Guest Editor: T. K. Lindhorst Beilstein J. Org. Chem.2012,8, 1831–1838.https://doi.org/10.3762/bjoc.8.210 Received 04 Aug 2012,
Accepted 24 Sep 2012,
Published 25 Oct 2012
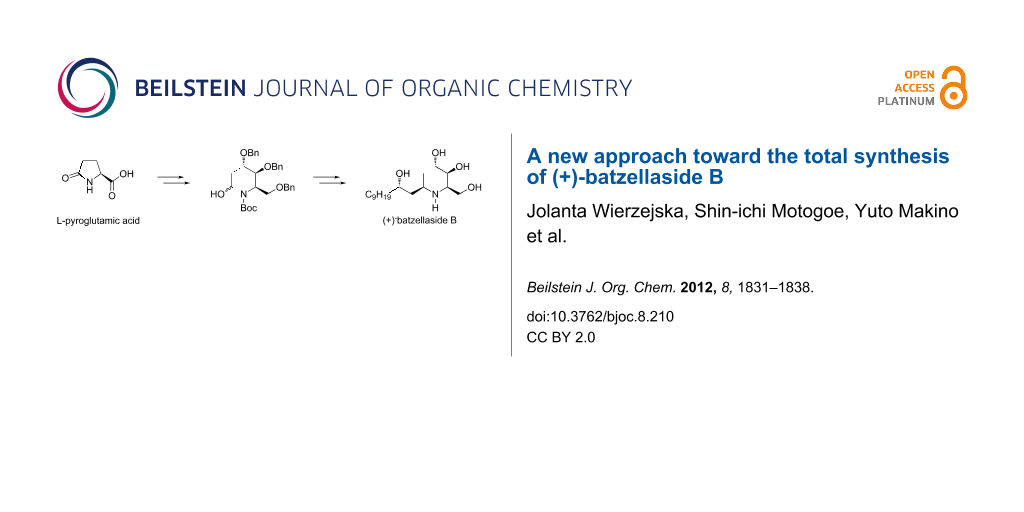
A new synthetic approach to (+)-batzellaside B from naturally abundant L-pyroglutamic acid is presented in this article. The key synthetic step involves Sharpless asymmetric dihydroxylation of an olefinic substrate functionalized with an acetoxy group to introduce two chiral centres diastereoselectively into the structure. Heterocyclic hemiaminal 4, which could be converted from the resulting product, was found to provide stereospecific access to enantiomerically enriched allylated intermediate, offering better prospects for the total synthesis of this natural product.
Iminosugars, monosaccharide analogues in which the endocyclic oxygen has been replaced by nitrogen, display beneficial therapeutic activity as sugar-mimicking glycosidase inhibitors [1-4]. Since the discovery of nojirimycin (Figure 1), which was isolated from Streptomyces roseochromogenes R-468 and S. lavendulae SF-425 in the 1960s [5], this class of compounds has attracted a great deal of interest in the medical community due to their promising pharmaceutical potential as antidiabetic [6], antitumor [7] and antiviral [8] agents. Undoubtedly, approval of Glyset [9] and Miglustat [10] (Figure 1) for treatment of type II diabetes and Gaucher disease, respectively, has imparted therapeutic applications of this class of natural products.
Figure 1:
Examples of naturally occurring iminosugars.
Figure 1:
Examples of naturally occurring iminosugars.
In this context, (+)-batzellasides A–C (1a–c) (Figure 2), C-alkylated azasugars isolated in 2004 from a sponge Batzella sp. collected off the west coast of Madagascar, represent the first example of iminosugars from a marine organism [11]. These naturally occurring products have been demonstrated to retain a remarkably high degree of potency against Staphylococcus epidermidis with MICs of ≤6.3 μg/mL, thus serving as new potent antibacterial agents [11]. As a part of our research program on the synthesis and investigation of biologically active natural products [12-18], we have pursued the synthetic elaboration of (+)-batzellaside B (1b) as a represent member of this new class of iminosugar alkaloids.
Figure 2:
The chemical structures of (+)-batzellasides A–C (1a–c).
Figure 2:
The chemical structures of (+)-batzellasides A–C (1a–c).
In a previous publication, we communicated the first total synthesis of (+)-batzellaside B (1b) by the use of L-arabinose, wherein the absolute stereochemistry of this natural product was completely established by the modified Mosher analysis of a synthetic intermediate prepared through a separate route (Scheme 1) [19]. In this approach involving nucleophilic cyclization of acyclic aldehyde in situ generated from olefin 2 for constructing the piperidine ring system, proper synthetic manipulation of the three inherent stereocenters contained in the chiral source was a key strategy for ensuring effective stereocontrol to achieve completion of the target-directed synthesis. The overall yield in 22 steps from 2,3,5-tri-O-benzyl-L-arabinose (3) was 3.9%. Although 1b and its related analogues are now accessible through the pathway established above, pursuing a new synthetic approach strongly appealed to us, because the existing route is still rather unsatisfactory mainly in terms of the insufficient supply and the time-consuming preparation of the tribenzyl ether 3, which is not commercially available [20]. We thus decided to explore new alternative strategies allowing for a more efficient and convenient access to this natural product and its derivatives. From a retrosynthetic point of view, L-pyroglutamic acid, whose rich natural abundance makes it a commercially and economically viable substrate [21], can be envisaged as a potentially practical starting material for this purpose (Scheme 1). Furthermore, it can be considered that the heterocyclic hemiaminal 4, a common precursor of the target molecule for both synthetic strategies, would be derived by an analogous cyclization of the acyclic aldehyde generated in situ from cyanide 5. In this proposed strategy, the key transformation will involve Sharpless asymmetric dihydroxylation to install stereoselectively the hydroxy groups at C3 and C4 positions of the olefinic substrate 6, and an intramolecular cyclization of aldehyde generated in situ from 5 to construct the piperidine ring system.
Scheme 1:
Our previous approach to (+)-batzellaside B and retrosynthetic analysis for the new synthetic strategy.
Scheme 1:
Our previous approach to (+)-batzellaside B and retrosynthetic analysis for the new synthetic strat...
The present publication describes the results of our continuing challenges associated with this targeted natural product on the basis of the above synthetic strategy. As a result, and in addition to improvement of the inefficient stereoselectivity observed in allylation process from 4 to 7, we established the new alternative synthetic route to (+)-batzellaside B from L-pyroglutamic acid, which offers the advantages of convenience and simplicity of total synthesis.
Results and Discussion
The synthesis began with the preparation of N-Boc-protected γ-lactam 8 by stepwise functionalization of L-pyroglutamic acid, using literature procedures (Scheme 2) [22]. As indicated previously, this compound underwent efficient ring opening upon treatment with sodium methoxide in methanol to provide acyclic ester 9 in 98% yield [23]. In the next step, the TBS protecting group in 9 was removed by exposure to methanolic p-TsOH to give the corresponding alcohol, which was then subjected to reaction with 2,2-dimethoxypropane (2,2-DMP) in the presence of BF3·Et2O [24] to produce N,O-acetonide 10 in 93% yield over two steps. For the preparation of the E-isomer of α,β-unsaturated ester 11, Wittig–Horner reaction employing Garner’s aldehyde has been well known [25,26]; however, we selected stereoselective olefination through deprotonation of 10 with LDA followed by the addition of phenylselenyl bromide [27] and subsequent oxidative elimination of the resulting phenylseleno group with m-CPBA according to our previous report [28], which gave E-isomer 11 in quantitative geometric purity and 90% yield over two steps. Then, the ester moiety of this compound was reduced with DIBAL-H to the corresponding hydroxymethyl functionality to afford 6a in 95% yield [29].
With this olefinic compound in hand, we examined the dihydroxylation of 6a, which was carried out with OsO4 and NMO in 50% aqueous t-BuOH at room temperature [30-33]. Under these conditions, 6a provided an approximately 1:3 ratio of the more- and less-polar diastereomeric triols 12a-A and 12a-B, respectively, in 30% yield. These diastereomers were separated by silica-gel column chromatography and then derivatized to trans-1,2-dibenzyloxy-substituted γ-lactone, which allows a precise assignment of the absolute configuration by comparison with the specific optical rotation value [34]. Accordingly, the primary hydroxy group of 12a-B was chemoselectively protected as the TBS ether [35] and the remaining diol moiety was then etherified with NaH and benzyl bromide, affording 13B in a crude form. Deprotection of the acetal and TBS groups of this product was carried out by stepwise reactions with p-TsOH and TFA to give a dihydroxyamine intermediate, which underwent spontaneous cyclization upon treatment with NaIO4[36] followed by PCC oxidation to form the corresponding trans-1,2-dibenzyloxy-substituted γ-lactone 14B in 17% yield over six steps. For comparison, optical rotation measurement was performed on a solution of 14B in CHCl3 at c 0.36. Indeed, this compound exhibited optical activity with an [α]D24 value of −51.6, indicative of an opposite sense of absolute configuration in comparison to the literature data given for (1S,2S)-dibenzyloxy γ-lactone ([α]D25 +60.1, c 1.0, CHCl3) [34]. From this, we can conclude, as seen in Scheme 2, that the more polar triol 12a-A obtained as a minor component of this dihydroxylation process could be identified as the (1S,2S)-isomer that should be supplied to advance our ongoing synthetic strategy, albeit with low yield for its preparation.
In an effort to improve selectivity of stereogenesis for the dihydroxylation step, we prepared three other olefinic substrates, in which the hydroxy group in 6a was replaced by tert-butyldimethylsilyloxy (TBSO), methoxymethyloxy (MOMO) and acetoxy (AcO) groups, according to standard procedures [37,38], to give moderate to high yields (68–99%) of 6b–d, respectively. Having the four different olefinic compounds 6a–d available, we turned to an asymmetric technique of dihydroxylation to synthesize the (1S,2S)-constituent in preference to another (Table 1). Indeed, the Sharpless methodology was initially applied to 6a, by carrying out the reactions at room temperature under a standard set of the asymmetric hydroxylation conditions [39-41]. Using AD-mix-α and -β, we obtained mixtures of 12a-A and 12a-B in 45:55 and 13:87 ratios with overall isolated yields of 32 and 77%, respectively (Table 1, entries 1 and 2). Analogously, the asymmetric dihydroxylations of 6b and 6c produced predominantly the undesired less-polar diastereomers 12b-B and 12c-B, which could be converted by acidic deprotection to 12a-B (Table 1, entries 3,4 and 6), while the reaction of 6c with AD-mix-α afforded a 50:50 mixture of diastereomers (Table 1, entry 5).
Table 1:
Asymmetric dihydroxylation of 6a–d.
Entry
6
R
Reagent (amount [mol %])
T
Yield [%]a (12-A/12-B)b
1
a
H
AD-mix-α (0.5), MeSO2NH2 (100)
rt
32 (45/55)
2
a
H
AD-mix-β (0.5), MeSO2NH2 (100)
rt
77 (13/87)
3
b
TBS
AD-mix-α (0.5), MeSO2NH2 (100)
rt
33 (14/86)
4
b
TBS
AD-mix-β (0.5), MeSO2NH2 (100)
rt
35 (40/60)
5
c
MOM
AD-mix-α (0.5), MeSO2NH2 (100)
rt
52 (50/50)
6
c
MOM
AD-mix-β (0.5), MeSO2NH2 (100)
rt
88 (0/100)
7
d
Ac
AD-mix-α (0.5), MeSO2NH2 (100)
rt
48 (69/31)
8
d
Ac
AD-mix-β (0.5), MeSO2NH2 (100)
rt
51 (9/91)
9
d
Ac
AD-mix-α (0.5), MeSO2NH2 (100)
0 °C
54 (78/22)
10
d
Ac
AD-mix-α (0.5)
0 °C
52 (84/16)
11
d
Ac
AD-mix-α (0.5), (DHQ)2PHAL (10)
0 °C
53 (83/17)
aIsolated yield. bDiastereomeric ratios were determined by 1H NMR (300 MHz).
A remarkable change in the product profile occurred when 6d was used for this reaction. By employing AD-mix-α under the above reaction conditions, 6d gave rise to a 69:31 mixture of 12d-A and 12d-B in 48% yield, leading to 12a-A and 12a-B through hydrolysis under basic conditions, respectively, whereas the use of AD-mix-β resulted in predominant formation of the undesired diastereomer (Table 1, entries 7 and 8). The above observations led us to explore the AD-mix-α-mediated reaction of 6d at lower temperatures. When the reaction was performed at 0 °C with the same set of the reagents, the product selectivity for 12d-A was slightly improved (Table 1, entry 9). Remarkably, 6d underwent slow reaction in the absence of MeSO2NH2 to give the product mixture in 52% yield, and the diastereomeric ratio could be further enriched to 84:16 (Table 1, entry 10). Additionally, a similar result was obtained by carrying out the reaction using 0.1 equiv of (DHQ)2PHAL (53%, 83:17 in Table 1, entry 11). The above observations clearly suggested a possibility of improving the product selectivity by lowering the reaction temperature and/or introducing an additional catalytic amount of chiral ligand.
Having established the optimized conditions for the preparation of 12d-A, our next objective was the construction of the piperidine ring system. As shown in Scheme 3, the acetyl group of the diastereomerically enriched mixture of 12d-A and 12d-B was removed by exposure to K2CO3 in methanol to give the separable mixture of alcohols 12a-A and 12a-B, respectively [42]. After purification by silica-gel column chromatography, 12a-A was subjected to the TBS protection–benzylation sequence, as illustrated for the preparation of 13B, to generate 13A in 50% yield over three steps. In the next step, deprotection of the TBS group with TBAF [37] and subsequent tosylation of the resulting hydroxy group with TsCl in the presence of pyridine was carried out to yield the corresponding tosylate, whose activated ester group could be displaced with NaCN in DMSO to provide cyanide 15 in 80% yield over three steps [43]. Then, the N,O-acetonide group of 15 was cleaved upon treatment with p-TsOH in methanol [44], and the released primary hydroxy group was subsequently protected as the benzyl ether to produce the key intermediate 5 in 67% yield over two steps. As expected, conversion of this compound into the heterocyclic hemiaminal 4 was achieved in one pot with DIBAL-H by the formation of the aldehyde followed by spontaneous intramolecular cyclization with a yield of 67%. The structural identity of this product was precisely confirmed by 1H NMR spectroscopic data, which proved to be in good agreement with those on record for 4[19]. Hence, we can conclude that a formal total synthesis of (+)-batzellasides B was accomplished, considering that the synthetic route from 4 to 1b has been established previously.
At this point, it was thus suggested that the remaining challenge was to improve the stereoselectivity of the allylation of 4. In the first approach of our previous work [19], we demonstrated that the heterocyclic hemiaminal 4 was allylated by following a protocol using allyltributylstannane (AllylSnBu3) and tert-butyldimethylsilyl triflate (TBSOTf) at −78 °C in toluene to furnish the product 7 as a 69:31 mixture of diastereomeric isomers in 96% yield (Table 2, entry 1). Despite the appealing performance observed in the above synthetic process, this reaction protocol has the major disadvantage of low stereoselectivity causing operational inconvenience associated with the laborious chromatographic separation of the two stereoisomers. From the practical considerations, we next explored a more efficient synthetic method for the stereoselective allylation of 4 using an appropriate combination of allylic reagent and Lewis acid to produce the desired diastereomer 7 preferentially. Beginning with a reaction employing indium chloride (InCl3) instead of TBSOTf at 0 °C in dichloromethane, we observed no substantial improvement in the stereoselectivity of the allylation, affording a 44:56 mixture of 7 and 7’ in quantitative yield (Table 2, entry 2). A much greater preference for the formation of 7 was observed in the cases where allyltrimethylsilane (AllylTMS) was used as an allylic reagent (Table 2, entries 3–5). In fact, the reaction carried out with zinc chloride (ZnCl2) at room temperature in toluene led to exclusive stereoselectivity for 7 with 98% de, albeit in low yield (24%, Table 2, entry 3). Furthermore, the use of TBSOTf as a Lewis acid resulted in significant enhancement of the reaction rate to give almost the same stereochemical outcome (98 and 92% de) with slightly and moderately higher yields (29 and 41%) for the periods of 2 and 3 h (Table 2, entries 4 and 5), respectively. In spite of its increased susceptibility, which should be discriminated from those of unsubstituted structural systems [45-50], it became apparent that this multiply functionalized hemiaminal 4 is well tolerated to undergo direct allylation with the silyl reagents. The results of the above investigations provide one particularly successful route that has the potential to allow direct asymmetric access to the advanced-stage intermediate 7 under precise stereochemical control as well as for circumventing the purification problems related to the diastereomeric impurity in this product.
Table 2:
Diastereoselective allylation of 4.
Entry
Reagent (amount [equiv])
Solvent
T
Time
Yield [%]a (7/7’)b
1
AllylSnBu3 (3.0), TBSOTf (1.5)
toluene
−78 °C
9 h
96 (69/31)
2
AllylSnBu3 (3.0), InCl3 (1.5)
CH2Cl2
0 °C
0.75 h
quant. (44/56)
3
AllylTMS (4.0), ZnCl2 (4.0)
toluene
rt
16 h
24 (99/1)
4
AllylTMS (4.0), TBSOTf (2.0)
CH2Cl2
−78 to −45 °C
2 h
29 (99/1)
5
AllylTMS (10.0), TBSOTf (1.5)
toluene
−78 °C
3 h
41 (96/4)
aIsolated yield. bDiastereomeric ratios were determined by 1H NMR (300 MHz).
Conclusion
We have described a new synthetic approach to (+)-batzellaside B from L-pyroglutamic acid. Starting from this chiral material, the formal total synthesis of the heterocyclic hemiaminal 4, a key intermediate elaborated commonly in the first total synthesis, has been achieved in an efficient 21-step protocol in 7.1% overall yield. Furthermore, the stereospecificity in the allylation of 4 has been exemplified by performing the procedures with AllylTMS and two types of Lewis acids, which allows for simpler synthetic operation due to the ease of purification of the products. The present study clearly demonstrates that L-pyroglutamic acid can be used as a versatile chiral source for synthesizing this class of biologically potent piperidine alkaloids and related analogues.
Supporting Information
Supporting Information File 1:
Full experimental details and characterization data.
We thank Ms. Manami Ohshima for partial support of this research. This research was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
Ganem, B. Acc. Chem. Res.1996,29, 340–347. doi:10.1021/ar9502184
Return to citation in text:
[1]
Stutz, A. E. Iminosugars as Glycosidase Inhibitors: Norjirimycin and Beyond; Wiley-VCH: New York, 1999.
Return to citation in text:
[1]
Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem.2003, 3693–3712. doi:10.1002/ejoc.200300193
Return to citation in text:
[1]
Segraves, N. L.; Crews, P. J. Nat. Prod.2005,68, 118–121. doi:10.1021/np049763g
Return to citation in text:
[1]
[2]
Sengoku, T.; Murata, Y.; Mitamura, H.; Takahashi, M.; Yoda, H. Tetrahedron Lett.2012,53, 435–437. doi:10.1016/j.tetlet.2011.11.066
Return to citation in text:
[1]
Sengoku, T.; Wierzejska, J.; Takahashi, M.; Yoda, H. Synlett2010, 2944–2946. doi:10.1055/s-0030-1259045
Return to citation in text:
[1]
Takahashi, M.; Suzuki, T.; Wierzejska, J.; Sengoku, T.; Yoda, H. Tetrahedron Lett.2010,51, 6767–6768. doi:10.1016/j.tetlet.2010.10.098
Return to citation in text:
[1]
Sengoku, T.; Satoh, Y.; Takahashi, M.; Yoda, H. Tetrahedron Lett.2009,50, 4937–4940. doi:10.1016/j.tetlet.2009.06.072
Return to citation in text:
[1]
Takahashi, M.; Maehara, T.; Sengoku, T.; Fujita, N.; Takabe, K.; Yoda, H. Tetrahedron2008,64, 5254–5261. doi:10.1016/j.tet.2008.03.029
Return to citation in text:
[1]
Matsuura, D.; Mitsui, T.; Sengoku, T.; Takahashi, M.; Yoda, H. Tetrahedron2008,64, 11686–11696. doi:10.1016/j.tet.2008.10.014
Return to citation in text:
[1]
Takahashi, M.; Takada, K.; Matsuura, D.; Takabe, K.; Yoda, H. Heterocycles2007,71, 2113–2118. doi:10.3987/COM-07-11119
Return to citation in text:
[1]
Wierzejska, J.; Ohshima, M.; Inuzuka, T.; Sengoku, T.; Takahashi, M.; Yoda, H. Tetrahedron Lett.2011,52, 1173–1175. doi:10.1016/j.tetlet.2011.01.018
Return to citation in text:
[1]
[2]
[3]
Tejima, S.; Fletcher, H. G., Jr. J. Org. Chem.1963,28, 2999–3004. doi:10.1021/jo01046a015
Return to citation in text:
[1]
Panday, S. K.; Prasad, J.; Dikshit, D. K. Tetrahedron: Asymmetry2009,20, 1581–1632. doi:10.1016/j.tetasy.2009.06.011
Return to citation in text:
[1]
Sengoku, T.; Satoh, Y.; Oshima, M.; Takahashi, M.; Yoda, H. Tetrahedron2008,64, 8052–8058. doi:10.1016/j.tet.2008.06.078
Return to citation in text:
[1]
[2]
Flynn, D. L.; Zelle, R. E.; Grieco, P. A. J. Org. Chem.1983,48, 2424–2426. doi:10.1021/jo00162a028
Return to citation in text:
[1]
Xu, Z.; Zhang, F.; Zhang, L.; Jia, Y. Org. Biomol. Chem.2011,9, 2512–2517. doi:10.1039/c0ob01115k
Return to citation in text:
[1]
Jako, I.; Uiber, P.; Mann, A.; Wermuth, C.-G.; Boulanger, T.; Norberg, B.; Evrard, G.; Durant, F. J. Org. Chem.1991,56, 5729–5733. doi:10.1021/jo00019a055
Return to citation in text:
[1]
Jako, I.; Uiber, P.; Mann, A.; Taddei, M.; Wermuth, C.-G. Tetrahedron Lett.1990,31, 1011–1014. doi:10.1016/S0040-4039(00)94416-2
Return to citation in text:
[1]
Silverman, R. B.; Invergo, B. J.; Mathew, J. J. Med. Chem.1986,29, 1840–1846. doi:10.1021/jm00160a007
Return to citation in text:
[1]
Yoda, H.; Shirai, T.; Katagiri, T.; Takabe, K.; Kimata, K.; Hosoya, K. Chem. Lett.1990,19, 2037–2038. doi:10.1246/cl.1990.2037
Return to citation in text:
[1]
Spangenberg, T.; Schoenfelder, A.; Breit, B.; Mann, A. Eur. J. Org. Chem.2010, 6005–6018. doi:10.1002/ejoc.201000865
Return to citation in text:
[1]
Krishna, P. R.; Reddy, P. S. Synlett2009, 209–212. doi:10.1055/s-0028-1087669
Return to citation in text:
[1]
Passiniemi, M.; Koskinen, A. M. P. Synthesis2010, 2816–2822. doi:10.1055/s-0029-1218843
Return to citation in text:
[1]
Upadhyay, P. K.; Kumar, P. Synthesis2010, 3063–3066. doi:10.1055/s-0030-1258185
Return to citation in text:
[1]
Pabba, J.; Rempel, B. P.; Withers, S. G.; Vasella, A. Helv. Chim. Acta2006,89, 635–666. doi:10.1002/hlca.200690066
Return to citation in text:
[1]
[2]
Reetz, M. T.; Kesseler, K. J. Org. Chem.1985,50, 5434–5436. doi:10.1021/jo00225a103
Return to citation in text:
[1]
House, H. O.; Berkowitz, W. F. J. Org. Chem.1963,28, 2271–2276. doi:10.1021/jo01044a028
Return to citation in text:
[1]
Corey, E. J.; Venkateswarlu, A. J. Am. Chem. Soc.1972,94, 6190–6191. doi:10.1021/ja00772a043
Return to citation in text:
[1]
[2]
Kluge, A. F.; Untch, K. G.; Fried, J. H. J. Am. Chem. Soc.1972,94, 7827–7832. doi:10.1021/ja00777a027
Return to citation in text:
[1]
VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett.1976,17, 1973–1976. doi:10.1016/S0040-4039(00)78093-2
Return to citation in text:
[1]
Sharpless, K. B.; Akashi, K. J. Am. Chem. Soc.1976,98, 1986–1987. doi:10.1021/ja00423a067
Return to citation in text:
[1]
Francais, A.; Bedel, O.; Haudrechy, A. Tetrahedron2008,64, 2495–2524. doi:10.1016/j.tet.2007.11.068
Return to citation in text:
[1]
Plattner, J. J.; Gless, R. D.; Rapoport, H. J. Am. Chem. Soc.1972,94, 8613–8615. doi:10.1021/ja00779a072
Return to citation in text:
[1]
Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem.–Eur. J.2005,11, 4667–4677. doi:10.1002/chem.200500282
Return to citation in text:
[1]
Wen, S.-J.; Zhang, H.-W.; Yao, Z.-J. Tetrahedron Lett.2002,43, 5291–5294. doi:10.1016/S0040-4039(02)01043-2
Return to citation in text:
[1]
Polniaszek, R. P.; Belmont, S. E.; Alvarez, R. J. Org. Chem.1990,55, 215–223. doi:10.1021/jo00288a036
Return to citation in text:
[1]
Butters, M.; Davies, C. D.; Elliott, M. C.; Hill-Cousins, J.; Kariuki, B. M.; Ooi, L.-l.; Wood, J. L.; Wordingham, S. V. Org. Biomol. Chem.2009,7, 5001–5009. doi:10.1039/b914744f
Return to citation in text:
[1]
Ukaji, Y.; Tsukamoto, K.; Nasada, Y.; Shimizu, M.; Fujisawa, T. Chem. Lett.1993,22, 221–224. doi:10.1246/cl.1993.221
Return to citation in text:
[1]
Pilli, R. A.; Robello, L. G. Synlett2005, 2297–2300. doi:10.1055/s-2005-872659
Return to citation in text:
[1]
Wang, Y.; Zhu, S.; Ma, D. Org. Lett.2011,13, 1602–1605. doi:10.1021/ol200004s
Return to citation in text:
[1]
Sakagami, H.; Kamikubo, T.; Ogasawara, K. Chem. Commun.1996, 1433–1434. doi:10.1039/cc9960001433
Return to citation in text:
[1]
Polniaszek, R. P.; Belmont, S. E.; Alvarez, R. J. Org. Chem.1990,55, 215–223. doi:10.1021/jo00288a036
46.
Butters, M.; Davies, C. D.; Elliott, M. C.; Hill-Cousins, J.; Kariuki, B. M.; Ooi, L.-l.; Wood, J. L.; Wordingham, S. V. Org. Biomol. Chem.2009,7, 5001–5009. doi:10.1039/b914744f
47.
Ukaji, Y.; Tsukamoto, K.; Nasada, Y.; Shimizu, M.; Fujisawa, T. Chem. Lett.1993,22, 221–224. doi:10.1246/cl.1993.221


![[1860-5397-8-210-1]](/bjoc/content/figures/1860-5397-8-210-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-210-2]](/bjoc/content/figures/1860-5397-8-210-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-210-i1]](/bjoc/content/inline/1860-5397-8-210-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-210-i2]](/bjoc/content/inline/1860-5397-8-210-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-210-i4.svg?max-width=637&scale=1.0)
![[1860-5397-8-210-i3]](/bjoc/content/inline/1860-5397-8-210-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-210-i5.svg?max-width=637&scale=1.0)