.; Wong, N.-B.; Tian, A. J. Mol. Struct. 2003, 639, 43. doi:10.1016/j.theochem.2003.07.007
Return to citation in text: [1] -
Priefer, R.; Martineau, E.; Harpp, D. N. J. Sulfur Chem. 2007, 28, 529. doi:10.1080/17415990701684701
Return to citation in text: [1]
| 33. | Braga, D.; Grepioni, F.; Tedesco, E.; Biradha, K.; Desiraju, G. R. Organometallics 1997, 16, 1846. doi:10.1021/om9608364 |
| 34. | Zhou, G.; Zhang, J.-L.; Wong, N.-B.; Tian, A. J. Mol. Struct. 2003, 639, 43. doi:10.1016/j.theochem.2003.07.007 |
| 35. | Priefer, R.; Martineau, E.; Harpp, D. N. J. Sulfur Chem. 2007, 28, 529. doi:10.1080/17415990701684701 |
| 1. | Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 962. doi:10.1021/ja01059a072 |
| 2. | Eaton, P. E.; Cole, T. W. J. Am. Chem. Soc. 1964, 86, 3157. doi:10.1021/ja01069a041 |
| 7. | Cheng, C.-Y.; Hsin, L.-W.; Lin, Y.-P.; Tao, P.-L.; Jong, T.-T. Bioorg. Med. Chem. 1996, 4, 73. doi:10.1016/0968-0896(95)00165-4 |
| 19. | Ingalsbe, M. L.; St Denis, J. D.; Gleason, J. L.; Savage, G. P.; Priefer, R. Synthesis 2010, 98. doi:10.1055/s-0029-1217083 |
| 6. | Gunosewoyo, H.; Guo, J. L.; Bennett, M. R.; Coster, M. J.; Kassiou, M. Bioorg. Med. Chem. Lett. 2008, 18, 3720. doi:10.1016/j.bmcl.2008.05.062 |
| 21. | Shibata, N.; Yoshimura, M.; Yamada, H.; Arakawa, R.; Sakaguchi, S. J. Org. Chem. 2012, 77, 4079. doi:10.1021/jo300472r |
| 22. | Matsumoto, Y.; Yamada, K.-i.; Tomioka, K. J. Org. Chem. 2008, 73, 4578. doi:10.1021/jo800613h |
| 5. | Camps, P.; Duque, M. D.; Vázquez, S.; Naesens, L.; De Clercq, E.; Sureda, F. X.; López-Querol, M.; Camins, A.; Pallàs, M.; Prathalingam, S. R.; Kelly, J. M.; Romero, V.; Ivorra, D.; Cortés, D. Bioorg. Med. Chem. 2008, 16, 9925. doi:10.1016/j.bmc.2008.10.028 |
| 19. | Ingalsbe, M. L.; St Denis, J. D.; Gleason, J. L.; Savage, G. P.; Priefer, R. Synthesis 2010, 98. doi:10.1055/s-0029-1217083 |
| 3. | Eaton, P. E.; Zhang, M.-X.; Gilardi, R.; Gelber, N.; Iyer, S.; Surapaneni, R. Propellants, Explos., Pyrotech. 2002, 27, 1. doi:10.1002/1521-4087(200203)27:1<1::AID-PREP1>3.0.CO;2-6 |
| 4. | Zhang, M.-X.; Eaton, P. E.; Gilardi, R. Angew. Chem., Int. Ed. 2000, 39, 401. doi:10.1002/(SICI)1521-3773(20000117)39:2<401::AID-ANIE401>3.0.CO;2-P |
| 20. | Della, E. W.; Hine, P. T.; Patney, H. K. J. Org. Chem. 1977, 42, 2940. doi:10.1021/jo00437a040 |
| 13. | Cole, T. W., Jr. Ph.D. Thesis, University of Chicago, 1966. |
| 14. | Hormann, R. E. Ph.D. Thesis, University of Chicago, 1987. |
| 16. | Carroll, V. M.; Harpp, D. N.; Priefer, R. Tetrahedron Lett. 2008, 49, 2677. doi:10.1016/j.tetlet.2008.02.161 |
| 17. | Griffiths, J. R.; Tsanaktsidis, J.; Savage, G. P.; Priefer, R. Thermochim. Acta 2010, 499, 15. doi:10.1016/j.tca.2009.10.015 |
| 12. | Eaton, P. E.; Cassar, L.; Halpern, J. J. Am. Chem. Soc. 1970, 92, 6366. doi:10.1021/ja00724a061 |
| 18. | Heaphy, P. J.; Griffiths, J. R.; Dietz, C. J.; Savage, G. P.; Priefer, R. Tetrahedron Lett. 2011, 52, 6359. doi:10.1016/j.tetlet.2011.09.037 |
| 11. | Cassar, L.; Eaton, P. E.; Halpern, J. J. Am. Chem. Soc. 1970, 92, 3515. doi:10.1021/ja00714a075 |
| 8. | Zhou, J. J. P.; Li, J.; Upadhyaya, S.; Eaton, P. E.; Silverman, R. B. J. Med. Chem. 1997, 40, 1165. doi:10.1021/jm9606249 |
| 9. | Silverman, R. B.; Zhou, J. J. P.; Eaton, P. E. J. Am. Chem. Soc. 1993, 115, 8841. doi:10.1021/ja00072a048 |
| 10. | Silverman, R. B. Acc. Chem. Res. 1995, 28, 335. doi:10.1021/ar00056a003 |
| 15. | Priefer, R.; Lee, Y. J.; Barrios, F.; Wosnick, J. H.; Lebuis, A.-M.; Farrell, P. G.; Harpp, D. N.; Sun, A.; Wu, S.; Snyder, J. P. J. Am. Chem. Soc. 2002, 124, 5626. doi:10.1021/ja025823y |
| 23. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi:10.1007/s00214-007-0310-x |
| 24. | Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157. doi:10.1021/ar700111a |
| 25. | Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. doi:10.1063/1.2370993 |
| 32. | Braga, D.; Grepioni, F.; Desiraju, G. R. Chem. Rev. 1998, 98, 1375. doi:10.1021/cr960091b |
| 31. | Jeffrey, G. A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, 1997. |
| 31. | Jeffrey, G. A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, 1997. |
| 29. | Brookhart, M.; Green, M. L. H. J. Organomet. Chem. 1983, 250, 395. doi:10.1016/0022-328X(83)85065-7 |
© 2012 Biegasiewicz et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)