Abstract
Hexahomotrioxacalix[3]arenes, commonly called oxacalix[3]arenes, were first reported in 1962. Since then, their chemistry has been expanded to include numerous derivatives and complexes. This review describes the syntheses of the parent compounds, their derivatives, and their complexation behaviour towards cations. Extraction data are presented, as are crystal structures of the macrocycles and their complexes with guest species. Applications in fields as diverse as ion selective electrode modifiers, fluorescence sensors, fullerene separations and biomimetic chemistry are described.

Graphical Abstract
Introduction
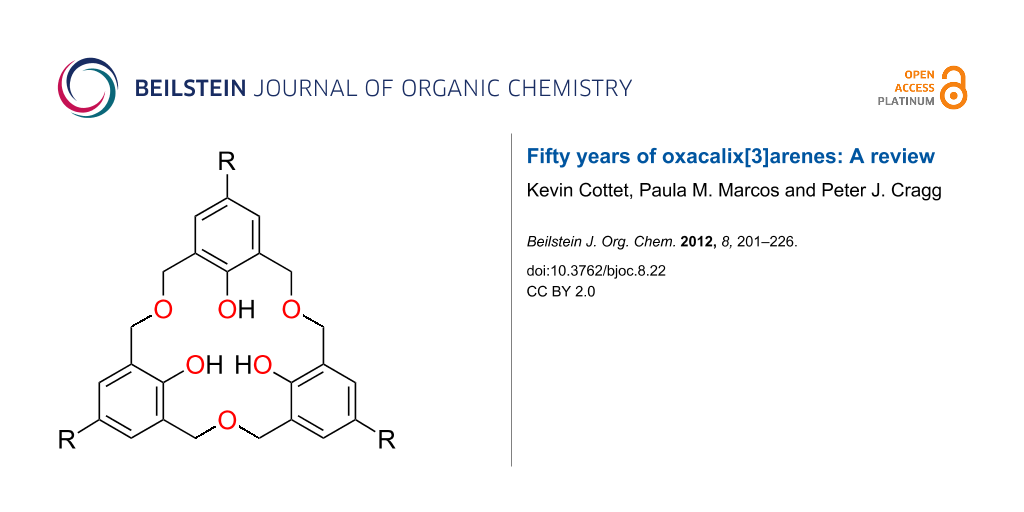
Calixarenes, macrocycles which are widely used in supramolecular chemistry, are 2,6-metacyclophanes with a methylene bridge between their phenolic groups, as shown in Figure 1 [1-3]. In 1994, the term “homocalixarene” was coined by Brodesser and Vögtle to describe analogues of calixarenes with two or more methylene groups between the aromatic moieties [4]. When one or more CH2 bridges are replaced by CH2OCH2 groups the macrocycles are known as homooxacalixarenes, or simply oxacalixarenes. The presence of the heteroatom is reflected in the name of the compound, for example, p-tert-butylcalix[4]arene (1) with a CH2OCH2 group instead of a CH2 bridge is p-tert-butyldihomooxacalix[4]arene (2) [5]. “Dihomo” implies two additional atoms in the bridge and “oxa” that one of them is oxygen. The remainder of the calixarene nomenclature denotes any substituents attached to the phenolic oxygens, known as the “lower rim”, and substituents found in the para-position of the phenols, also known as the “upper rim” (Figure 2). For the purposes of this review the term “oxacalix[n]arene” will be used as a generalization for this class of compounds.
![[1860-5397-8-22-1]](/bjoc/content/figures/1860-5397-8-22-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Calixarenes and expanded calixarenes: p-tert-Butylcalix[4]arene (1), p-tert-butyldihomooxacalix[4]arene (2), p-tert-butylhexahomotrioxacalix[3]arene (3a).
Figure 1: Calixarenes and expanded calixarenes: p-tert-Butylcalix[4]arene (1), p-tert-butyldihomooxacalix[4]a...
![[1860-5397-8-22-2]](/bjoc/content/figures/1860-5397-8-22-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Conventional nomenclature for oxacalix[n]arenes.
Figure 2: Conventional nomenclature for oxacalix[n]arenes.
Although some aspects of homooxacalixarene chemistry have been reviewed [4,6-8], notably by Shokova and Kovalev in 2004 [9,10], it is timely for the 50th anniversary of Hultzsch’s discovery of p-tert-butylhexahomotrioxacalix[3]arene (3a) [11] to reflect on the history of these compounds and assess recent advances in the field. Many other expanded calix[n]arenes are now known, including the methyl ethers of dihomooxa-, tetrahomodioxa-, hexahomotrioxa- and octahomotetraoxacalix[4]arenes, which have been described in detail by Masci [12]. Despite these advances, the oxacalix[3]arenes have remained the main focus of attention for researchers and are the subject of this review.
Review
1 Synthesis of parent oxacalix[3]arenes
1.1 Thermal dehydration
The first oxacalix[n]arenes to be reported were the hexahomotrioxacalix[3]arenes, and these remain the most-studied members of the class. p-tert-Butylhexahomotrioxacalix[3]arene (3a), initially reported by Hultzsch in 1962, was isolated in less than 1% yield by heating 2,6-bis(hydroxymethyl)-4-tert-butylphenol [11]. Elemental analysis gave an empirical formula of C12H16O2 and molecular weight determinations gave values corresponding to a trimer. Despite interest in novel phenol–formaldehyde polymers and macrocycles and characterization of 3a in 1979 [13], it took a further 20 years for a reproducible synthesis to be published. In 1983, Gutsche reported that the thermally induced dehydration of 2,6-bis(hydroxymethyl)phenols in xylene under reflux gave rise to the formation of homooxacalixarenes, some of them in reasonable yields, as shown in Scheme 1 [14].
![[1860-5397-8-22-i1]](/bjoc/content/inline/1860-5397-8-22-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of oxacalix[3]arenes: (i) Formaldehyde (37% aq), NaOH (aq), 1,4-dioxane; glacial acetic acid, acetone; (ii) refluxing o-xylene [14] or Na2SO4, MsOH, in refluxing DME [15].
Scheme 1: Synthesis of oxacalix[3]arenes: (i) Formaldehyde (37% aq), NaOH (aq), 1,4-dioxane; glacial acetic a...
Although not discussed by Gutsche, both cyclotrimers and tetramers are usually formed by this method and, in 1991, Vicens and Zerr performed a thermal dehydration of 2,6-bis(hydroxymethyl)-4-tert-butylphenol in xylene under reflux allowing them to isolate p-tert-butyloctahomotetraoxacalix[4]arene (4a), illustrated in Figure 3, along with 3a [16].
![[1860-5397-8-22-3]](/bjoc/content/figures/1860-5397-8-22-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: p-tert-Butyloctahomotetraoxacalix[4]arene (4a) [16].
Figure 3: p-tert-Butyloctahomotetraoxacalix[4]arene (4a) [16].
To finally prove that the main product from thermal dehydration was indeed a trimer, Vicens reported the X-ray crystal structure of 3a in 1992 (Figure 4) demonstrating it to be exclusively in the bowl-shaped cone conformation [17].
![[1860-5397-8-22-4]](/bjoc/content/figures/1860-5397-8-22-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of 3a showing phenolic hydrogen bonding (IUCr ID AS0508) [17].
Figure 4: X-ray crystal structure of 3a showing phenolic hydrogen bonding (IUCr ID AS0508) [17].
In 1994, Hampton et al. used an alternative acid-catalyzed procedure to prepare 3a and developed a method that improved its purity through the formation of the Na+ salt and its subsequent neutralization with acid [15]. The process separated 3a from the cyclic tetramer; the former precipitates as the sodium salt in dry methanol due to complementarity between the arrangement of phenolic groups and the preferred coordination environment of Na+. Removal of the tert-butyl groups through a conventional AlCl3 driven retro-Friedel–Crafts de-tert-butylation reaction, as seen in other calixarenes, is unsuccessful in the case of oxacalixarenes, therefore different para-substituents must be introduced through the starting phenol in order to obtain derivatives with different groups at the upper rim. A number of other para-substituted bis(hydroxymethyl)phenols were therefore also cyclized in the presence of methanesulfonic acid (MsOH) or para-toluenesulfonic acid (TsOH) and Na2SO4. The corresponding oxacalixarenes were isolated in varying yields: t-Bu (3a) 32%; Me (3b) 21%; Et (3c) 21%; iPr (3d) 30%; Cl (3e)12% [15].
Although conditions were not necessarily optimal, the principles of oxacalix[3]arene syntheses had been established. Monomers react to give the cyclic trimer, predominantly, when heated under reflux in high-boiling-point organic solvents along with an organic acid. Water formed in the dehydration process must be removed through reaction with anhydrous drying agents or be collected in a Dean–Stark trap. In Gutsche’s report, and presumably in the work of Hultzsch too, the bis(hydroxymethyl)phenol monomer was isolated as the sodium salt and neutralized with acetic acid. Upon removal of solvent, traces of the acid presumably remained and were taken through to the cyclization step. Cragg noted that acid had to be present for the cyclization to occur, as carefully purified monomers formed calix[4]arenes or dihomooxacalix[4]arenes rather than oxacalix[3]arenes when subjected to standard synthetic methods [18]. To test this theory, the synthesis of 3a was attempted in o-xylene under reflux by using either the freshly prepared crude monomer or the recrystallized monomer. The formation of 3a was observed in the reaction of the unpurified monomer, but not under acid-free conditions. Moreover, in separate experiments MsOH, TsOH or glacial acetic acid (AcOH) were added to reactions involving the recrystallized monomer. MsOH or TsOH, having complementary threefold symmetry with the lower rim of oxacalix[3]arenes, were expected to increase the yields, but AcOH appeared to be just as effective. Notably, the addition of TsOH gave the oxacalix[3]arene as the sole product.
1.2 Other synthetic methods
Since the initial reports of oxacalix[3]arene syntheses, several procedures have been developed to improve both the reaction conditions and the range of derivatives that can be prepared. The initial strategy to make oxacalix[3]arenes was a single step condensation, which can only lead to C3-symmetric compounds bearing the same para-substituted phenol; however, in host–guest chemistry an asymmetric macrocycle can provide a site for enantioselective molecular recognition. In the case of p-tert-butylcalix[n]arenes the tert-butyl substituent can be removed, as mentioned previously, through a retro-Friedel–Crafts acylation, and replaced by other groups, but the dibenzyl ether bridge in the oxacalixarenes is too fragile for this to be successful. In 1998, Fuji proposed a stepwise synthesis of asymmetric oxacalix[3]arenes based on linear precursors protected with a combination of isopropylidene and methoxymethyl groups [19]. As shown in Scheme 2, the phenolic position of a monomeric precursor is protected with methyl chloromethyl ether (MOMCl). A different monomer is then protected with 2,2-dimethoxypropane, in the presence of TsOH. This links one methylol group to the phenol, leaving the second open to bromination with CBr4 and PPh3. The linear trimer is formed between one methoxymethyl protected monomer and two benzyl bromide derivatives in DMF with NaH as the base. Intramolecular cyclization was achieved in 4 h at room temperature with 60% HClO4 in CHCl3 under high-dilution conditions. Pretreatment of the solvent with water was found to be necessary to remove the ethanol stabilizer and to aid deprotection. Yields were up to 50%, and, interestingly, there was no template effect from any alkali metals. An analogous strategy was developed by Georghiou in 2001 to prepare asymmetric oxacalix[3]naphthalene derivatives [20], and this is discussed in greater detail below.
![[1860-5397-8-22-i2]](/bjoc/content/inline/1860-5397-8-22-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Stepwise synthesis of asymmetric oxacalix[3]arenes: (i) MOMCl, Adogen®464; (ii) 2,2-dimethoxypropane, p-TsOH; (iii) CBr4, PPh3, CH2Cl2; (iv) NaH, DMF; (v) HClO4 (aq), wet CHCl3 [19].
Scheme 2: Stepwise synthesis of asymmetric oxacalix[3]arenes: (i) MOMCl, Adogen®464; (ii) 2,2-dimethoxypropan...
In a later communication, Fuji reported the crystal structure of an unusual byproduct of the reaction, a heptahomotetraoxacalix[3]arene 5 with t-Bu, Et and H upper-rim substituents (Figure 5) [21].
![[1860-5397-8-22-5]](/bjoc/content/figures/1860-5397-8-22-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray crystal structure of heptahomotetraoxacalix[3]arene 5 (CCDC ID 166088) [21].
Figure 5: X-ray crystal structure of heptahomotetraoxacalix[3]arene 5 (CCDC ID 166088) [21].
In 2001, Komatsu proposed a different way to access compounds in which two, or all three, units are identical [22]. The method was based on the reductive coupling of silylated derivatives of 2,6-hydroxymethylphenols, in which R is t-Bu, Me, benzyl (Bz), phenyl (Ph), or a halide, as shown in Scheme 3. The reaction takes place under conditions of high dilution at −78 °C to favour intramolecular cyclization over polymerization. Coupling reactions are successful, whether the groups in the para-position are the same or different, and this method also gives access to oxacalix[4]arenes in modest yields up to 42% for the p-tert-butyl derivative.
![[1860-5397-8-22-i3]](/bjoc/content/inline/1860-5397-8-22-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Oxacalix[3]arene synthesis by reductive coupling: (i) Me3SiOTf, Et3SiH, CH2Cl2; R1, R2 = I, Br, benzyl, n-octyl (x = 1 or 2) [22].
Scheme 3: Oxacalix[3]arene synthesis by reductive coupling: (i) Me3SiOTf, Et3SiH, CH2Cl2; R1, R2 = I, Br, ben...
1.3 Oxacalix[3]naphthalenes
The oxacalix[3]naphthalenes, e.g., 6a and 6b reported by Georghiou, have extended aromatic groups with H or t-Bu groups in the 6-position and can be considered as close relatives of the oxacalix[3]arenes [20]. The synthesis, shown in Scheme 4, is analogous to Fuji’s method for oxacalix[3]arenes [19]. As noted below, this extended aromatic surface is oriented perfectly for C60 inclusion [23].
![[1860-5397-8-22-i4]](/bjoc/content/inline/1860-5397-8-22-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxacalix[3]naphthalene: (i) HClO4 (aq), wet CHCl3 (R = tert-butyl, 6a, H, 6b) [20].
Scheme 4: Oxacalix[3]naphthalene: (i) HClO4 (aq), wet CHCl3 (R = tert-butyl, 6a, H, 6b) [20].
2 Conformational properties
Oxacalix[3]arenes have received significant attention as receptors, mainly due to their structural features: A cavity formed by a 18-membered ring, only two basic conformations (cone and partial-cone), and a C3-symmetry [24]. This last feature can provide a suitable binding site for species that require trigonal-planar, tetrahedral or octahedral coordination environments. The flexibility of the macrocycles can allow them to establish ideal bond distances and angles to bind such species. In common with other calix[n]arenes, oxacalix[3]arenes containing free OH groups are conformationally mobile, leading to cone and partial-cone conformers (Figure 6). Without lower-rim substituents there is free rotation of each phenolic unit through the macrocyclic annulus; however, the presence of a hydrogen-bond motif in the cone conformer makes it the more stable form.
![[1860-5397-8-22-6]](/bjoc/content/figures/1860-5397-8-22-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
In 1985, Gutsche investigated the conformational flexibility of parent calix[n]arenes (n = 4–8) and oxacalixarenes by temperature-dependent 1H NMR [5]. The through-the-annulus rotation barrier for oxacalix[3]arenes was calculated to be much lower than that for other calixarenes, either in non-coordinating or in polar solvents, such as CDCl3 or pyridine, respectively. The 1H NMR spectrum of 3a in CDCl3/CS2 only showed a singlet for the CH2 resonance, even at −90 °C, and the ∆G≠ barrier for conformational inversion in CDCl3 was <38 kJ mol−1, in contrast with 66 kJ mol−1 for the calix[4]arene analogue. To freeze the oxacalix[3]arene conformer, through-the-annulus rotation must be prevented. This can be achieved by the introduction of sufficiently large groups on the lower rim of the macrocycle. Upper-rim inversion is less likely to occur when, as in the case of 3a, it is hindered by the tert-butyl group.
3 Oxacalix[3]arene derivatives
3.1 Lower-rim derivatives
Oxacalix[3]arene derivatization at the lower rim has been achieved through alkylation reactions with simple alkyl halides or with functionalized alkylating agents. Lower-rim derivatization is relatively straightforward, but conformational control is harder to achieve. The main drawback of lower-rim substitution is that statistically only 25% of the product is formed in the cone conformation, as shown in Scheme 5 [25,26].
![[1860-5397-8-22-i5]](/bjoc/content/inline/1860-5397-8-22-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Origin of the 25:75 cone:partial-cone statistical distribution of O-substituted oxacalix[3]arenes (p = probability) [25,26].
Scheme 5: Origin of the 25:75 cone:partial-cone statistical distribution of O-substituted oxacalix[3]arenes (p...
3.1.1 Alkyl ethers: Classical O-alkylation of oxacalix[3]arenes was first achieved by Shinkai et al. in 1993 [24]. Treatment of 3a with the corresponding alkyl halides in DMF in the presence of NaH afforded Me (7), Et (8), n-Pr (9) and n-Bu (10) derivatives (Scheme 6). Under these conditions, 8 was obtained in the partial-cone conformation only. When the reaction was performed in the presence of t-BuOK a 1:4 mixture of cone and partial-cone was obtained and even with Cs2CO3 the cone conformer could be detected. It seems that K+ and Cs+ favourably interact with the three phenolic oxygen atoms placed on the same side, whereas Na+ preferentially interacts with them across the ring.
![[1860-5397-8-22-i6]](/bjoc/content/inline/1860-5397-8-22-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of alkyl ethers 7–10: (i) Alkyl halide, NaH, DMF [24].
Scheme 6: Synthesis of alkyl ethers 7–10: (i) Alkyl halide, NaH, DMF [24].
Introduction of heteroatoms, such as nitrogen, into the oxacalix[3]arene lower rims can also be achieved by O-alkylation. Pyridine is known to be a good ligand towards metals and is widely employed in transition-metal coordination chemistry; therefore, in an attempt to incorporate these binding sites into oxacalix[3]arenes, Yamato [27] and Cragg [26] independently reacted 3a with 2-(chloromethyl)pyridine, as shown in Scheme 7. The presence of Cs2CO3 leads to the formation of the partial-cone conformer, whereas K2CO3 and NaH increase the yield of the cone conformer of 11a to about 25%. 1H NMR analysis of the cone conformer indicates that the nitrogen atoms point away from the macrocyclic cavity [27].
![[1860-5397-8-22-i7]](/bjoc/content/inline/1860-5397-8-22-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of a pyridyl derivative 11a: (i) Picolyl chloride hydrochloride, NaH, DMF [26,27].
Scheme 7: Synthesis of a pyridyl derivative 11a: (i) Picolyl chloride hydrochloride, NaH, DMF [26,27].
When 4-(chloromethyl)pyridine was used instead, NaH was ineffectual as a deprotonating agent. Na2CO3 yielded the disubstituted product only, K2CO3 gave both cone (8%) and partial-cone (68%) conformers, and the only isolated product with Cs2CO3 was the partial-cone conformer (75%) [28].
The X-ray structure of the partial-cone conformer (Figure 7), reported by Cragg, shows one pyridyl group to be included within the macrocyclic cavity and the remaining two with their nitrogen atoms pointing away from it [26].
![[1860-5397-8-22-7]](/bjoc/content/figures/1860-5397-8-22-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: X-ray crystal structure of partial-cone 11a (CCDC ID 150580) [26].
Figure 7: X-ray crystal structure of partial-cone 11a (CCDC ID 150580) [26].
3.1.2 Functionalized alkyl ethers: Functionalized alkyl halides of the type XCH2Y, where X is a leaving group and Y is a functional group, have also been used to introduce a variety of groups into the lower rim of oxacalix[3]arenes. Thus, derivatives containing carbonyl groups (ester, acid, amide and ketone) and heteroatoms, such as nitrogen and phosphorous, have been obtained.
In 1993, Shinkai et al. [29] reported the synthesis of the first ethyl ester derivative 12a. In the belief that the alkali-metal template effect would lead preferentially to the cone conformer with NaH, the reaction of excess ethyl bromoacetate with 3a was carried out in acetone under reflux (Scheme 8). The partial-cone conformer of 12a was formed exclusively when weaker bases, K2CO3 or Cs2CO3, were used. NaH or t-BuOK in THF gave a mixture of products, but the yield of the cone conformer never exceeded 22%. An experiment with the oxacalix[3]naphthalene analogue was performed in 2003 by Georghiou [30], which also gave the cone conformer in 25% yield.
![[1860-5397-8-22-i8]](/bjoc/content/inline/1860-5397-8-22-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Lower-rim ethyl ester synthesis: (i) Ethyl bromoacetate, NaH, t-BuOK or alkali metal carbonate, THF or acetone [29].
Scheme 8: Lower-rim ethyl ester synthesis: (i) Ethyl bromoacetate, NaH, t-BuOK or alkali metal carbonate, THF...
Cone-12a was used by Shinkai as the starting point from which to construct the chiral capped oxacalix[3]arene 13 as shown in Scheme 9 [31]. The parent compound was cleaved to form the tris(acid) 14a, which then reacted with S-phenylalanine methyl ester. Deprotection of the methyl ester followed by reduction with LiBH4 gave the chiral amide 15, which reacted with 1,3,5-benzenetricarbonyl chloride to form the capped species 13. Compound 13 was shown to bind primary ammonium cations better than an uncapped ester analogue.
![[1860-5397-8-22-i9]](/bjoc/content/inline/1860-5397-8-22-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Forming chiral receptor 13: (i) Ethyl bromoacetate, NaH, THF; (ii) NaOH, H2O/1,4-dioxane; (iii) S-PheOMe∙HCl, DCC, HOBt, NEt3, CH2Cl2; (iv) LiBH4, THF; (v) 1,3,5-benzenetricarbonyl chloride, pyridine, THF [31].
Scheme 9: Forming chiral receptor 13: (i) Ethyl bromoacetate, NaH, THF; (ii) NaOH, H2O/1,4-dioxane; (iii) S-P...
In 1995, Vicens reported the crystal structure of a partial-cone triethyl ester derivative of 4-phenyloxacalix[3]arene illustrated as 16 in Figure 8 [32]. Few synthetic details were given; however, it was reported that cyclization of bis(2,5-methylol)-4-phenylphenol to give 3g was followed by reaction with ethylbromoacetate, but no mention of the yield or isolation of a cone conformer was made.
![[1860-5397-8-22-8]](/bjoc/content/figures/1860-5397-8-22-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: X-ray crystal structure of 16 (IUCr ID PA1110) [32].
Figure 8: X-ray crystal structure of 16 (IUCr ID PA1110) [32].
The first amide derivative was reported by Shinkai in 1995 [25] through the reaction of 3a with N,N-diethylchloroacetamide (Scheme 10). Heating under reflux in THF, with NaH as base, gave cone amide 17a as the only isolated product in 23% yield. Using the same conditions, Cragg reported an improved yield of 44% through a slight modification of the previous procedure (recrystallization from MeCN instead of MeOH) [26], and Yamato later reported a 90% yield [33]. This is in stark contrast to the maximum yield of 25% for the esterification reaction discussed above and points to a subtle, yet essential, difference between the interaction modes of the oxacalixarene, cation and alkylating agent. Despite much speculation, the reason for this is not yet understood. As with the esterification reaction, use of K2CO3 or Cs2CO3 in place of NaH, and with acetone as the solvent, reverses the conformer preference with partial-cone-17a isolated in 45% yield with only a trace of the cone conformer. This suggests a template effect for both K+ and Cs+ that occurs whether an amide or ester is formed, and a function for Na+ beyond that of a mere template.
![[1860-5397-8-22-i10]](/bjoc/content/inline/1860-5397-8-22-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Lower rim N,N-diethylamide 17a: (i) N,N-Diethylchloroacetamide, NaH, t-BuOK or alkali metal carbonate, THF or DMF or acetone [25,26,33].
Scheme 10: Lower rim N,N-diethylamide 17a: (i) N,N-Diethylchloroacetamide, NaH, t-BuOK or alkali metal carbona...
One consequence of this work is that the preferred route to C3 symmetric cone derivatives is through tris(amide) derivative 17a, which can readily be cleaved by hydrolysis employing sodium hydroxide in 1,4-dioxane/water to give cone-14a. In 2001 Yamato used cone-14a to form a C3 symmetric hydrophobic receptor 18 in 13% yield through reaction with 1,3,5-tris(bromomethyl)benzene in the presence of Na2CO3 (Scheme 11) [33]. As the reaction failed to work when K2CO3 was used, the authors suggested that Na+ may play a templating role in addition to that of a deprotonating agent.
![[1860-5397-8-22-i11]](/bjoc/content/inline/1860-5397-8-22-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Capping the lower rim: (i) N,N-Diethylchloroacetamide, NaH, THF; (ii) NaOH, H2O/1,4-dioxane; (iii) 1,3,5-tris(bromomethyl)benzene, Na2CO3, DMF [33].
Scheme 11: Capping the lower rim: (i) N,N-Diethylchloroacetamide, NaH, THF; (ii) NaOH, H2O/1,4-dioxane; (iii) ...
The X-ray crystal structure of the product (Figure 9) shows that the carbonyl oxygen atoms point away from the cavity to create a large hydrophobic cavity. Extraction studies indicated a slight, and statistically insignificant, preference for K+ over Cs+ and Ag+, with a much lower affinity for Na+. The highest affinity was reserved for n-BuNH3+.
![[1860-5397-8-22-9]](/bjoc/content/figures/1860-5397-8-22-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: X-ray crystal structure of 18 (CCDC ID 142599) [33].
Figure 9: X-ray crystal structure of 18 (CCDC ID 142599) [33].
An analogue of 18, which showed little affinity for metal cations, was prepared with three 4-methylbenzyl substituents on the lower rim (19).
In 2001, Yamato reported an oxacalix[3]arene with pendant pyridines linked by amide bonds [34]. The intramolecular hydrogen bonds between neighbouring amide groups enforced a flattened-cone conformer for the macrocycle, which prevented binding to both metal cations and, to a large extent, alkyl ammonium cations. Extending the link between the macrocycle and aromatic termini did not disrupt the strong amide interactions, although binding was detected for Ag+, as the triflate, and for n-BuNH3+, as the chloride salt [35]. Further work on this class of derivatives showed some anion selectivity in the presence of n-BuNH3+ through intermolecular hydrogen bonding with amide hydrogens [36].
In 2006, the same group used a similar route in order to synthesize the extended, uncapped derivative 20 incorporating three (phenylcarbamoyl)methylcarbamate substituents (Scheme 12), to mimic the binding sites in a protein, complete with hydrophobic region [37]. These amides were designed to act as heteroditopic receptors, capable of binding anions and cations separately and simultaneously in a cooperative way, and were shown to bind n-BuNH3+ halide salts in this manner.
![[1860-5397-8-22-i12]](/bjoc/content/inline/1860-5397-8-22-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Extending the lower rim: (i) Glycine methyl ester, HOBt, dicyclohexycarbodiimide (DCC), CH2Cl2; (ii) NaOH, H2O/1,4-dioxane; (iii) p-toluidine, HOBt, DCC, CH2Cl2 [37].
Scheme 12: Extending the lower rim: (i) Glycine methyl ester, HOBt, dicyclohexycarbodiimide (DCC), CH2Cl2; (ii...
N-Hydroxypyrazinones are known to function as bidentate ligands for metals such as iron or gallium that require an octahedral geometry. Katoh coupled N-hydroxypyrazinone substituents to cone-14a in order to prepare 23 (Scheme 13). Binding Ga3+ with remote lower-rim groups induced the cooperative binding of alkyl ammonium cations by the macrocycle [38].
![[1860-5397-8-22-i13]](/bjoc/content/inline/1860-5397-8-22-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of N-hydroxypyrazinone derivative 23: (i) 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride, HOBt, Et3N; (ii) H2, 10% Pd-C, MeOH [38].
Scheme 13: Synthesis of N-hydroxypyrazinone derivative 23: (i) 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide...
Recently, Marcos reported the synthesis of an oxacalix[3]arene ketone derivative (Scheme 14) [39]. Treatment of 3a with 1-adamantyl bromomethyl ketone and NaH in THF under reflux afforded adamantyl ketone 24 in the cone conformation only.
![[1860-5397-8-22-i14]](/bjoc/content/inline/1860-5397-8-22-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of 24: (i) 1-Adamantyl bromomethyl ketone, NaH, THF [39].
Scheme 14: Synthesis of 24: (i) 1-Adamantyl bromomethyl ketone, NaH, THF [39].
3.1.3 Phosphorus derivatives: Complete phosphorylation of 3a was reported by Matt in 1999 and was achieved through reaction with NaH and Ph2P(O)CH2OTs in toluene at 90 °C for three days (Scheme 15) [40]. The reaction resulted in the formation of a 4:1 mixture of the cone and partial-cone diphenylphosphine oxide derivatives 25: The preference for the cone formation is highly atypical but may be due to the templating effect of Na+. Separation by column chromatography afforded the cone conformer in 72% yield although the partial-cone was never obtained in a pure form. Reduction by phenylsilane (PhSiH3) gave the corresponding cone and partial-cone phosphines 26 quantitatively.
![[1860-5397-8-22-i15]](/bjoc/content/inline/1860-5397-8-22-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of 25 and 26: (i) (Diphenylphosphino)methyl tosylate, NaH, toluene; (ii) phenylsilane, toluene [40].
Scheme 15: Synthesis of 25 and 26: (i) (Diphenylphosphino)methyl tosylate, NaH, toluene; (ii) phenylsilane, to...
3.1.4 Silyl derivatives: In 1996, Hampton investigated the selectivity of silylation on oxacalix[3]arenes to determine the influence of the group in the para-position, the nature of the silylating agent and the reaction conditions [41]. Unsurprisingly, the formation of the partial-cone was favoured for all oxacalix[3]arenes, with small upper-rim substituents having the highest partial-cone:cone ratio (e.g., 100:1 for the Cl derivative) when bis(trimethylsilyl)trifluoroacetamide was used as the silylating reagent. When 1-(trimethylsilyl)imidazole was used, the ratios were 30 to 45:1 and were independent of the group in the para-position. A silylated p-tert-butyloxacalix[3]arene 27 was characterized by X-ray crystallography to confirm that it was in the partial-cone conformation as shown in Figure 10. These derivatives could serve as reaction intermediates, due to the ease with which the silicon–oxygen bond can be cleaved in the presence of fluoride, although this chemistry has yet to be explored.
![[1860-5397-8-22-10]](/bjoc/content/figures/1860-5397-8-22-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: X-ray crystal structure of 27 in the partial-cone conformer (CCDC ID SUP 90399) [41].
Figure 10: X-ray crystal structure of 27 in the partial-cone conformer (CCDC ID SUP 90399) [41].
3.1.5 Intramolecularly bridged derivatives: Linking two or more phenolic calixarene oxygen atoms together is a common method to improve selectivity and complex stability, and derivatives such as calixcrowns have been known for a considerable time [42]. Amido-di-O-bridged oxacalix[3]arenes were reported by Chen in 2005 through reaction of 3a with N,N’-bis(chloroacetyl)-α,ω-alkylenediamines in refluxing acetone with K2CO3 as the base (Scheme 16) [43,44]. Those compounds linked by two (28) or three (29) methylene groups had a binding affinity for linear primary alkyl ammonium ions from n-BuNH3+ to n-HexNH3+.
![[1860-5397-8-22-i16]](/bjoc/content/inline/1860-5397-8-22-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of strapped oxacalix[3]arene derivatives 28 and 29: (i) N,N’-Bis(chloroacetyl)-1,2-ethylenediamine or N,N’-bis(chloroacetyl)-1,3-propylenediamine, K2CO3, acetone [43].
Scheme 16: Synthesis of strapped oxacalix[3]arene derivatives 28 and 29: (i) N,N’-Bis(chloroacetyl)-1,2-ethyle...
3.2 Upper-rim derivatives
Although the lower rim has many advantages as a binding site for guests, not least in the relative ease with which substituents can be attached, the upper rim can also function as a molecular recognition centre. The cavity created by the lipophilic phenolic units, particularly when held in place through allosteric effects of lower-rim substituents bound to metals, can accommodate a number of quaternary ammonium ions or buckminsterfullerene, C60. Consequently, the ability to vary the upper rim functional groups after cyclization is of some interest.
3.2.1 Asymmetric oxacalix[3]arenes: Using the synthetic routes described by Gutsche or Hampton it is possible to create oxacalixarenes with a range of upper-rim groups [14,15]; however, these methods can only yield threefold symmetric oxacalix[3]arenes. In order to introduce other groups and create asymmetric derivatives it is necessary to go through a stepwise synthetic route. Fortunately the strategy described by Fuji in 1998 [19] can be used to prepare linear trimers in which two or three different substituents are present. Using this method it was possible to prepare chiral oxacalix[3]arenes incorporating t-Bu, iPr, Et or H in the para-position of the phenolic moieties, as seen in example 30 in Figure 11 [45].
![[1860-5397-8-22-11]](/bjoc/content/figures/1860-5397-8-22-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: A chiral oxacalix[3]arene [45].
Figure 11: A chiral oxacalix[3]arene [45].
The enantiomers can be separated by a chiral HPLC column and give opposite circular dichroic spectra, and can be crystallized out for structural characterization. X-ray crystallography was again able to determine the structure of compound 30 (Figure 12).
![[1860-5397-8-22-12]](/bjoc/content/figures/1860-5397-8-22-12.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: X-ray crystal structure of asymmetric oxacalix[3]arene 30 incorporating t-Bu, iPr and Et groups (CCDC ID 108839) [19].
Figure 12: X-ray crystal structure of asymmetric oxacalix[3]arene 30 incorporating t-Bu, iPr and Et groups (CC...
The work was extended in 2001 [21], and expanded in 2002 [46] to include a single Br substituent (31), which led to an important advance in oxacalix[3]arene chemistry as debromination of 31 allowed the introduction of new groups in the vacant para-position via the mono-unsubstituted derivative 32 as shown in Scheme 17. The route introduced nitro (33), azide (34), imidazole (35), phthalimide (36), cyano (37) and methoxyether (38) groups, linked to one of the oxacalix[3]arene rings by a methylene spacer.
![[1860-5397-8-22-i17]](/bjoc/content/inline/1860-5397-8-22-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Reactions of an oxacalix[3]arene incorporating an upper-rim Br atom with (i) Pd(OAc)2, PPh3, HCO2H, Et3N; (ii) NH4NO3, acetic anhydride; (iii) Et2NH, H2CO (aq), AcOH, MeI; (iv) NaN3; (v) imidazole; (vi) potassium phthalimide; (vii) NaCN; (viii) NaOMe [46].
Scheme 17: Reactions of an oxacalix[3]arene incorporating an upper-rim Br atom with (i) Pd(OAc)2, PPh3, HCO2H,...
As noted earlier, in Scheme 3, Komatsu’s diformylphenol approach also generates symmetric and asymmetric oxacalix[n]arenes, where n = 3 or 4 [22].
3.2.2 Upper-rim esters and their reactions: Formation of the oxacalix[3]arene 3k with an upper-rim ester [47-49] makes further derivatives accessible by cleavage of the ester to leave the carboxylic acid 39 as shown in Scheme 18.
![[1860-5397-8-22-i18]](/bjoc/content/inline/1860-5397-8-22-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of acid 39: (i) NaOH, EtOH/H2O, HCl (aq) [47].
Scheme 18: Synthesis of acid 39: (i) NaOH, EtOH/H2O, HCl (aq) [47].
Shinkai used this methodology to prepare dimeric oxacalix[3]arene capsules linked by 1,4-xylylenediamine spacers. Derivatives of 39, protected at the lower rim by methyl or N,N-diethylamide groups, were coupled to mono-t-Boc-protected 1,4-xylylenediamine. Subsequent deprotection and reaction with a second equivalent of the oxacalixarene acid gave the dimeric compound (capsule-40) shown in Figure 13. A nonencapsulating analogue was prepared through reaction of the acid derivative with benzylamine. The overall yield from the oxacalix[3]arene is less than 5%, but, given that the dimerization proceeds in only 14%, this is nevertheless quite impressive. However, in addition to the formation of the molecular capsule, a self-threaded dimer (rotaxane-40) was also isolated, which had resulted from an upper-rim substituent threading through the central cavity during dimerization. The existence of the rotaxane structure was deduced from the complexity of the patterns observed in the 1H NMR spectrum compared to that of the capsule. A similar strategy was adopted to incorporate porphyrin linkers between two oxacalix[3]arenes, but, due to the size of the porphyrins and their rigidity, only the capsular form was found [50]. Treatment with zinc(II) acetate introduced three equivalents of the metal, one for each porphyrin unit.
![[1860-5397-8-22-13]](/bjoc/content/figures/1860-5397-8-22-13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: Two forms of dimeric oxacalix[3]arene 40 [47].
Figure 13: Two forms of dimeric oxacalix[3]arene 40 [47].
3.2.3 Capping the upper rim: Capping the upper rim is also possible, as shown by Araki in 2000, through a complex synthetic pathway starting from bromooxacalix[3]arene [51]. As shown in Scheme 19, oxacalix[3]arene 3f was treated with methyl iodide in the presence of NaH in THF at reflux to afford its methyl ether 41 in 41% yield. With the lower rim protected, the upper rim was converted to the aldehyde 42 and then reduced to the methylol 43. Reaction with 1,3,5-tris(bromomethyl)benzene in a boiling suspension of NaH in THF/DMF afforded the upper-rim capped compound 44 in 26% yield. The sulfur-bridged analogue 45 was prepared in 36% yield by bromination of the methylol-terminated oxacalix[3]arene, employing PBr3, and coupling with 1,3,5-tris(methanethiol)benzene in the presence of Cs2CO3 in THF.
![[1860-5397-8-22-i19]](/bjoc/content/inline/1860-5397-8-22-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Capping the upper rim: (i) t-BuLi, THF, −78 °C; (ii) NaBH4, THF/EtOH; (iii) 1,3,5-tris(bromomethyl)benzene, Na2CO3, DMF [51].
Scheme 19: Capping the upper rim: (i) t-BuLi, THF, −78 °C; (ii) NaBH4, THF/EtOH; (iii) 1,3,5-tris(bromomethyl)...
3.2.4 Upper-rim coordination chemistry: The functionalization of the upper rims of oxacalix[3]arenes has also been achieved through classical inorganic coordination chemistry. Shinkai reported that the reactions of 4- and 3-pyridyloxacalix[3]arenes, protected on the lower rims by esters or methyl ethers, with [1,3-(diphenylphosphine)propane]palladium(II) salts gave dimeric capsules linked by three Pd(II) ions at the 4-pyridyl groups (46, Figure 14) or 3-pyridyl groups (47) [52,53]. The twist inherent in pyridylphenols, and by extension oxacalix[3]arenes incorporating these motifs, was expected to result in two chiral (M and P) forms of the capsules. The addition of Na+ appeared to enhance the twisting of capsule 46, presumably through an allosteric effect that occurred when the cations bound to the lower-rim esters, as indicated by increasingly complex 1H NMR patterns. When 46 bound to S-2-methylbutylammonium triflate, the presence of a chiral complex was confirmed by circular dichroism [53].
![[1860-5397-8-22-14]](/bjoc/content/figures/1860-5397-8-22-14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: Oxacalix[3]arene capsules 46 and 47 formed through coordination chemistry [52,53].
Figure 14: Oxacalix[3]arene capsules 46 and 47 formed through coordination chemistry [52,53].
4 Oxacalix[3]arene complexes
4.1 Complexation by parent oxacalix[3]arenes
4.1.1 Receptors for ammonium cations: The symmetric cavity of the oxacalix[3]arenes, with three CH2OCH2 bridges and electron-rich aromatic groups, makes them attractive macrocycles to bind ammonium cations. The affinity of 3a for acetylcholine and several other quaternary ammonium ions was investigated by Masci in 1995 [54] who found that Kassoc values in CDCl3 were modest, ranging from 38 M−1 for N,N,N-trimethylanilinium to 90 M−1 for N,N-dimethylpyrrolidinium, but significantly greater than those of the dihomocalix[4]arene and tetrahomooxacalix[4]arene analogues.
4.1.2 Alkali-metal complexes: The parent oxacalix[3]arenes (calixarenes with free OH groups) show little ability to bind alkali metals, and extraction studies from water to CH2Cl2 showed that this ability was enhanced only in the presence of strong bases [15]. Hampton’s purification of 3a involved the formation and precipitation of the Na+ salt, which would seem to indicate a significant affinity for metal cations. Surprisingly, only para-chlorooxacalix[3]arene, 3e, was found to bind alkali metals and then only when triethylamine was used to promote salt formation. The binding constants were determined by 1H NMR as 0.39 M−1 for Na+, 0.32 M−1 for K+ and 0.11 M−1 for Li+ in the presence of 10 equiv of the triflate salts. However, those oxacalixarenes form stronger complexes with transition, lanthanide and uranyl cations.
Cragg employed the quartz-crystal-microbalance technique to investigate binding by Na+, K+ and Ca2+ to 3a and 3k [48]. Again, Na+ was bound preferentially, with computer models suggesting that this was due to the depth to which the cation was drawn into the macrocyclic cavity when in the cone conformer.
4.1.3 Transition-metal complexes: The first example of transition-metal binding to an oxacalix[3]arene was Hampton’s variable temperature 1H NMR investigation of the interactions between titanium(IV) species and 3a [55]. In the absence of crystallographic evidence the NMR splitting patterns were compared to simulated spectra. At ambient temperature the NMR-derived symmetry was C3v, matching that of the macrocycle, but upon cooling an asymmetric Cs symmetry emerged. It was proposed that rapid interconversion between isomers occurred by a “turnstile” or Berry-pseudorotation mechanism. A subsequent paper from the group reported the crystal structure of the titanium(IV) isopropoxide (Ti(iPrO)4) complex [56]. The structure was dimeric; each macrocycle was present as the trianion bound to the titanium by all three oxygens and pulled slightly into the cavity by iPrO−. The paper also reported the result of a reaction between the lithium salt of 3b and vanadyl chloride (VOCl3). Based on powder diffraction and 51V NMR data it was proposed that the VO group bound within the macrocyclic cavity, by analogy to the Ti(IV) complex, and that these units formed linear aggregates held together by V=O···V=O interactions. Ten years later, Redshaw was able to prove Hampton’s assertion regarding the structure by X-ray analysis of the VO complex shown in Figure 15 [57].
![[1860-5397-8-22-15]](/bjoc/content/figures/1860-5397-8-22-15.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: X-ray crystal structure of the 3b-vanadyl complex (CCDC ID 240185) [57].
Figure 15: X-ray crystal structure of the 3b-vanadyl complex (CCDC ID 240185) [57].
Katz used calixarenes to disperse reactive titanium on silica in order to prepare a catalytically active surface [58]. While p-tert-butylcalix[4]arene appeared to work successfully, oxacalix[3]arene 3a first bound titanium and was then cleaved to give an acyclic surface-bound product with free methyl and aldehyde termini (Scheme 20).
![[1860-5397-8-22-i20]](/bjoc/content/inline/1860-5397-8-22-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Effect of Ti(IV)/SiO2 on 3a: (i) Ti(OiPr)4, toluene; (ii) triphenylsilanol, toluene; (iii) partially dehydroxylated silica gel, toluene [58].
Scheme 20: Effect of Ti(IV)/SiO2 on 3a: (i) Ti(OiPr)4, toluene; (ii) triphenylsilanol, toluene; (iii) partiall...
Klufers prepared complexes of 3b, 3d and 3k through reaction of the macrocycles with (Et3N)2[Re(CO)3Br3] in acetonitrile [49]. The X-ray crystal structures of the complexes with 3b and 3d showed binding by Re(CO)3 to two deprotonated phenolic oxygen atoms as shown in Figure 16. Reaction with ester derivative 3k at 85 °C resulted in decomposition of the macrocycle.
![[1860-5397-8-22-16]](/bjoc/content/figures/1860-5397-8-22-16.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 16: X-ray crystal structures of oxacalix[3]arene complexes with rhenium: 3b∙Re(CO)3 (CCDC ID 620981, left) and 3d∙Re(CO)3 (CCDC ID 620982, right) [49].
Figure 16: X-ray crystal structures of oxacalix[3]arene complexes with rhenium: 3b∙Re(CO)3 (CCDC ID 620981, le...
4.1.4 Lanthanide complexation: The first study of the binding affinities of lanthanides for oxacalix[3]arenes was in 1995 when Hampton reported the crystal structure and dynamic behaviour of a scandium(III) complex of 3a [59]. Later, the X-ray structures of lanthanum, lutetium and yttrium complexes with the same macrocycle showed 2:2 complexes between the cations and macrocycles [60]. In these structures the lanthanides are either six-coordinate, with distorted octahedral metal centres, or eight-coordinate, as in the structure illustrated in Figure 17.
![[1860-5397-8-22-17]](/bjoc/content/figures/1860-5397-8-22-17.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 17: X-ray crystal structure of the La2·3a2 complex (CSD ID TIXXUT) [60].
Figure 17: X-ray crystal structure of the La2·3a2 complex (CSD ID TIXXUT) [60].
The same group calculated the apparent binding constants of metal triflates with 3a and 3e [61]. Results showed that the binding constants for 3e were slightly higher than 3a and that the strength of binding increased in the sequence Ca2+, Na+, Li+ < Mg2+ < La3+ << Y3+ < Lu3+ << Sc3+. To reinforce this, the transporting ability of the oxacalixarenes was investigated. Aqueous/organic/aqueous liquid-membrane transport experiments were undertaken with both oxacalix[3]arenes in order to determine their cation selectivities. No transport of Li+ or Mg2+ was observed, but 3e transported 44% of the Sc3+ over 24 h when a mixture of three cations (Sc3+, Mg2+ and Li+) was used as the source phase.
4.1.5 Chelating behaviour with uranium: Complexation of the uranyl cation by oxacalix[3]arenes has been ongoing since 1999 when Thuéry reported a complex of uranyl (UO22+) and 3a [62]. The X-ray crystal structure showed that the cation was threaded through a single macrocycle in what was, at the time, an unprecedented pseudotrigonal geometry, which included a weak interaction between the nitrogen of Et3N and a uranyl oxygen (Figure 18). Masci and Thuéry later reported more tetrahedrally and pentagonally distorted structures with 3a and 3b [63]. The nature of the alkylammonium counterion appeared to be influential in determining the final geometry around the uranium centre, yet in some cases it did not interact with the uranyl moiety (Figure 18).
![[1860-5397-8-22-18]](/bjoc/content/figures/1860-5397-8-22-18.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 18: X-ray crystal structures of [3a∙UO2]− with a cavity-bound cation (CCDC ID 135575, left) and without a coordinated cation (CCDC ID 181042, right) [62,63].
Figure 18: X-ray crystal structures of [3a∙UO2]− with a cavity-bound cation (CCDC ID 135575, left) and without...
Replacing the alkylammonium cations with protonated [2.2.2]cryptand resulted in 1:1 and 2:1 complexes in which the uranyl–oxacalix[3]arene moiety acts as a recognition site for the [2.2.2]cryptand [64]. Figure 19 shows the crystal structure of the 2:1 complex.
![[1860-5397-8-22-19]](/bjoc/content/figures/1860-5397-8-22-19.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 19: X-ray crystal structure of a supramolecule comprising two [3g·UO2]− complexes that encapsulate a diprotonated cryptand (CCDC ID 181044) [64].
Figure 19: X-ray crystal structure of a supramolecule comprising two [3g·UO2]− complexes that encapsulate a di...
4.2 Binding properties of oxacalix[3]arene derivatives
One of the most important features of calixarenes in general and oxacalix[3]arenes in particular is their vast ability to selectively bind and carry ions and neutral species. This is achieved mainly with lower-rim derivatives in solution.
4.2.1 Receptors for ammonium cations: Although the simple parent oxacalix[3]arene 3a is able to bind quaternary ammonium ions (as described above), several derivatives have also been studied with respect to these and other ammonium ions. Extraction studies from alkaline aqueous picrate solutions into CH2Cl2 indicated that the n-butyl ether derivative 10 showed a high affinity for n-BuNH3+ (82% E) as postulated by the authors, because both host and guest possess the same C3-symmetry [24]. Ethyl ester 12a was more efficient at extracting n-BuNH3+ picrate from water into CH2Cl2 than its calix[4]arene analogue was, in both the cone (77% vs 24% E) and partial-cone (42% vs 6% E) conformers [29]. In a wider study, Yamato determined extraction data for 17a with n-BuNH3+ picrate (98% E cone vs 93% E partial-cone), iBuNH3+ picrate (48% E cone vs 37% E partial-cone) and t-BuNH3+ picrate (35% E cone vs 14% E partial-cone) [34]. The hexaamide derivative 20 bound n-BuNH3+ well, and an anion dependence was determined; Kassoc values in CDCl3 were 536 ± 32 M−1 for Cl− and 230 ± 17 M−1 for Br− [37].
Studies of the C3 symmetrically capped triamide 13 reported that this derivative acts as a well-preorganized host for binding primary ammonium ions, such as phenylalanine methyl ester [31]. Chiral recognition of optically active primary alkyl ammonium ions was also obtained with an ether derivative of oxacalix[3]arene 3a with one methyl and two n-butyl lower-rim substituents 49, as shown in Figure 20 [65]. The compound was shown to exist in (+) and (−) enantiomers, and in a partial-cone conformation, proof of which came from X-ray crystallography. The compound bound to α-amino acid ethyl esters and 1-arylethylamines with the methoxy and one n-butoxy oxygen. The (−)-4-tert-butyloxacalix[3]arene derivative bound L-alanine ethyl ester and L-phenylalanine ethyl ester better than their enantiomers, with association constants of 4500 M−1 and 2000 M−1, respectively. (R)-1-Phenylethylamine and (R)-1-naphthylethylamine cations were bound more strongly by the (+)-enantiomer.
![[1860-5397-8-22-20]](/bjoc/content/figures/1860-5397-8-22-20.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 20: X-ray crystal structure of oxacalix[3]arene 49 capable of chiral selectivity (CSD ID HIGMUF) [65].
Figure 20: X-ray crystal structure of oxacalix[3]arene 49 capable of chiral selectivity (CSD ID HIGMUF) [65].
Allosteric effects can also be employed to affect the binding of ammonium cations. Katoh’s N-hydroxypyrazinone-containing oxacalix[3]arene 23 extracted n-BuNH3+ picrate and t-BuNH3+ picrate better in the presence of Ga3+, indicating cooperation between the two binding sites [38]. The association constant for n-HexNH3+ picrate was found to be 4375 M−1, but when Ga3+ was present this dropped to 2833 M−1, suggesting that the macrocyclic cavity, while preorganized for the smaller cations, was too rigid for the extended ammonium cation.
One of the more unusual derivatives to have been prepared, 50, incorporates an N-pyridinium dye on one of the upper-rim positions, which, in combination with the phenolic unit of the macrocycle, forms a proton-ionizable Reichardt dye, illustrated in Figure 21 [66]. The other p-tert-butyl substituted phenols are blocked from ionization, as are the methyl ethers. The native oxacalix[3]arene dye is pale green and gives no response to benzylamine (BzNH2) or triethylamine (Et3N), but cyclohexylamine (c-HexNH2) and n-butylamine (n-BuNH2) bind with a concomitant colour change to blue.
![[1860-5397-8-22-21]](/bjoc/content/figures/1860-5397-8-22-21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 21: The structure of derivative 50 incorporating a Reichardt dye [66].
Figure 21: The structure of derivative 50 incorporating a Reichardt dye [66].
4.2.2 Alkali metals: The ionophoric properties of the conformationally mobile ethyl ether of 3a (8) and both cone and partial-cone n-butyl ether 10 derivatives toward alkali-metal cations were estimated by extraction experiments from alkaline aqueous picrate solutions to CH2Cl2 [24], with the latter showing some preference for K+ (59% E) over Na+ (6%) and Cs+ (35%).
Replacement of the alkyl groups by residues with additional binding sites, such as the carbonyl group, strongly affects the binding ability of calixarene derivatives. Thus, the binding properties of derivatives containing esters, amides and ketones, have been assessed. Extraction studies performed under the same conditions as described above reported that cone ester 12a shows high selectivity for Na+ whereas the partial-cone conformer shows K+ selectivity (Table 1) [29]. Similar extraction experiments performed with amide 17a [34] reported that this derivative is a better phase-transfer agent than 12a, but shows the same trend as 12a: cone-17a exhibits the highest preference for Na+, while partial-cone-17a prefers K+ (Table 1).
Table 1: Percentage extraction of alkali-metal picrates into CH2Cl2.a,b
| Li+ | Na+ | K+ | Cs+ | |
|---|---|---|---|---|
| cone-12a | 7 | 79 | 64 | 49 |
| paco-12ab | 0 | 26 | 88 | 82 |
| cone-17a | – | 93 | 72 | – |
| paco-17a | – | 28 | 73 | – |
aData adapted from references [29] and [34]. bpartial-cone denoted as paco.
The association constants, Kassoc, for both derivatives (12a and 17a) were determined in THF/CHCl3 (1:1) at 25 °C by UV absorption spectrophotometry (Table 2) [25].
Table 2: Association constants (log Kassoc) of alkali- and alkaline-earth-metal complexes.a,b.
| Na+ | K+ | Rb+ | Cs+ | Mg2+ | Ca2+ | Ba2+ | |
|---|---|---|---|---|---|---|---|
| cone-12a | 4 | 4.7 | 4.2 | 3.9 | <2 | <2 | <2 |
| cone-17a | >7 | 5.9 | 5.5 | 5.2 | 4.9 | >7 | >7 |
| paco-17ab | 5.1 | 6.2 | 6.0 | 5.5 | – | – | – |
aData adapted from reference [25]. bpartial-cone denoted as paco.
Marcos [39,67] reported binding data for alkali- and alkaline-earth-metal cations with 17a and 24 (Table 3). Extraction studies performed under different conditions than the previous ones (neutral aqueous picrate solutions to CH2Cl2), indicated that both derivatives show similar extraction profiles, although 17a is a much stronger binder than 24. Both exhibit highest selectivity for Na+ (50 and 20% E for 17a and 24, respectively) and 17a is also a good extractant for Ba2+ (55% E).
Table 3: Percentage extraction of alkali and alkaline earth metal picrates into CH2Cl2.
| Li+ | Na+ | K+ | Rb+ | Cs+ | Mg2+ | Ca2+ | Sr2+ | Ba2+ | |
|---|---|---|---|---|---|---|---|---|---|
| cone-17aa | 25 | 50 | 32 | 27 | 20 | 17 | 34 | 41 | 55 |
| cone-24b | 4 | 20 | 5 | 6 | 6 | 2 | 4 | 4 | 4 |
aData adapted from references [67]. bData adapted from reference [39].
Derivatives with heteroatoms on the lower rim have also been tested as cation chelators. The binding properties of 2-pyridylmethyloxy derivative 11a in both conformations, have been established [27,68]. Extraction studies from neutral aqueous picrate solutions to CH2Cl2 showed that, among all the cations studied, the partial-cone conformer is a better extractant than the cone.
As well as simple oxacalix[3]arenes and their derivatives, capped compounds have also been investigated. Association constants for several metal cations were determined for Yamato’s lower-rim-capped derivative 18 [33]. Values were found for Na+ (log Kassoc 5.3), K+ (log Kassoc 6.7) and Cs+ (log Kassoc 5.8). This contrasts with log Kassoc of 7.6 for n-BuNH3+ picrate. The extractability of metals from aqueous solution into CH2Cl2 by Araki’s upper-rim-capped derivatives was also determined [51]. The complementary cavity size and the rigid structure of the cage molecule 44 probably led to the high Cs+ selectivity (≈45% E) compared to negligible amounts of Na+, K+ or Rb+ (<5% E); however, the sulfur-linked compound 45 failed to extract any cations.
4.2.3 Transition metals: The tris(diphenylphosphine) derivative 26 prepared by Matt [40] was reacted with [Mo(CO)3(cycloheptatriene)] to give a complex that was determined to be the symmetrically bound Mo(CO)3 complex involving all three of the phosphorus donors. The oxacalix[3]arene also formed a complex with rhodium. Elemental analysis supported a composition incorporating the H–Rh–C=O fragment. 1H NMR indicated that this was threaded through the macrocyclic annulus, based on the presence of a peak at −9.70 ppm, and infrared analysis showed a carbonyl absorption band at 1977 cm−1. This suggested an orientation in which the hydrogen was endo, and the carbonyl exo, to the macrocyclic cavity. Gold(I) and silver(I) complexes also form with the cations most likely adopting a trigonal planar C3v geometry, as shown in Figure 22, based on the symmetric 31P NMR pattern at ambient temperature. At lower temperatures, however, the A3X pattern seen for the silver(I) complex changes to an A2BX pattern, indicating that the apparent symmetry is a time-averaged effect.
![[1860-5397-8-22-22]](/bjoc/content/figures/1860-5397-8-22-22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 22: Phosphorylated oxacalix[3]arene complexes with transition metals: (Left to right) 26∙Au, 26∙Mo(CO)3 and 26∙RhH(CO) [40].
Figure 22: Phosphorylated oxacalix[3]arene complexes with transition metals: (Left to right) 26∙Au, 26∙Mo(CO)3...
Cragg reported the reaction of 17a with mercury(II) chloride and the X-ray crystal structure of the product (Figure 23) [69]. The structure revealed that a [HgCl2]2 fragment bridged between two macrocycles through coordination to one amide group of each. The cations were thus exo to the macrocyclic cavity and represented the first example in which a cation was not bound within the annulus.
![[1860-5397-8-22-23]](/bjoc/content/figures/1860-5397-8-22-23.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 23: X-ray crystal structure of [17a·HgCl2]2 (CCDC ID 168653) [69].
Figure 23: X-ray crystal structure of [17a·HgCl2]2 (CCDC ID 168653) [69].
Marcos reported on the binding properties and theoretical studies of 17a [67] and 24 [39] with transition and heavy metals. Extraction studies from neutral aqueous picrate solutions to CH2Cl2 indicated that amide 17a is a good extractant for Ni2+, Co2+, and Ag+, and mainly for Pb2+ with 80% E. The data in Table 4 also shows that ketone 24 is a weak extracting agent, with a slight preference for Ag+. This is in agreement with the higher basicity of the carbonyl oxygen in the amide group compared with the ketone group.
Table 4: Percentage extraction of transition- and heavy-metal picrates into CH2Cl2.
| Mn2+ | Fe2+ | Co2+ | Ni2+ | Cu2+ | Zn2+ | Ag+ | Cd2+ | Pb2+ | |
|---|---|---|---|---|---|---|---|---|---|
| 17aa | 19 | 19 | 39 | 45 | 24 | 15 | 40 | 37 | 80 |
| 24b | 2 | 5 | 2 | 4 | 4 | 3 | 13 | 3 | 5 |
aData adapted from reference [67]. bData adapted from reference [39].
4.2.4 Lanthanides: Marcos investigated the lanthanide extraction by both 17a [70] and 24 [39] using the same conditions as described above (Table 5). Ketone 24 is a poor phase-transfer agent (% E ranges from 5 to 7), while amide 17a clearly discriminates between the light and heavy lanthanides. The lower-weight cations, such as Ce3+, Pr3+ and Nd3+ (34% E) are preferred over the heavier, such as Er3+ and Yb3+ (13% E). The stability constants for the 1:1 complexes with 17a were also determined by UV absorption spectrophotometry in methanol at 25 °C, by using chloride salts. The same positive discrimination for the light lanthanides was observed (log β = 5.5 and 3.4 for La3+ and Yb3+, respectively).
Table 5: Percentage extraction of lanthanide-metal picrates into CH2Cl2.
| La3+ | Ce3+ | Pr3+ | Nd3+ | Sm3+ | Eu3+ | Gd3+ | Dy3+ | Er3+ | Yb3+ | |
|---|---|---|---|---|---|---|---|---|---|---|
| 17aa | 28 | 34 | 34 | 34 | 31 | 30 | 17 | 18 | 13 | 13 |
| 24b | 6 | 5 | 5 | 6 | 6 | 6 | 6 | 5 | 6 | 7 |
aData adapted from reference [70]. bData adapted from reference [39].
The complexing ability of the ionizable tricarboxylic acid 14a towards lanthanides Pr3+, Eu3+ and Yb3+ and actinide Th4+ was established in methanol, by potentiometry measurements [71]. Results showed that the complex formed with Th4+ was more stable than the complexes of lanthanides (log β values are 20.5, 19.6, 21.3 and 23.1, respectively).
5 Other applications
5.1 Hosts for fullerenes
One of the remarkable characteristics of calixarenes is the bowl shape of the molecule. In the case of oxacalix[3]arenes, the bowl is quite shallow, which indicates that they may be good hosts for spherical guests and immediately suggests binding to fullerenes. Furthermore, the macrocyclic bowl is the perfect size for C60 and has a complementary threefold-symmetry element.
Based on the knowledge that p-tert-butylcalix[8]arene was able to complex C60 [72,73] Shinkai investigated the interaction of C60 with 3a in 1997 by UV–vis spectroscopy [74]. In a later full paper, UV–vis absorption spectra of C60 were recorded with calix[n]arenes and oxacalix[3]arenes. The interaction of fullerenes with calixarenes affected the spectra between 420 and 450 nm [75]. By using the Benesi–Hildebrand method, 3a was shown to bind to C60 with a Kassoc of 35.5 M−1 in toluene at 25 °C; however, when methylated on the lower rim, no binding was observed. Molecular modelling was employed to illustrate how the shallow cavity of 3a allowed for optimum interactions between the oxacalix[3]arene aromatic rings and C60.
While subtle spectroscopic features and computer models appeared to indicate fullerene binding, structural evidence was to be more compelling. In 1998, Fuji reported the solid-state structure of 3f with C60 as proof of 1:1 binding [76]. Alignment of the oxacalix[3]arene C3 axis with the same symmetry axis of the fullerenes is observed. This arrangement maximizes the number of points of contact within the supramolecular complex, thereby enhancing the van der Waals interactions. In the same paper, the association constants of several oxacalix[3]arenes were calculated by the Rose–Drago method based on absorption features at 425 or 430 nm in toluene. The strongest binding was observed for 3a (35.6 M−1) and the weakest for 3h (9.1 M−1).
Although spectroscopic methods are widely used to determine host–guest association constants, Georghiou has argued persuasively that spectral changes in solution may be due to a combination of several factors, of which host–C60 complex formation is only one [77]. Consequently, reported Kassoc values determined by this method should be treated with some caution.
Fullerene derivatives that lack some of the symmetry of the parent compound have been shown to bind to oxacalix[3]arenes, as in Fuji’s X-ray structure of 1,4-bis(9-fluorenyl)-1,4-dihydro[60]fullerene with 3f shown in Figure 24, in which the oxacalix[3]arene binds to the C60 derivative with the fluorenyl substituents oriented away from the macrocycle [78].
![[1860-5397-8-22-24]](/bjoc/content/figures/1860-5397-8-22-24.png?scale=1.4&max-width=1024&background=FFFFFF)
Figure 24: X-ray crystal structures of 3f with C60 (CCDC ID 182801, left) [76] and a 1,4-bis(9-fluorenyl) C60 derivative (CCDC ID 139793, right) [78].
Figure 24: X-ray crystal structures of 3f with C60 (CCDC ID 182801, left) [76] and a 1,4-bis(9-fluorenyl) C60 deri...
Raston reported that p-benzyloxacalix[3]arene (3i) formed a 2:1 complex with C60 in toluene [79]. The X-ray crystal structure showed how the two oxacalix[3]arenes bound on opposite sides of the fullerene, with their benzyl arms interdigitated. When the complex was isolated and added to CH2Cl2 then the fullerene was released. The method could be used to separate C60 from fullerite (a mixture of fullerenes of different sizes) in greater than 99.5% purity. A similar experiment was undertaken by Georghiou with 6a leading to much higher association constants of 296 M−1 (toluene) and 441 M−1 (benzene) [23]. Crystallography revealed a similar interdigitated 2:1 complex to that observed by Raston for 3i (Figure 25).
![[1860-5397-8-22-25]](/bjoc/content/figures/1860-5397-8-22-25.png?scale=1.56&max-width=1024&background=FFFFFF)
Figure 25: X-Ray crystal structure of 3i and 6a encapsulating C60 (CCDC ID 102473 and 166077) [23,79].
Figure 25: X-Ray crystal structure of 3i and 6a encapsulating C60 (CCDC ID 102473 and 166077) [23,79].
One area of interest has been the selective separation of C70 from a mixture of fullerenes. Komatsu proposed a method for the preferential precipitation of C70 over C60 with p-halooxacalix[3]arenes [80]. p-Iodooxacalix[3]arene (3j) was able to achieve 90% extraction with a selectivity approaching 90%.
An unexpected effect of fullerene complexation was that a water-soluble capsule formed from two p-tert-butyloxacalix[3]arenes with trimethylammonium groups on the lower rims, 51, which bound C60, was able to cleave DNA (Figure 26) [81]. The capsule was solubilized as the MsO− salt and applied to a supercoiled form of DNA. In the absence of light, no change was seen, but in the presence of visible light the DNA became “nicked”, that is, a phosphodiester bond in one strand was broken. The authors speculated that the cationic complex was able to bind to the anionic DNA whereupon 1O2 generated by photoinduced electron transfer from guanine and C60, or alternatively photochemically by C60 alone, cleaves the DNA strand. Ikeda later advanced this line of research to carbohydrate-containing oxacalix[3]arenes that functioned in water [82].
![[1860-5397-8-22-26]](/bjoc/content/figures/1860-5397-8-22-26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 26: A C60 complexing cationic oxacalix[3]arene 51 [81].
Figure 26: A C60 complexing cationic oxacalix[3]arene 51 [81].
The same cationic complex was deposited as a monolayer onto an alkylsulfonate coated gold surface and elicited both a redox response, as determined by cyclic voltammetry, and a photochemical response to visible light [83,84]. The optical response was studied further [85], and a transient band was observed at 545 nm, which was not present in the spectrum of C60 alone. The origin of the band was ascribed to C60-capsule triplet–triplet absorption.
As discussed above, oxacalix[3]arenes with pyridine in the para-position and ethyl esters on the lower rim are able to form capsules through coordination to palladium [53]. Capsule 46 was shown to bind to C60 by the presence of two peaks in the 13C NMR spectrum, which did not coalesce even at 90 °C. 1H NMR was used to determine an association constant of 54 M−1 in Cl2CDCDCl2 at 60 °C. An asymmetric capsule incorporating an oxacalix[3]arene and three Zn(II)porphyrin moieties, 52, was also able to bind C60 in a similar fashion with an association constant of 60 M−1 in toluene-d8 at −30 °C [86].
Another strategy to promote fullerene inclusion in an oxacalix[3]arene was to link the two by a triethylene glycol tether to form a molecular cup-and-ball 53 [87]. In addition to self-inclusion, the authors also proposed the formation of higher order oligomers arising from C60 inclusion in a neighbouring oxacalixarene through the change in conformation illustrated in Figure 27.
![[1860-5397-8-22-27]](/bjoc/content/figures/1860-5397-8-22-27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 27: An oxacalix[3]arene-C60 self-associating system 53 [87].
Figure 27: An oxacalix[3]arene-C60 self-associating system 53 [87].
5.2 Fluorescent chemosensors
In order to determine the equilibrium constants with quaternary ammonium ions, Shinkai [88] prepared an oxacalixarene with pendent pyrene groups, 54, which fluoresced at 480 nm. Oxacalix[3]arene fluorescence was significantly quenched in the presence of n-hexyl ammonium cations (n-HexNH3+), but only in the partial-cone conformation, as the ammonium cation forced the lower-rim pyrene groups apart. The same cation had a much higher affinity for cone-54 through its complementary binding sites, but approached these from the upper rim, leaving the excimer fluorescence unaffected. Yamato also pursued this path, preparing a tris(pyrenyl) derivative 55 in the cone conformer by employing “click” chemistry (Scheme 21) [89]. One interesting aspect of the synthesis was that the tris(propargyl) click precursor crystallized as a mixture of cone and partial-cone conformers, yet addition of n-BuNH3+ClO4− to the conformers in solution pushed the equilibrium towards the cone. Cone-55 gave a response to Pb2+ through the enhancement of minor fluorescence peaks between 370 and 400 nm, which were unaffected by other metal guests. The group also reported that the fluorescence intensity at 396 nm increased linearly when Zn2+ was added and that the 1:1 complex of this macrocycle gave an increasing linear response at 485 nm to H2PO4− [90].
![[1860-5397-8-22-i21]](/bjoc/content/inline/1860-5397-8-22-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of fluorescent pyrene derivative 55: (i) Propargyl bromide, acetone; (ii) CuI, 1-azidomethylpyrene, THF/H2O [89].
Scheme 21: Synthesis of fluorescent pyrene derivative 55: (i) Propargyl bromide, acetone; (ii) CuI, 1-azidomet...
Rhodamine substituents can be introduced to the lower rim of the cone-14a through the ethylamine derivative of the dye (Scheme 22) [91]. Fluorescence enhancement was observed between 500 nm and 600 nm upon addition of Fe3+, Ni2+ and Sb3+ to 57, turning the colourless solution fluorescent orange–yellow, together with a colourless-to-magenta colourimetric response.
![[1860-5397-8-22-i22]](/bjoc/content/inline/1860-5397-8-22-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of responsive rhodamine derivative 57: (i) DCC, CH2Cl2 [91].
Scheme 22: Synthesis of responsive rhodamine derivative 57: (i) DCC, CH2Cl2 [91].
Kang found that the reaction of 3a with 1-bromo-4-nitrobenzyl acetate gave the trisubstituted nitrobenzene derivative 58 in 40% yield (Scheme 23) as the partial-cone conformer [92]. When a range of fluorescent ammonium cations incorporating pyrene, anthracene or naphthalene groups was tested, quenching was observed. Association constants were determined to be in the range of 1850 M−1 to 78000 M−1. The uncharged pyrenemethylamine was not bound at all, and a trimethylpyrenium cation was weakly bound (Kassoc = 300 M−1).
![[1860-5397-8-22-i23]](/bjoc/content/inline/1860-5397-8-22-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of nitrobenzyl derivative 58: (i) 1-Bromo-4-nitrobenzyl acetate, K2CO3, refluxing acetone, 3 h [92].
Scheme 23: Synthesis of nitrobenzyl derivative 58: (i) 1-Bromo-4-nitrobenzyl acetate, K2CO3, refluxing acetone...
5.3 Ion-selective electrodes
Given the apparent oxacalix[3]arene selectivity for Na+ and certain protonated amines it is likely that they can act as ion-selective agents in electrodes. This aspect of oxacalix[3]arene research demonstrates that they are not limited to fluorescent sensor applications but can also function in the electrochemical sphere.
5.3.1 Dopamine recognition: The first example of oxacalixarenes being used as electrode modifiers was in 1999 when Odashima incorporated cone p-tert-butyloxacalix[3]arene tri(n-butyl ether) (10) in a PVC matrix liquid membrane [93]. The electrode displayed excellent selectivity for dopamine over biologically important alkali-metal cations K+, by a factor of 150, and Na+, by a factor of 1600. Selectivity for dopamine against other catecholamine neurotransmitters, such as adrenaline and noradrenaline, was also greater by a factor of at least 100. This selectivity obtained with 10 is a very promising result with the potential to be developed into a dopamine sensor for use under physiological conditions. The dopamine selectivity of the trimethyl ether analogue 7 was investigated by Arrigan at the interface between water and 1,2-dichloroethane using cyclic voltammetry [94]. The log Kassoc value obtained was 8.3, which was significantly higher than those for Na+ and K+. The log Kassoc comparative data for dibenzo-18-crown-6 were 7.6 for the dopamine complex and 10.1 for the K+ complex, indicating that not only was 7 a better host for dopamine but also that K+ would not be bound preferentially as is the case for the crown ether.
5.3.2 Sensing Pb2+: In 2007, Yaftian incorporated Matt’s phosphorylated derivative 26 in a membrane solution, prepared by dissolving PVC, NaBF4, a plasticizer and the oxacalixarene in THF, which was then used to coat a graphite electrode [95]. This electrode gave a good Nernstian response of 29.7 mV/decade, over a concentration range of 1 × 10−8 M to 1 × 10−4 M of Pb2+ ions, with a detection limit of 0.4 × 10−8 M. When tested in mixtures of several competing cations (such as alkali, alkaline earth, transition, heavy metal, lanthanide and Th4+ ions) the electrode was able to determine the concentration of Pb2+ correctly within 5%, even when other ions were present in tenfold excess.
Diethylacetamide 17a was also used as an active material in ion-selective electrodes to check the detection of different types of cations [96]. Optimization of the PVC membrane composition was achieved by using different plasticizers (DEHA, o-NPOE and BBPA). The performance of the ISE incorporating 17a indicated a high affinity for Pb2+ and the use of DEHA as the best plasticizer.
5.4 Biological models
The crystal structure of the complex of 17a with NaPF6 (Figure 28) shows how the lower-rim binding site, composed of phenolic oxygen and amide nitrogen atoms, is predisposed to bind Na+ in its ideal octahedral environment [97]. The compound has been proposed to be an artificial analogue for the filter region in cation channels formed by naturally occurring transmembrane proteins and has been shown to have some activity on transmembrane ion transport in cells.
![[1860-5397-8-22-28]](/bjoc/content/figures/1860-5397-8-22-28.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 28: X-ray crystal structure of [Na2∙17a](PF6)2 (CCDC ID 116656) [97].
Figure 28: X-ray crystal structure of [Na2∙17a](PF6)2 (CCDC ID 116656) [97].
Conclusion
Since their origins in the phenol-formaldehyde chemistry of the 1960s, oxacalix[3]arenes and their analogues have shown themselves to be interesting and useful additions to the large array of artificial macrocycles that has been developed by supramolecular chemists. The C3 symmetry of oxacalix[3]arenes, commonly encountered in nature but relatively rare in synthetic host molecules, has made them valuable members of the calixarene family, with an affinity for guests with complementary binding requirements. While the parent compounds do not form particularly strong complexes with metal ions, their O-alkylated derivatives are easy to prepare and can show very efficient and selective cation binding, extending to alkyl ammonium salts. Advances in upper-rim functionalization allow for the formation of molecular capsules and chiral recognition sites, and applications have been found in fluorescence sensors, ion-selective electrodes and the extraction of pure C60 and C70 from crude fullerite. Fifty years on from their discovery by Hultzsch, oxacalix[3]arenes and their derivatives are still able to amaze chemists with their elegant symmetry and fascinating complexes.
References
-
Gutsche, C. D. Calixarenes: An Introduction; Monographs in Supramolecular Chemistry, 2nd ed.; Royal Society of Chemistry: Cambridge, U.K., 2008.
Return to citation in text: [1] -
Sliwa, W.; Kozlowski, C. Calixarenes and Resorcinarenes: Synthesis, Properties and Applications; Wiley VCH: Weinheim, Germany, 2009.
Return to citation in text: [1] -
Böhmer, V. Angew. Chem., Int. Ed. Engl. 1995, 34, 713–745. doi:10.1002/anie.199507131
Return to citation in text: [1] -
Brodesser, G.; Vögtle, F. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 19, 111–135. doi:10.1007/BF00708978
Return to citation in text: [1] [2] -
Gutsche, C. D.; Bauer, L. J. J. Am. Chem. Soc. 1985, 107, 6052–6059. doi:10.1021/ja00307a038
Return to citation in text: [1] [2] -
Cragg, P. J. Homocalixarenes. In Encyclopedia of Supramolecular Chemistry; Atwood, J. L.; Steel, J. W., Eds.; Marcel Dekker: New York, 2004; pp 649–657.
Return to citation in text: [1] -
Späth, A.; König, B. Beilstein J. Org. Chem. 2010, 6, No. 32. doi:10.3762/bjoc.6.32
Return to citation in text: [1] -
Yamato, D. J. J. Inclusion Phenom. Mol. Recognit. Chem. 1998, 32, 195–207. doi:10.1023/A:1008011410507
Return to citation in text: [1] -
Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2004, 40, 607–643. doi:10.1023/B:RUJO.0000043707.97314.52
Return to citation in text: [1] -
Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2004, 40, 1547–1578. doi:10.1007/s11178-005-0062-9
Return to citation in text: [1] -
Hultzsch, K. Kunststoffe 1962, 52, 19–24.
Return to citation in text: [1] [2] -
Masci, B.; Finelli, M.; Varrone, M. Chem.–Eur. J. 1998, 4, 2018–2030. doi:10.1002/(SICI)1521-3765(19981002)4:10<2018::AID-CHEM2018>3.0.CO;2-I
Return to citation in text: [1] -
Mukoyama, Y.; Tanno, T. Org. Coat. Plast. Chem. 1979, 40, 894–897.
Return to citation in text: [1] -
Dhawan, D.; Gutsche, C. D. J. Org. Chem. 1983, 48, 1536–1539. doi:10.1021/jo00157a033
Return to citation in text: [1] [2] [3] -
Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026
Return to citation in text: [1] [2] [3] [4] [5] -
Zerr, P.; Mussrabi, M.; Vicens, J. Tetrahedron Lett. 1991, 32, 1879–1880. doi:10.1016/S0040-4039(00)85986-9
Return to citation in text: [1] [2] -
Suzuki, K.; Minami, H.; Yamagata, Y.; Fuji, S.; Tomita, K.-I.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1992, 48, 350–352. doi:10.1107/S0108270191010399
Return to citation in text: [1] [2] -
Miah, M.; Romanov, N. N.; Cragg, P. J. J. Org. Chem. 2002, 67, 3124–3126. doi:10.1021/jo025597a
Return to citation in text: [1] -
Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a
Return to citation in text: [1] [2] [3] [4] [5] -
Ashram, M.; Mizyed, S.; Georghiou, P. E. J. Org. Chem. 2001, 66, 1473–1479. doi:10.1021/jo001518o
Return to citation in text: [1] [2] [3] -
Tsubaki, K.; Morimoto, T.; Otsubo, T.; Kinoshita, T.; Fuji, K. J. Org. Chem. 2001, 66, 4083–4086. doi:10.1021/jo0100502
Return to citation in text: [1] [2] [3] -
Komatsu, N. Tetrahedron Lett. 2001, 42, 1733–1736. doi:10.1016/S0040-4039(00)02336-4
Return to citation in text: [1] [2] [3] -
Mizyed, S.; Ashram, M.; Miller, D. O.; Georghiou, P. E. J. Chem. Soc., Perkin Trans. 2 2001, 1916–1919. doi:10.1039/B105019M
Return to citation in text: [1] [2] [3] -
Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7
Return to citation in text: [1] [2] [3] [4] [5] -
Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Yamato, T.; Haraguchi, H.; Nishikawa, J.-I.; Ide, S.; Tsuzuki, H. Can. J. Chem. 1998, 76, 989–996. doi:10.1139/v98-102
Return to citation in text: [1] [2] [3] [4] -
Yamato, T.; Zhang, F.; Sato, T.; Ide, S. J. Chem. Res., Synop. 2000, 10–12. doi:10.3184/030823400103165707
Return to citation in text: [1] -
Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021
Return to citation in text: [1] [2] [3] [4] [5] -
Ashram, M.; Mizyed, S.; Georghiou, P. E. Org. Biomol. Chem. 2003, 1, 599–603. doi:10.1039/b209046p
Return to citation in text: [1] -
Takeshita, M.; Inokuchi, F.; Shinkai, S. Tetrahedron Lett. 1995, 36, 3341–3344. doi:10.1016/0040-4039(95)00536-L
Return to citation in text: [1] [2] [3] -
Khrifi, S.; Guelzim, A.; Baert, F.; Mussrabi, M.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1995, 51, 153–157. doi:10.1107/S0108270194008036
Return to citation in text: [1] [2] -
Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Yamato, T.; Zhang, F. L. J. Inclusion Phenom. Macrocyclic Chem. 2001, 39, 55–64. doi:10.1023/A:1008196612553
Return to citation in text: [1] [2] [3] [4] -
Takimoto, M.; Ni, X.-L.; Rahman, S.; Xi, Z.; Yamato, T. J. Inclusion Phenom. Macrocyclic Chem. 2011, 70, 69–80. doi:10.1007/s10847-010-9863-8
Return to citation in text: [1] -
Ni, X.-L.; Rahman, S.; Zeng, X.; Hughes, D. L.; Redshaw, C.; Yamato, T. Org. Biomol. Chem. 2011, 9, 6535–6541. doi:10.1039/c1ob05564j
Return to citation in text: [1] -
Yamato, T.; Rahman, S.; Zeng, X.; Kitajima, F.; Gil, J. T. Can. J. Chem. 2006, 84, 58–64. doi:10.1139/v05-260
Return to citation in text: [1] [2] [3] -
Ohkanda, J.; Shibui, H. Chem. Commun. 1998, 375–376. doi:10.1039/a706869g
Return to citation in text: [1] [2] [3] -
Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Dieleman, C. B.; Matt, D.; Neda, I.; Schmutzler, R.; Harriman, A.; Yaftian, R. Chem. Commun. 1999, 1911–1912. doi:10.1039/a905677g
Return to citation in text: [1] [2] [3] [4] -
Hampton, P. D.; Daitch, C. E.; Duesler, E. N. New J. Chem. 1996, 20, 427–432.
Return to citation in text: [1] [2] -
Casnati, A.; Pochini, A.; Ungaro, R.; Ugozzoli, F.; Arnaud, F.; Fanni, S.; Schwing, M.-J.; Egberink, R. J. M.; de Jong, F.; Reinhoudt, D. N. J. Am. Chem. Soc. 1995, 117, 2767–2777. doi:10.1021/ja00115a012
Return to citation in text: [1] -
Liu, S.-L.; Gong, S.-L.; Chen, Y.-Y. Chin. J. Chem. 2005, 23, 1651–1654. doi:10.1002/cjoc.200591651
Return to citation in text: [1] [2] -
Liu, S.-L.; Gong, S.-L.; Zheng, Q.; Chen, Y.-Y.; Wu, X.-J. J. Chem. Res., Synop. 2005, 126–129. doi:10.3184/0308234054497218
Return to citation in text: [1] -
Tsubaki, K.; Otsubo, T.; Kinoshita, T.; Kawada, M.; Fuji, K. Chem. Pharm. Bull. 2001, 49, 507–509. doi:10.1248/cpb.49.507
Return to citation in text: [1] [2] -
Tsubaki, K.; Otsubo, T.; Morimoto, T.; Maruoka, H.; Furukawa, M.; Momose, Y.; Shang, M.; Fuji, K. J. Org. Chem. 2002, 67, 8151–8156. doi:10.1021/jo026152p
Return to citation in text: [1] [2] -
Zhong, Z.; Ikeda, A.; Shinkai, S. J. Am. Chem. Soc. 1999, 121, 11906–11907. doi:10.1021/ja9925002
Return to citation in text: [1] [2] [3] -
Miah, M.; Pavey, K. D.; Gun’ko, V. M.; Sheehan, R.; Cragg, P. J. Supramol. Chem. 2004, 16, 185–192. doi:10.1080/10610270310001644473
Return to citation in text: [1] [2] -
Hinrichs, M.; Hofbauer, F. R.; Klüfers, P.; Suhanji, M. Inorg. Chem. 2006, 45, 6688–6693. doi:10.1021/ic0603048
Return to citation in text: [1] [2] [3] -
Kawaguchi, M.; Ikeda, A.; Shinkai, S. Tetrahedron Lett. 2001, 42, 3725–3728. doi:10.1016/S0040-4039(01)00463-4
Return to citation in text: [1] -
Araki, K.; Hayashida, H. Tetrahedron Lett. 2000, 41, 1807–1810. doi:10.1016/S0040-4039(00)00034-4
Return to citation in text: [1] [2] [3] -
Ikeda, A.; Yoshimura, M.; Tani, F.; Naruta, Y.; Shinkai, S. Chem. Lett. 1998, 27, 587–588. doi:10.1246/cl.1998.587
Return to citation in text: [1] [2] -
Ikeda, A.; Udzu, H.; Zhong, Z.; Shinkai, S.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 2001, 123, 3872–3877. doi:10.1021/ja003269r
Return to citation in text: [1] [2] [3] [4] -
Masci, B. Tetrahedron 1995, 51, 5459–5464. doi:10.1016/0040-4020(95)00207-O
Return to citation in text: [1] -
Hampton, P. D.; Daitch, C. E.; Alam, T. M.; Bencze, Z.; Rosay, M. Inorg. Chem. 1994, 33, 4750–4758. doi:10.1021/ic00099a028
Return to citation in text: [1] -
Hampton, P. D.; Daitch, C. E.; Alam, T. M.; Pruss, E. A. Inorg. Chem. 1997, 36, 2879–2883. doi:10.1021/ic9611195
Return to citation in text: [1] -
Redshaw, C.; Rowan, M. A.; Warford, L.; Homden, D. M.; Arbaoui, A.; Elsegood, M. R. J.; Dale, S. D.; Yamato, T.; Peréz Casas, C.; Matsui, S.; Matsuura, S. Chem.–Eur. J. 2007, 13, 1090–1107. doi:10.1002/chem.200600679
Return to citation in text: [1] [2] -
Notestein, J. M.; Andrini, L. R.; Kalchenko, V. I.; Requejo, F. G.; Katz, A.; Iglesia, E. J. Am. Chem. Soc. 2007, 129, 1122–1131. doi:10.1021/ja065830c
Return to citation in text: [1] [2] -
Hampton, P. D.; Daitch, C. E.; Duesler, E. N. Inorg. Chem. 1995, 34, 5641–5645. doi:10.1021/ic00126a038
Return to citation in text: [1] -
Daitch, C. E.; Hampton, P. D.; Duesler, E. N.; Alam, T. M. J. Am. Chem. Soc. 1996, 118, 7769–7773. doi:10.1021/ja9605984
Return to citation in text: [1] [2] -
Hampton, P. D.; Daitch, C. E.; Shachter, A. M. Inorg. Chem. 1997, 36, 2956–2959. doi:10.1021/ic961445k
Return to citation in text: [1] -
Thuéry, P.; Nierlich, M.; Masci, B.; Asfari, Z.; Vicens, J. J. Chem. Soc., Dalton Trans. 1999, 3151–3152.
Return to citation in text: [1] [2] -
Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 120–128. doi:10.1039/b108947c
Return to citation in text: [1] [2] -
Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 766–774. doi:10.1039/b200734g
Return to citation in text: [1] [2] -
Araki, K.; Inada, K.; Shinkai, S. Angew. Chem., Int. Ed. Engl. 1996, 35, 72–74. doi:10.1002/anie.199600721
Return to citation in text: [1] [2] -
Tsubaki, K.; Morimoto, T.; Otsubo, T.; Fuji, K. Org. Lett. 2002, 4, 2301–2304. doi:10.1021/ol026019j
Return to citation in text: [1] [2] -
Marcos, P. M.; Ascenso, J. R.; Cragg, P. J. Supramol. Chem. 2007, 19, 199–206. doi:10.1080/10610270601026594
Return to citation in text: [1] [2] [3] [4] -
Marcos, P. M. et al., manuscript in preparation.
Return to citation in text: [1] -
Cragg, P. J.; Miah, M.; Steed, J. W. Supramol. Chem. 2002, 14, 75–78. doi:10.1080/10610270290006592
Return to citation in text: [1] [2] -
Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Cragg, P. J.; Michel, S.; Hubscher-Bruder, V.; Arnaud-Neu, F. Supramol. Chem. 2011, 23, 93–101. doi:10.1080/10610278.2010.510562
Return to citation in text: [1] [2] -
Arnaud-Neu, F.; Cremin, S.; Harris, S.; McKervey, M. A.; Schwing-Weill, M.-J.; Schwinté, P.; Walker, A. J. Chem. Soc., Dalton Trans. 1997, 329–334. doi:10.1039/A602631A
Return to citation in text: [1] -
Atwood, J. L.; Koutsantonis, G. A.; Raston, C. L. Nature 1994, 368, 229–231. doi:10.1038/368229a0
Return to citation in text: [1] -
Suzuki, T.; Nakashima, K.; Shinkai, S. Chem. Lett. 1994, 23, 699–702. doi:10.1246/cl.1994.699
Return to citation in text: [1] -
Ikeda, A.; Yoshimura, M.; Shinkai, S. Tetrahedron Lett. 1997, 38, 2107–2110. doi:10.1016/S0040-4039(97)00318-3
Return to citation in text: [1] -
Ikeda, A.; Suzuki, Y.; Yoshimura, M.; Shinkai, S. Tetrahedron 1998, 54, 2497–2508. doi:10.1016/S0040-4020(98)00012-X
Return to citation in text: [1] -
Tsubaki, K.; Tanaka, K.; Kinoshita, T.; Fuji, K. Chem. Commun. 1998, 895–896. doi:10.1039/a800078f
Return to citation in text: [1] [2] -
Georghiou, P. E.; Tran, A. H.; Stroud, S. S.; Thompson, D. W. Tetrahedron 2006, 62, 2036–2044. doi:10.1016/j.tet.2005.09.151
Return to citation in text: [1] -
Tsubaki, K.; Murata, Y.; Komatsu, K.; Kinoshita, T.; Fuji, K. Heterocycles 1999, 51, 2553–2556. doi:10.3987/COM-99-8650
Return to citation in text: [1] [2] -
Atwood, J. L.; Barbour, L. J.; Nichols, P. J.; Raston, C. L.; Sandoval, C. A. Chem.–Eur. J. 1999, 5, 990–996. doi:10.1002/(SICI)1521-3765(19990301)5:3<990::AID-CHEM990>3.0.CO;2-4
Return to citation in text: [1] [2] -
Komatsu, N. Org. Biomol. Chem. 2003, 1, 204–209. doi:10.1039/b208107e
Return to citation in text: [1] -
Ikeda, A.; Hanato, T.; Kawaguchi, M.; Suenaga, H.; Shinkai, S. Chem. Commun. 1999, 1403–1404. doi:10.1039/a903872h
Return to citation in text: [1] [2] -
Ikeda, A.; Ejima, A.; Nishiguchi, K.; Kikuchi, J.-i.; Matsumoto, T.; Hatano, T.; Shinkai, S.; Goto, M. Chem. Lett. 2005, 34, 308–309. doi:10.1246/cl.2005.308
Return to citation in text: [1] -
Hatano, T.; Ikeda, A.; Akiyama, T.; Yamada, S.; Sano, M.; Kanekiyo, Y.; Shinkai, S. J. Chem. Soc., Perkin Trans. 2 2000, 909–912. doi:10.1039/b000022l
Return to citation in text: [1] -
Ikeda, A.; Hanato, T.; Shinkai, S.; Akiyama, T.; Yamada, S. J. Am. Chem. Soc. 2001, 123, 4855–4856. doi:10.1021/ja015596k
Return to citation in text: [1] -
Islam, S. D.-M.; Fujitsuka, M.; Ito, O.; Ikeda, A.; Hanato, T.; Shinkai, S. Chem. Lett. 2000, 29, 78–79. doi:10.1246/cl.2000.78
Return to citation in text: [1] -
Ikeda, A.; Sonoda, K.; Shinkai, S. Chem. Lett. 2000, 29, 1220–1221. doi:10.1246/cl.2000.1220
Return to citation in text: [1] -
Ikeda, A.; Nobukuni, S.; Udzu, H.; Zhong, Z.; Shinkai, S. Eur. J. Org. Chem. 2000, 3287–3293. doi:10.1002/1099-0690(200010)2000:19<3287::AID-EJOC3287>3.0.CO;2-R
Return to citation in text: [1] [2] -
Takeshita, M.; Shinkai, S. Chem. Lett. 1994, 23, 125–128. doi:10.1246/cl.1994.125
Return to citation in text: [1] -
Ni, X.-L.; Wang, S.; Zeng, X.; Tao, Z.; Yamato, T. Org. Lett. 2011, 13, 552–555. doi:10.1021/ol102914t
Return to citation in text: [1] [2] -
Ni, X.-L.; Zeng, X.; Redshaw, C.; Yamato, T. J. Org. Chem. 2011, 76, 5696–5702. doi:10.1021/jo2007303
Return to citation in text: [1] -
Wu, C.; Zhang, W.-J.; Zeng, X.; Mu, L.; Xue, S.-F.; Tao, Z.; Yamato, T. J. Inclusion Phenom. Macrocyclic Chem. 2010, 66, 125–131. doi:10.1007/s10847-009-9665-z
Return to citation in text: [1] [2] -
Kang, J.-M.; Cheong, N.-Y. Bull. Korean Chem. Soc. 2002, 23, 995–997. doi:10.5012/bkcs.2002.23.7.995
Return to citation in text: [1] [2] -
Odashima, K.; Yagi, K.; Tohda, K.; Umezawa, Y. Bioorg. Med. Chem. Lett. 1999, 9, 2375–2378. doi:10.1016/S0960-894X(99)00395-9
Return to citation in text: [1] -
Herzog, G.; McMahon, B.; Lefoix, M.; Mullins, N. D.; Collins, C. J.; Moynihan, H. A.; Arrigan, D. W. M. J. Electroanal. Chem. 2008, 622, 109–114. doi:10.1016/j.jelechem.2008.05.006
Return to citation in text: [1] -
Yaftian, M. R.; Parinejad, M.; Matt, D. J. Chin. Chem. Soc. 2007, 54, 1535–1542.
Return to citation in text: [1] -
Bocheńska, M.; Cragg, P. J.; Guziński, M.; Jasiński, A.; Kulesza, J.; Marcos, P. M.; Pomećko, R. Supramol. Chem. 2009, 21, 732–737. doi:10.1080/10610270902853043
Return to citation in text: [1] -
Cragg, P. J.; Allen, M. C.; Steed, J. W. Chem. Commun. 1999, 553–554. doi:10.1039/a808492k
Return to citation in text: [1] [2]
| 43. | Liu, S.-L.; Gong, S.-L.; Chen, Y.-Y. Chin. J. Chem. 2005, 23, 1651–1654. doi:10.1002/cjoc.200591651 |
| 14. | Dhawan, D.; Gutsche, C. D. J. Org. Chem. 1983, 48, 1536–1539. doi:10.1021/jo00157a033 |
| 15. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 42. | Casnati, A.; Pochini, A.; Ungaro, R.; Ugozzoli, F.; Arnaud, F.; Fanni, S.; Schwing, M.-J.; Egberink, R. J. M.; de Jong, F.; Reinhoudt, D. N. J. Am. Chem. Soc. 1995, 117, 2767–2777. doi:10.1021/ja00115a012 |
| 43. | Liu, S.-L.; Gong, S.-L.; Chen, Y.-Y. Chin. J. Chem. 2005, 23, 1651–1654. doi:10.1002/cjoc.200591651 |
| 44. | Liu, S.-L.; Gong, S.-L.; Zheng, Q.; Chen, Y.-Y.; Wu, X.-J. J. Chem. Res., Synop. 2005, 126–129. doi:10.3184/0308234054497218 |
| 95. | Yaftian, M. R.; Parinejad, M.; Matt, D. J. Chin. Chem. Soc. 2007, 54, 1535–1542. |
| 41. | Hampton, P. D.; Daitch, C. E.; Duesler, E. N. New J. Chem. 1996, 20, 427–432. |
| 94. | Herzog, G.; McMahon, B.; Lefoix, M.; Mullins, N. D.; Collins, C. J.; Moynihan, H. A.; Arrigan, D. W. M. J. Electroanal. Chem. 2008, 622, 109–114. doi:10.1016/j.jelechem.2008.05.006 |
| 92. | Kang, J.-M.; Cheong, N.-Y. Bull. Korean Chem. Soc. 2002, 23, 995–997. doi:10.5012/bkcs.2002.23.7.995 |
| 91. | Wu, C.; Zhang, W.-J.; Zeng, X.; Mu, L.; Xue, S.-F.; Tao, Z.; Yamato, T. J. Inclusion Phenom. Macrocyclic Chem. 2010, 66, 125–131. doi:10.1007/s10847-009-9665-z |
| 21. | Tsubaki, K.; Morimoto, T.; Otsubo, T.; Kinoshita, T.; Fuji, K. J. Org. Chem. 2001, 66, 4083–4086. doi:10.1021/jo0100502 |
| 93. | Odashima, K.; Yagi, K.; Tohda, K.; Umezawa, Y. Bioorg. Med. Chem. Lett. 1999, 9, 2375–2378. doi:10.1016/S0960-894X(99)00395-9 |
| 92. | Kang, J.-M.; Cheong, N.-Y. Bull. Korean Chem. Soc. 2002, 23, 995–997. doi:10.5012/bkcs.2002.23.7.995 |
| 45. | Tsubaki, K.; Otsubo, T.; Kinoshita, T.; Kawada, M.; Fuji, K. Chem. Pharm. Bull. 2001, 49, 507–509. doi:10.1248/cpb.49.507 |
| 90. | Ni, X.-L.; Zeng, X.; Redshaw, C.; Yamato, T. J. Org. Chem. 2011, 76, 5696–5702. doi:10.1021/jo2007303 |
| 19. | Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a |
| 89. | Ni, X.-L.; Wang, S.; Zeng, X.; Tao, Z.; Yamato, T. Org. Lett. 2011, 13, 552–555. doi:10.1021/ol102914t |
| 19. | Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a |
| 91. | Wu, C.; Zhang, W.-J.; Zeng, X.; Mu, L.; Xue, S.-F.; Tao, Z.; Yamato, T. J. Inclusion Phenom. Macrocyclic Chem. 2010, 66, 125–131. doi:10.1007/s10847-009-9665-z |
| 45. | Tsubaki, K.; Otsubo, T.; Kinoshita, T.; Kawada, M.; Fuji, K. Chem. Pharm. Bull. 2001, 49, 507–509. doi:10.1248/cpb.49.507 |
| 89. | Ni, X.-L.; Wang, S.; Zeng, X.; Tao, Z.; Yamato, T. Org. Lett. 2011, 13, 552–555. doi:10.1021/ol102914t |
| 96. | Bocheńska, M.; Cragg, P. J.; Guziński, M.; Jasiński, A.; Kulesza, J.; Marcos, P. M.; Pomećko, R. Supramol. Chem. 2009, 21, 732–737. doi:10.1080/10610270902853043 |
| 47. | Zhong, Z.; Ikeda, A.; Shinkai, S. J. Am. Chem. Soc. 1999, 121, 11906–11907. doi:10.1021/ja9925002 |
| 61. | Hampton, P. D.; Daitch, C. E.; Shachter, A. M. Inorg. Chem. 1997, 36, 2956–2959. doi:10.1021/ic961445k |
| 50. | Kawaguchi, M.; Ikeda, A.; Shinkai, S. Tetrahedron Lett. 2001, 42, 3725–3728. doi:10.1016/S0040-4039(01)00463-4 |
| 60. | Daitch, C. E.; Hampton, P. D.; Duesler, E. N.; Alam, T. M. J. Am. Chem. Soc. 1996, 118, 7769–7773. doi:10.1021/ja9605984 |
| 22. | Komatsu, N. Tetrahedron Lett. 2001, 42, 1733–1736. doi:10.1016/S0040-4039(00)02336-4 |
| 63. | Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 120–128. doi:10.1039/b108947c |
| 47. | Zhong, Z.; Ikeda, A.; Shinkai, S. J. Am. Chem. Soc. 1999, 121, 11906–11907. doi:10.1021/ja9925002 |
| 48. | Miah, M.; Pavey, K. D.; Gun’ko, V. M.; Sheehan, R.; Cragg, P. J. Supramol. Chem. 2004, 16, 185–192. doi:10.1080/10610270310001644473 |
| 49. | Hinrichs, M.; Hofbauer, F. R.; Klüfers, P.; Suhanji, M. Inorg. Chem. 2006, 45, 6688–6693. doi:10.1021/ic0603048 |
| 62. | Thuéry, P.; Nierlich, M.; Masci, B.; Asfari, Z.; Vicens, J. J. Chem. Soc., Dalton Trans. 1999, 3151–3152. |
| 46. | Tsubaki, K.; Otsubo, T.; Morimoto, T.; Maruoka, H.; Furukawa, M.; Momose, Y.; Shang, M.; Fuji, K. J. Org. Chem. 2002, 67, 8151–8156. doi:10.1021/jo026152p |
| 49. | Hinrichs, M.; Hofbauer, F. R.; Klüfers, P.; Suhanji, M. Inorg. Chem. 2006, 45, 6688–6693. doi:10.1021/ic0603048 |
| 46. | Tsubaki, K.; Otsubo, T.; Morimoto, T.; Maruoka, H.; Furukawa, M.; Momose, Y.; Shang, M.; Fuji, K. J. Org. Chem. 2002, 67, 8151–8156. doi:10.1021/jo026152p |
| 49. | Hinrichs, M.; Hofbauer, F. R.; Klüfers, P.; Suhanji, M. Inorg. Chem. 2006, 45, 6688–6693. doi:10.1021/ic0603048 |
| 60. | Daitch, C. E.; Hampton, P. D.; Duesler, E. N.; Alam, T. M. J. Am. Chem. Soc. 1996, 118, 7769–7773. doi:10.1021/ja9605984 |
| 59. | Hampton, P. D.; Daitch, C. E.; Duesler, E. N. Inorg. Chem. 1995, 34, 5641–5645. doi:10.1021/ic00126a038 |
| 58. | Notestein, J. M.; Andrini, L. R.; Kalchenko, V. I.; Requejo, F. G.; Katz, A.; Iglesia, E. J. Am. Chem. Soc. 2007, 129, 1122–1131. doi:10.1021/ja065830c |
| 51. | Araki, K.; Hayashida, H. Tetrahedron Lett. 2000, 41, 1807–1810. doi:10.1016/S0040-4039(00)00034-4 |
| 97. | Cragg, P. J.; Allen, M. C.; Steed, J. W. Chem. Commun. 1999, 553–554. doi:10.1039/a808492k |
| 52. | Ikeda, A.; Yoshimura, M.; Tani, F.; Naruta, Y.; Shinkai, S. Chem. Lett. 1998, 27, 587–588. doi:10.1246/cl.1998.587 |
| 53. | Ikeda, A.; Udzu, H.; Zhong, Z.; Shinkai, S.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 2001, 123, 3872–3877. doi:10.1021/ja003269r |
| 97. | Cragg, P. J.; Allen, M. C.; Steed, J. W. Chem. Commun. 1999, 553–554. doi:10.1039/a808492k |
| 47. | Zhong, Z.; Ikeda, A.; Shinkai, S. J. Am. Chem. Soc. 1999, 121, 11906–11907. doi:10.1021/ja9925002 |
| 51. | Araki, K.; Hayashida, H. Tetrahedron Lett. 2000, 41, 1807–1810. doi:10.1016/S0040-4039(00)00034-4 |
| 62. | Thuéry, P.; Nierlich, M.; Masci, B.; Asfari, Z.; Vicens, J. J. Chem. Soc., Dalton Trans. 1999, 3151–3152. |
| 63. | Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 120–128. doi:10.1039/b108947c |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 65. | Araki, K.; Inada, K.; Shinkai, S. Angew. Chem., Int. Ed. Engl. 1996, 35, 72–74. doi:10.1002/anie.199600721 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 31. | Takeshita, M.; Inokuchi, F.; Shinkai, S. Tetrahedron Lett. 1995, 36, 3341–3344. doi:10.1016/0040-4039(95)00536-L |
| 32. | Khrifi, S.; Guelzim, A.; Baert, F.; Mussrabi, M.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1995, 51, 153–157. doi:10.1107/S0108270194008036 |
| 65. | Araki, K.; Inada, K.; Shinkai, S. Angew. Chem., Int. Ed. Engl. 1996, 35, 72–74. doi:10.1002/anie.199600721 |
| 1. | Gutsche, C. D. Calixarenes: An Introduction; Monographs in Supramolecular Chemistry, 2nd ed.; Royal Society of Chemistry: Cambridge, U.K., 2008. |
| 2. | Sliwa, W.; Kozlowski, C. Calixarenes and Resorcinarenes: Synthesis, Properties and Applications; Wiley VCH: Weinheim, Germany, 2009. |
| 3. | Böhmer, V. Angew. Chem., Int. Ed. Engl. 1995, 34, 713–745. doi:10.1002/anie.199507131 |
| 29. | Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021 |
| 24. | Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7 |
| 37. | Yamato, T.; Rahman, S.; Zeng, X.; Kitajima, F.; Gil, J. T. Can. J. Chem. 2006, 84, 58–64. doi:10.1139/v05-260 |
| 34. | Yamato, T.; Zhang, F. L. J. Inclusion Phenom. Macrocyclic Chem. 2001, 39, 55–64. doi:10.1023/A:1008196612553 |
| 9. | Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2004, 40, 607–643. doi:10.1023/B:RUJO.0000043707.97314.52 |
| 10. | Shokova, E. A.; Kovalev, V. V. Russ. J. Org. Chem. 2004, 40, 1547–1578. doi:10.1007/s11178-005-0062-9 |
| 35. | Takimoto, M.; Ni, X.-L.; Rahman, S.; Xi, Z.; Yamato, T. J. Inclusion Phenom. Macrocyclic Chem. 2011, 70, 69–80. doi:10.1007/s10847-010-9863-8 |
| 4. | Brodesser, G.; Vögtle, F. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 19, 111–135. doi:10.1007/BF00708978 |
| 6. | Cragg, P. J. Homocalixarenes. In Encyclopedia of Supramolecular Chemistry; Atwood, J. L.; Steel, J. W., Eds.; Marcel Dekker: New York, 2004; pp 649–657. |
| 7. | Späth, A.; König, B. Beilstein J. Org. Chem. 2010, 6, No. 32. doi:10.3762/bjoc.6.32 |
| 8. | Yamato, D. J. J. Inclusion Phenom. Mol. Recognit. Chem. 1998, 32, 195–207. doi:10.1023/A:1008011410507 |
| 5. | Gutsche, C. D.; Bauer, L. J. J. Am. Chem. Soc. 1985, 107, 6052–6059. doi:10.1021/ja00307a038 |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 64. | Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 766–774. doi:10.1039/b200734g |
| 4. | Brodesser, G.; Vögtle, F. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 19, 111–135. doi:10.1007/BF00708978 |
| 34. | Yamato, T.; Zhang, F. L. J. Inclusion Phenom. Macrocyclic Chem. 2001, 39, 55–64. doi:10.1023/A:1008196612553 |
| 64. | Masci, B.; Nierlich, M.; Thuéry, P. New J. Chem. 2002, 26, 766–774. doi:10.1039/b200734g |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 12. | Masci, B.; Finelli, M.; Varrone, M. Chem.–Eur. J. 1998, 4, 2018–2030. doi:10.1002/(SICI)1521-3765(19981002)4:10<2018::AID-CHEM2018>3.0.CO;2-I |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 37. | Yamato, T.; Rahman, S.; Zeng, X.; Kitajima, F.; Gil, J. T. Can. J. Chem. 2006, 84, 58–64. doi:10.1139/v05-260 |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 36. | Ni, X.-L.; Rahman, S.; Zeng, X.; Hughes, D. L.; Redshaw, C.; Yamato, T. Org. Biomol. Chem. 2011, 9, 6535–6541. doi:10.1039/c1ob05564j |
| 37. | Yamato, T.; Rahman, S.; Zeng, X.; Kitajima, F.; Gil, J. T. Can. J. Chem. 2006, 84, 58–64. doi:10.1139/v05-260 |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 67. | Marcos, P. M.; Ascenso, J. R.; Cragg, P. J. Supramol. Chem. 2007, 19, 199–206. doi:10.1080/10610270601026594 |
| 34. | Yamato, T.; Zhang, F. L. J. Inclusion Phenom. Macrocyclic Chem. 2001, 39, 55–64. doi:10.1023/A:1008196612553 |
| 29. | Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021 |
| 34. | Yamato, T.; Zhang, F. L. J. Inclusion Phenom. Macrocyclic Chem. 2001, 39, 55–64. doi:10.1023/A:1008196612553 |
| 29. | Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021 |
| 66. | Tsubaki, K.; Morimoto, T.; Otsubo, T.; Fuji, K. Org. Lett. 2002, 4, 2301–2304. doi:10.1021/ol026019j |
| 40. | Dieleman, C. B.; Matt, D.; Neda, I.; Schmutzler, R.; Harriman, A.; Yaftian, R. Chem. Commun. 1999, 1911–1912. doi:10.1039/a905677g |
| 24. | Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7 |
| 41. | Hampton, P. D.; Daitch, C. E.; Duesler, E. N. New J. Chem. 1996, 20, 427–432. |
| 66. | Tsubaki, K.; Morimoto, T.; Otsubo, T.; Fuji, K. Org. Lett. 2002, 4, 2301–2304. doi:10.1021/ol026019j |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 40. | Dieleman, C. B.; Matt, D.; Neda, I.; Schmutzler, R.; Harriman, A.; Yaftian, R. Chem. Commun. 1999, 1911–1912. doi:10.1039/a905677g |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 19. | Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a |
| 23. | Mizyed, S.; Ashram, M.; Miller, D. O.; Georghiou, P. E. J. Chem. Soc., Perkin Trans. 2 2001, 1916–1919. doi:10.1039/B105019M |
| 67. | Marcos, P. M.; Ascenso, J. R.; Cragg, P. J. Supramol. Chem. 2007, 19, 199–206. doi:10.1080/10610270601026594 |
| 20. | Ashram, M.; Mizyed, S.; Georghiou, P. E. J. Org. Chem. 2001, 66, 1473–1479. doi:10.1021/jo001518o |
| 69. | Cragg, P. J.; Miah, M.; Steed, J. W. Supramol. Chem. 2002, 14, 75–78. doi:10.1080/10610270290006592 |
| 40. | Dieleman, C. B.; Matt, D.; Neda, I.; Schmutzler, R.; Harriman, A.; Yaftian, R. Chem. Commun. 1999, 1911–1912. doi:10.1039/a905677g |
| 51. | Araki, K.; Hayashida, H. Tetrahedron Lett. 2000, 41, 1807–1810. doi:10.1016/S0040-4039(00)00034-4 |
| 69. | Cragg, P. J.; Miah, M.; Steed, J. W. Supramol. Chem. 2002, 14, 75–78. doi:10.1080/10610270290006592 |
| 40. | Dieleman, C. B.; Matt, D.; Neda, I.; Schmutzler, R.; Harriman, A.; Yaftian, R. Chem. Commun. 1999, 1911–1912. doi:10.1039/a905677g |
| 27. | Yamato, T.; Haraguchi, H.; Nishikawa, J.-I.; Ide, S.; Tsuzuki, H. Can. J. Chem. 1998, 76, 989–996. doi:10.1139/v98-102 |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 67. | Marcos, P. M.; Ascenso, J. R.; Cragg, P. J. Supramol. Chem. 2007, 19, 199–206. doi:10.1080/10610270601026594 |
| 24. | Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7 |
| 33. | Yamato, T.; Zhang, F.; Tsuzuki, H.; Miura, Y. Eur. J. Org. Chem. 2001, 1069–1075. doi:10.1002/1099-0690(200103)2001:6<1069::AID-EJOC1069>3.0.CO;2-R |
| 24. | Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7 |
| 27. | Yamato, T.; Haraguchi, H.; Nishikawa, J.-I.; Ide, S.; Tsuzuki, H. Can. J. Chem. 1998, 76, 989–996. doi:10.1139/v98-102 |
| 68. | Marcos, P. M. et al., manuscript in preparation. |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 25. | Matsumoto, H.; Nishio, S.; Takeshita, M.; Shinkai, S. Tetrahedron 1995, 51, 4647–4654. doi:10.1016/0040-4020(95)00165-5 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 24. | Araki, K.; Inada, K.; Otsuka, H.; Shinkai, S. Tetrahedron 1993, 49, 9465–9478. doi:10.1016/S0040-4020(01)80216-7 |
| 5. | Gutsche, C. D.; Bauer, L. J. J. Am. Chem. Soc. 1985, 107, 6052–6059. doi:10.1021/ja00307a038 |
| 27. | Yamato, T.; Haraguchi, H.; Nishikawa, J.-I.; Ide, S.; Tsuzuki, H. Can. J. Chem. 1998, 76, 989–996. doi:10.1139/v98-102 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 27. | Yamato, T.; Haraguchi, H.; Nishikawa, J.-I.; Ide, S.; Tsuzuki, H. Can. J. Chem. 1998, 76, 989–996. doi:10.1139/v98-102 |
| 28. | Yamato, T.; Zhang, F.; Sato, T.; Ide, S. J. Chem. Res., Synop. 2000, 10–12. doi:10.3184/030823400103165707 |
| 74. | Ikeda, A.; Yoshimura, M.; Shinkai, S. Tetrahedron Lett. 1997, 38, 2107–2110. doi:10.1016/S0040-4039(97)00318-3 |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 70. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Cragg, P. J.; Michel, S.; Hubscher-Bruder, V.; Arnaud-Neu, F. Supramol. Chem. 2011, 23, 93–101. doi:10.1080/10610278.2010.510562 |
| 72. | Atwood, J. L.; Koutsantonis, G. A.; Raston, C. L. Nature 1994, 368, 229–231. doi:10.1038/368229a0 |
| 73. | Suzuki, T.; Nakashima, K.; Shinkai, S. Chem. Lett. 1994, 23, 699–702. doi:10.1246/cl.1994.699 |
| 71. | Arnaud-Neu, F.; Cremin, S.; Harris, S.; McKervey, M. A.; Schwing-Weill, M.-J.; Schwinté, P.; Walker, A. J. Chem. Soc., Dalton Trans. 1997, 329–334. doi:10.1039/A602631A |
| 31. | Takeshita, M.; Inokuchi, F.; Shinkai, S. Tetrahedron Lett. 1995, 36, 3341–3344. doi:10.1016/0040-4039(95)00536-L |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 32. | Khrifi, S.; Guelzim, A.; Baert, F.; Mussrabi, M.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1995, 51, 153–157. doi:10.1107/S0108270194008036 |
| 67. | Marcos, P. M.; Ascenso, J. R.; Cragg, P. J. Supramol. Chem. 2007, 19, 199–206. doi:10.1080/10610270601026594 |
| 29. | Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021 |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 31. | Takeshita, M.; Inokuchi, F.; Shinkai, S. Tetrahedron Lett. 1995, 36, 3341–3344. doi:10.1016/0040-4039(95)00536-L |
| 70. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Cragg, P. J.; Michel, S.; Hubscher-Bruder, V.; Arnaud-Neu, F. Supramol. Chem. 2011, 23, 93–101. doi:10.1080/10610278.2010.510562 |
| 29. | Araki, K.; Hashimoto, N.; Otsuka, H.; Shinkai, S. J. Org. Chem. 1993, 58, 5958–5963. doi:10.1021/jo00074a021 |
| 30. | Ashram, M.; Mizyed, S.; Georghiou, P. E. Org. Biomol. Chem. 2003, 1, 599–603. doi:10.1039/b209046p |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 39. | Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2009, 65, 496–503. doi:10.1016/j.tet.2008.11.005 |
| 26. | Cragg, P. J.; Drew, M. G. B.; Steed, J. W. Supramol. Chem. 1999, 11, 5–15. doi:10.1080/10610279908048711 |
| 55. | Hampton, P. D.; Daitch, C. E.; Alam, T. M.; Bencze, Z.; Rosay, M. Inorg. Chem. 1994, 33, 4750–4758. doi:10.1021/ic00099a028 |
| 56. | Hampton, P. D.; Daitch, C. E.; Alam, T. M.; Pruss, E. A. Inorg. Chem. 1997, 36, 2879–2883. doi:10.1021/ic9611195 |
| 15. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 48. | Miah, M.; Pavey, K. D.; Gun’ko, V. M.; Sheehan, R.; Cragg, P. J. Supramol. Chem. 2004, 16, 185–192. doi:10.1080/10610270310001644473 |
| 52. | Ikeda, A.; Yoshimura, M.; Tani, F.; Naruta, Y.; Shinkai, S. Chem. Lett. 1998, 27, 587–588. doi:10.1246/cl.1998.587 |
| 53. | Ikeda, A.; Udzu, H.; Zhong, Z.; Shinkai, S.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 2001, 123, 3872–3877. doi:10.1021/ja003269r |
| 23. | Mizyed, S.; Ashram, M.; Miller, D. O.; Georghiou, P. E. J. Chem. Soc., Perkin Trans. 2 2001, 1916–1919. doi:10.1039/B105019M |
| 79. | Atwood, J. L.; Barbour, L. J.; Nichols, P. J.; Raston, C. L.; Sandoval, C. A. Chem.–Eur. J. 1999, 5, 990–996. doi:10.1002/(SICI)1521-3765(19990301)5:3<990::AID-CHEM990>3.0.CO;2-4 |
| 53. | Ikeda, A.; Udzu, H.; Zhong, Z.; Shinkai, S.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 2001, 123, 3872–3877. doi:10.1021/ja003269r |
| 23. | Mizyed, S.; Ashram, M.; Miller, D. O.; Georghiou, P. E. J. Chem. Soc., Perkin Trans. 2 2001, 1916–1919. doi:10.1039/B105019M |
| 79. | Atwood, J. L.; Barbour, L. J.; Nichols, P. J.; Raston, C. L.; Sandoval, C. A. Chem.–Eur. J. 1999, 5, 990–996. doi:10.1002/(SICI)1521-3765(19990301)5:3<990::AID-CHEM990>3.0.CO;2-4 |
| 17. | Suzuki, K.; Minami, H.; Yamagata, Y.; Fuji, S.; Tomita, K.-I.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1992, 48, 350–352. doi:10.1107/S0108270191010399 |
| 78. | Tsubaki, K.; Murata, Y.; Komatsu, K.; Kinoshita, T.; Fuji, K. Heterocycles 1999, 51, 2553–2556. doi:10.3987/COM-99-8650 |
| 17. | Suzuki, K.; Minami, H.; Yamagata, Y.; Fuji, S.; Tomita, K.-I.; Asfari, Z.; Vicens, J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1992, 48, 350–352. doi:10.1107/S0108270191010399 |
| 77. | Georghiou, P. E.; Tran, A. H.; Stroud, S. S.; Thompson, D. W. Tetrahedron 2006, 62, 2036–2044. doi:10.1016/j.tet.2005.09.151 |
| 16. | Zerr, P.; Mussrabi, M.; Vicens, J. Tetrahedron Lett. 1991, 32, 1879–1880. doi:10.1016/S0040-4039(00)85986-9 |
| 78. | Tsubaki, K.; Murata, Y.; Komatsu, K.; Kinoshita, T.; Fuji, K. Heterocycles 1999, 51, 2553–2556. doi:10.3987/COM-99-8650 |
| 16. | Zerr, P.; Mussrabi, M.; Vicens, J. Tetrahedron Lett. 1991, 32, 1879–1880. doi:10.1016/S0040-4039(00)85986-9 |
| 76. | Tsubaki, K.; Tanaka, K.; Kinoshita, T.; Fuji, K. Chem. Commun. 1998, 895–896. doi:10.1039/a800078f |
| 14. | Dhawan, D.; Gutsche, C. D. J. Org. Chem. 1983, 48, 1536–1539. doi:10.1021/jo00157a033 |
| 58. | Notestein, J. M.; Andrini, L. R.; Kalchenko, V. I.; Requejo, F. G.; Katz, A.; Iglesia, E. J. Am. Chem. Soc. 2007, 129, 1122–1131. doi:10.1021/ja065830c |
| 15. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 57. | Redshaw, C.; Rowan, M. A.; Warford, L.; Homden, D. M.; Arbaoui, A.; Elsegood, M. R. J.; Dale, S. D.; Yamato, T.; Peréz Casas, C.; Matsui, S.; Matsuura, S. Chem.–Eur. J. 2007, 13, 1090–1107. doi:10.1002/chem.200600679 |
| 76. | Tsubaki, K.; Tanaka, K.; Kinoshita, T.; Fuji, K. Chem. Commun. 1998, 895–896. doi:10.1039/a800078f |
| 14. | Dhawan, D.; Gutsche, C. D. J. Org. Chem. 1983, 48, 1536–1539. doi:10.1021/jo00157a033 |
| 57. | Redshaw, C.; Rowan, M. A.; Warford, L.; Homden, D. M.; Arbaoui, A.; Elsegood, M. R. J.; Dale, S. D.; Yamato, T.; Peréz Casas, C.; Matsui, S.; Matsuura, S. Chem.–Eur. J. 2007, 13, 1090–1107. doi:10.1002/chem.200600679 |
| 75. | Ikeda, A.; Suzuki, Y.; Yoshimura, M.; Shinkai, S. Tetrahedron 1998, 54, 2497–2508. doi:10.1016/S0040-4020(98)00012-X |
| 18. | Miah, M.; Romanov, N. N.; Cragg, P. J. J. Org. Chem. 2002, 67, 3124–3126. doi:10.1021/jo025597a |
| 15. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 15. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 87. | Ikeda, A.; Nobukuni, S.; Udzu, H.; Zhong, Z.; Shinkai, S. Eur. J. Org. Chem. 2000, 3287–3293. doi:10.1002/1099-0690(200010)2000:19<3287::AID-EJOC3287>3.0.CO;2-R |
| 87. | Ikeda, A.; Nobukuni, S.; Udzu, H.; Zhong, Z.; Shinkai, S. Eur. J. Org. Chem. 2000, 3287–3293. doi:10.1002/1099-0690(200010)2000:19<3287::AID-EJOC3287>3.0.CO;2-R |
| 88. | Takeshita, M.; Shinkai, S. Chem. Lett. 1994, 23, 125–128. doi:10.1246/cl.1994.125 |
| 22. | Komatsu, N. Tetrahedron Lett. 2001, 42, 1733–1736. doi:10.1016/S0040-4039(00)02336-4 |
| 85. | Islam, S. D.-M.; Fujitsuka, M.; Ito, O.; Ikeda, A.; Hanato, T.; Shinkai, S. Chem. Lett. 2000, 29, 78–79. doi:10.1246/cl.2000.78 |
| 20. | Ashram, M.; Mizyed, S.; Georghiou, P. E. J. Org. Chem. 2001, 66, 1473–1479. doi:10.1021/jo001518o |
| 83. | Hatano, T.; Ikeda, A.; Akiyama, T.; Yamada, S.; Sano, M.; Kanekiyo, Y.; Shinkai, S. J. Chem. Soc., Perkin Trans. 2 2000, 909–912. doi:10.1039/b000022l |
| 84. | Ikeda, A.; Hanato, T.; Shinkai, S.; Akiyama, T.; Yamada, S. J. Am. Chem. Soc. 2001, 123, 4855–4856. doi:10.1021/ja015596k |
| 21. | Tsubaki, K.; Morimoto, T.; Otsubo, T.; Kinoshita, T.; Fuji, K. J. Org. Chem. 2001, 66, 4083–4086. doi:10.1021/jo0100502 |
| 86. | Ikeda, A.; Sonoda, K.; Shinkai, S. Chem. Lett. 2000, 29, 1220–1221. doi:10.1246/cl.2000.1220 |
| 22. | Komatsu, N. Tetrahedron Lett. 2001, 42, 1733–1736. doi:10.1016/S0040-4039(00)02336-4 |
| 53. | Ikeda, A.; Udzu, H.; Zhong, Z.; Shinkai, S.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 2001, 123, 3872–3877. doi:10.1021/ja003269r |
| 19. | Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a |
| 81. | Ikeda, A.; Hanato, T.; Kawaguchi, M.; Suenaga, H.; Shinkai, S. Chem. Commun. 1999, 1403–1404. doi:10.1039/a903872h |
| 21. | Tsubaki, K.; Morimoto, T.; Otsubo, T.; Kinoshita, T.; Fuji, K. J. Org. Chem. 2001, 66, 4083–4086. doi:10.1021/jo0100502 |
| 19. | Tsubaki, K.; Otsubo, T.; Tanaka, K.; Fuji, K.; Kinoshita, T. J. Org. Chem. 1998, 63, 3260–3265. doi:10.1021/jo971945a |
| 81. | Ikeda, A.; Hanato, T.; Kawaguchi, M.; Suenaga, H.; Shinkai, S. Chem. Commun. 1999, 1403–1404. doi:10.1039/a903872h |
| 20. | Ashram, M.; Mizyed, S.; Georghiou, P. E. J. Org. Chem. 2001, 66, 1473–1479. doi:10.1021/jo001518o |
| 82. | Ikeda, A.; Ejima, A.; Nishiguchi, K.; Kikuchi, J.-i.; Matsumoto, T.; Hatano, T.; Shinkai, S.; Goto, M. Chem. Lett. 2005, 34, 308–309. doi:10.1246/cl.2005.308 |
© 2012 Cottet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)