Guest Editor: T. K. Lindhorst Beilstein J. Org. Chem.2012,8, 2067–2071.https://doi.org/10.3762/bjoc.8.232 Received 01 Sep 2012,
Accepted 01 Nov 2012,
Published 26 Nov 2012
Safety-catch linkers are useful for solid-phase oligosaccharide synthesis as they are orthogonal to many common protective groups. A new acylsulfonamide safety-catch linker was designed, synthesized and employed during glycosylations using an automated carbohydrate synthesizer. The analysis of the cleavage products revealed shortcomings for oligosaccharide synthesis.
Solid-phase oligosaccharide synthesis [1,2] has been automated [3-5] to rapidly assemble complex oligosaccharides. Key to the success of solid-phase syntheses is the linker that connects the first carbohydrate building block to the solid support [6]. This linker has to remain stable throughout oligosaccharide synthesis but must be cleaved at the end of the reaction sequence to release the oligosaccharide and reveal a functional group for ready conjugation to array surfaces and carrier proteins. Since the first successful automated system for solid-phase oligosaccharide synthesis was introduced [7], alternative linker strategies have been explored [8-12]. Recently, a bifunctional amino–ester linker 1[13,14] (Figure 1) has been developed, which can be readily cleaved from the resin by basic methanolysis. The released chromophore-containing part, such as aromatic benzyl ether protecting groups, facilitates the purification of the synthetic oligosaccharides by HPLC. Final deprotection of the product provides a terminal amine at the reducing end of the oligosaccharide for the fabrication of carbohydrate microarrays.
Figure 1:
Structures of two novel linkers on different resins.
Figure 1:
Structures of two novel linkers on different resins.
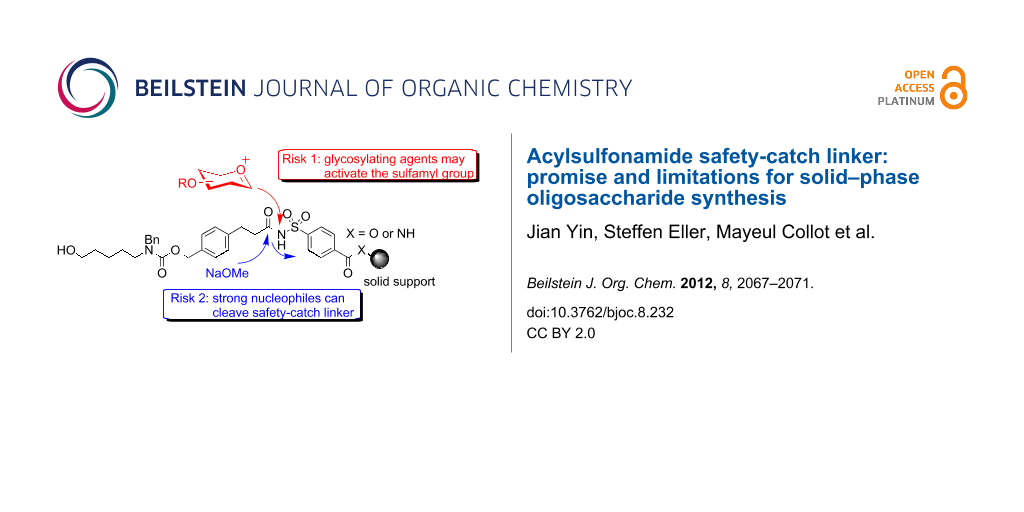
An acylsulfonamide safety-catch linker 2 (Figure 1), was developed in combination with TentaGel resin to provide orthogonality to temporary ester protecting groups. This linker was employed in the successful synthesis of a sialyl LewisX tetrasaccharide [15] and a sialyl Tn antigen [16]. In the search for a linker suitable for the solid-phase synthesis of complex glycosaminoglycans (GAGs) [17,18], we designed a new acylsulfonamide safety-catch linker that combined the advantageous features of linker 1, the amino–ester bifunctional linker, and linker 2, the safety-catch linker, to create a connection to the solid support that remains stable under conditions for cleaving temporary ester protective groups. Furthermore, the safety-catch linker should enable a variety of different reaction conditions on solid support since cleavage only occurs following preactivation. Thus, different modification reactions, such as Staudinger reduction, ester saponification or sulfation, necessary for the synthesis of GAGs, can be performed on solid support in an automated carbohydrate synthesizer. In the process of evaluating the performance of this linker in solid-phase glycosylation reactions, the potential as well as some severe limitations became apparent.
Linker 10 is the newly designed acylsulfonamide safety-catch linker (Scheme 1). The safety-catch linkage to the resin permits methanolysis of temporary ester protecting groups. This linker can only be cleaved after activation with TMSCH2N2 or ICH2CN, afterwards, the subsequent aromatic protection of the masked amine enables UV detection during HPLC purification of the resultant oligosaccharide [13]. Finally, the terminal amine group revealed during final deprotection serves in the formation of glycoconjugates and glycan microarrays [19].
Scheme 1:
Synthesis of a new acylsulfonamide safety-catch linker. Reagents and conditions: (a) benzaldehyde, Na2SO4, DCM; (b) NaBH4, EtOH; 82% over two steps; (c) H2, 10% Pd/C, EtOH, DIPEA; (d) cat. H2SO4, MeOH, 88% over two steps; (e) DSC, Et3N, DCM/CH3CN, 85%; (f) DHP, p-TsOH·H2O, DCM, 86%; (g) LiOH·H2O, THF/H2O, reflux, 95%; (h) 4-sulfamoylbenzoic acid, DCC, DMAP, DCM/DMF, 60%.
Scheme 1:
Synthesis of a new acylsulfonamide safety-catch linker. Reagents and conditions: (a) benzaldehyde, ...
Synthesis of 10 relied on key intermediates 4 and 6 (Scheme 1). Monobenzylated amine 4 was prepared by reductive amination [20]. An established three-step synthesis starting with hydrocinnamic acid generated ester 6 with an overall yield of only 28% [21,22]. In contrast, when commercially available 4-formylcinnamic acid (5) served as starting material, ester 6 was prepared in just two steps and with an increased overall yield of 88% [23]. Reaction of alcohol 6 and disuccinimidyl carbonate (DSC) in the presence of NEt3 afforded a crude carbonate, which was smoothly reacted with amine 4 to provide carbamate 7 with 85% yield. Carbamate 7 was converted to the free acid 9 by simple protection to form THP ether 8 followed by saponification. Finally, coupling acid 9 with 4-sulfamoylbenzoic acid [15] afforded linker 10.
To support oligosaccharide synthesis, the safety-catch linker was first coupled to different resins (Scheme 2). In addition, since the activation and cleavage of safety-catch linkers is typically quite slow, a second, base-labile (Zemplén [24]) cleavage site was integrated to facilitate the fast release and analysis of glycosides. Linker-functionalized resin 11 contains both cleavage sites, and was accessed by coupling the cesium salt of linker 10 with Merrifield chloride resin prior to capping and deprotection (Scheme 2). A second resin 12, containing only the safety-catch cleavage site, was created by installing linker 10 on Merrifield amine resin by treatment with DIC and HOBt (Scheme 2) [15]. After capping with acetic anhydride and removing the THP protecting group with p-TsOH·H2O, the functionalized resin 12 was ready for use. Finally, a third linker-functionalized resin 16 was created with TentaGel resin [15,16], which swells in water, because encouraging results have been achieved previously with this resin. 4-(Chloromethyl)benzoic acid (13) served as an additional spacer [25] between 10 and TentaGel amino resin to incorporate the Zemplén cleavage site. Treatment of acid 13 with oxalyl chloride yielded acyl chloride 14, which, in turn, was coupled to linker 10 under the conditions established for the construction of resins 11 and 12, and afforded modified TentaGel resin 16 with both cleavage sites (Scheme 2).
Functionalized resins 11 and 16, both containing acylsulfonamide safety-catch linker 10 plus the Zemplén cleavage site, were employed in glycosylation reactions on a solid phase using an automated synthesizer [13], subsequently cleaved with NaOMe, and the products were analyzed by HPLC (Scheme 3, see Supporting Information File 1). Glycosylations were performed with either glucosamine thioglycoside 17 and glucosamine trichloroacetimidate 18 or perbenzylated thioglycoside 19, which are both important building blocks for the synthesis of heparin and heparan sulfate. Three repetitions of a glycosylation using each three equivalents trichloroacetimidate 18 activated by TMSOTf were conducted, and followed by Zemplén cleavage. Similarly, three equivalents of thioglycosides 17 and 19 were added three times (triple coupling) for each glycosylation employing DMTST or NIS/TfOH as an activator. In both instances, surprisingly, only N-glycoside 20 (minor product) and glycosylated linker 21 (major product) rather than the desired product 22 were found (Scheme 3). N-Glycosidic sulfonamides were previously used during the synthesis of inhibitors of hepatocellular carcinoma cells [26]. This observation illustrates a limitation of the linker system since these undesired reactions result in the preactivation of the safety-catch linker, which can lead to cleavage in presence of nucleophiles. Similar results were observed when the experiments were repeated. The desired product 22 was detected in trace amounts only in the case of the coupling of thioglycoside 17 activated by NIS/TfOH to Merrifield resin 11, as determined by HPLC (see Supporting Information File 1).
Scheme 3:
Glycosylation and cleavage reactions for analysis. Reagents and conditions: (a) automated glycosylations using 17, 18 or 19; (b) NaOMe, MeOH.
Scheme 3:
Glycosylation and cleavage reactions for analysis. Reagents and conditions: (a) automated glycosyla...
Based on these unexpected findings, we hypothesized that excess glycosylating agent may alkylate the sulfamyl group during glycosylation, such that subsequent cleavage provides products 20 and 21. To test this, the amount of glycosylating agent was decreased during a reaction, which resulted in the production of glycosylated linkers 21 and 22. It was evident that two unexpected reactions had occurred. First, the use of excess glycosylating agent, common practice for solid-phase-synthesis protocols, leads to activation of the sulfamyl group of the acylsulfonamide safety-catch linker, permitting cleavage at this location. This reaction had not been reported previously [15,16], because the architecture of this safety-catch linker means that the modified sulfamyl group remains on the resin after cleavage and release of the product, and this was not examined. As the new bifunctional resins 11 and 16 contain the additional Zemplén cleavage site, the product of this dominant but undesired reaction was evident. Second, the observation that reducing the concentration of the glycosylation agent caused an increase in the production of glycosylated linker 21, implied that sodium methoxide may directly cleave safety-catch linkers without prior activation.
To examine linker reactivity in more detail, additional automated glycosylations were performed by using thioglycoside 19 activated with NIS/TfOH, on Merrifield resin 12 containing only the safety-catch cleavage site (Scheme 4). The presence of glycosylated linker 23 as the main product confirmed that premature linker cleavage had occurred. Even nonglycosylated resin 12 was cleaved by treatment with sodium methoxide to afford linker 7 (Scheme 4). A possible explanation is provided by Unverzagt and co-workers [27], who reported, when using another acylsulfonamide safety-catch linker on a solid support, that capping the remaining amino group using acetic anhydride preactivates the sulfamyl group. To test this hypothesis, linker 10 was transformed by benzylation and deprotection to afford 24 prior to treatment with sodium methoxide (Scheme 4). Surprisingly, linker 7 was again isolated as the major product as determined by TLC and NMR, indicating that the nucleophilicity or basicity of NaOMe was responsible for cleavage. Other reagents, such as aqueous NaOH, hydrazine acetate, pyridine and benzylamine failed to cleave the linker. Even the strongly basic and weakly nucleophilic t-BuOK was not sufficient to induce premature cleavage. Thus, it is the nucleophilic action of sodium methoxide that is strong enough to induce cleavage of the safety-catch linker (Scheme 4).
Scheme 4:
Further investigations of safety-catch linker. Reagents and conditions: (a) NaOMe, MeOH; (b) 19, NIS, TfOH, DCM; (c) DCC, BnOH, DCM; (d) p-TsOH·H2O, MeOH; 55% over two steps.
Scheme 4:
Further investigations of safety-catch linker. Reagents and conditions: (a) NaOMe, MeOH; (b) 19, NI...
In conclusion, we described a new acylsulfonamide safety-catch linker with an additional Zemplén cleavage site, designed for automated solid-phase synthesis of glycosaminoglycans. With this novel linker, inherent but previously unknown limitations of the safety-catch concept for solid-phase oligosaccharide synthesis were discovered. The sulfamyl group can be attacked by excess glycosylating agent to give the unexpected resin-bound N-glycoside, which may block additional reaction sequences and extension of the oligosaccharide [28]. In addition, sodium methoxide can directly cleave the sulfamyl group, prohibiting its use in conjunction with safety-catch linkers in general. While safety-catch linkers offer many attractive features for the solid-phase synthesis of complex molecules, particularly peptides, they should be used with great caution for oligosaccharide assembly.
Supporting Information
Supporting Information File 1:
Experimental section.
We thank the Max Planck Society for very generous support, and the European Research Council (ERC Advanced Grant AUTOHEPARIN to PHS) is also gratefully acknowledged. We thank Dr. V. Mountain for critically editing the manuscript.
References
Seeberger, P. H.; Haase, W.-C. Chem. Rev.2000,100, 4349. doi:10.1021/cr9903104
Return to citation in text:
[1]
Seeberger, P. H., Ed. Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries; John Wiley & Sons Inc.: New York, 2001. doi:10.1002/0471220442
Return to citation in text:
[1]
Seeberger, P. H. Chem. Commun.2003, 1115. doi:10.1039/b210230g
Return to citation in text:
[1]
Seeberger, P. H.; Werz, D. B. Nature2007,446, 1046. doi:10.1038/nature05819
Return to citation in text:
[1]
Seeberger, P. H. Chem. Soc. Rev.2008,37, 19. doi:10.1039/b511197h
Return to citation in text:
[1]
Guillier, F.; Orain, D.; Bradley, M. Chem. Rev.2000,100, 2091. doi:10.1021/cr980040+
Return to citation in text:
[1]
Plante, O. J.; Palmacci, E. R.; Seeberger, P. H. Science2001,291, 1523. doi:10.1126/science.1057324
Return to citation in text:
[1]
Love, K. R.; Seeberger, P. H. Angew. Chem., Int. Ed.2004,43, 602. doi:10.1002/anie.200352539
Return to citation in text:
[1]
Werz, D. B.; Castagner, B.; Seeberger, P. H. J. Am. Chem. Soc.2007,129, 2770. doi:10.1021/ja069218x
Return to citation in text:
[1]
Codée, J. D. C.; Kröck, L.; Castagner, B.; Seeberger, P. H. Chem.–Eur. J.2008,14, 3987. doi:10.1002/chem.200701864
Return to citation in text:
[1]
Bindschädler, P. Different aspects of automeated heparin synthesis: de novo synthesis of building blocks, a new linker, and synthesis of heparin oligosaccharides. Ph.D. Thesis, Eidgenössische Technische Hochschule Zürich, Switzerland, 2008.
Return to citation in text:
[1]
Czechura, P.; Guedes, N.; Kopitzki, S.; Vazquez, N.; Martin-Lomas, M.; Reichardt, N.-C. Chem. Commun.2011,47, 2390. doi:10.1039/c0cc04686h
Return to citation in text:
[1]
Kröck, L.; Esposito, D.; Castagner, B.; Wang, C.-C.; Bindschädler, P.; Seeberger, P. H. Chem. Sci.2012,3, 1617. doi:10.1039/c2sc00940d
Return to citation in text:
[1]
[2]
[3]
Christ, W.; Kröck, L.; Plante, O. J.; Castagner, B.; Seeberger, P. H. Automated Oligosaccharide Synthesizer. WO Patent 2010011828, Jan 28, 2010.
Return to citation in text:
[1]
Kanemitsu, T.; Wong, C.-H.; Kanie, O. J. Am. Chem. Soc.2002,124, 3591. doi:10.1021/ja0167396
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Kanemitsu, T.; Daikoku, S.; Kanie, O. J. Carbohydr. Chem.2006,25, 361. doi:10.1080/07328300600778785
Return to citation in text:
[1]
[2]
[3]
Yeung, B. K. S.; Chong, P. Y. C.; Petillo, P. A. J. Carbohydr. Chem.2002,21, 799. doi:10.1081/CAR-120016490
Return to citation in text:
[1]
Karst, N. A.; Linhardt, R. J. Curr. Med. Chem.2003,10, 1993. doi:10.2174/0929867033456891
Return to citation in text:
[1]
Yin, J.; Seeberger, P. H. Methods Enzymol.2010,478, 197. doi:10.1016/S0076-6879(10)78009-5
and references therein.
Return to citation in text:
[1]
De Michelis, C.; Rocheblave, L.; Priem, G.; Chermann, J. C.; Kraus, J. L. Bioorg. Med. Chem.2000,8, 1253. doi:10.1016/S0968-0896(00)00055-9
Return to citation in text:
[1]
Arshady, R.; Atherton, E.; Clive, D. L. J.; Sheppard, R. C. J. Chem. Soc., Perkin Trans. 11981, 529. doi:10.1039/P19810000529
Return to citation in text:
[1]
Párkányi, C.; Yeh Huang, L. S.-C.; Chu, S. G.; Jeffries, A. T., III. Collect. Czech. Chem. Commun.1996,61, 342. doi:10.1135/cccc19960342
Return to citation in text:
[1]
Bolli, M.; Lehmann, D.; Mathys, B.; Mueller, C.; Nayler, O.; Velker, J.; Weller, T. Novel thiophene derivatives. PCT Int. Appl. WO 2006/100635 A2, Sept 28, 2006.
Return to citation in text:
[1]
Zemplén, G.; Kunz, A. Ber. Dtsch. Chem. Ges.1923,56, 1705. doi:10.1002/cber.19230560736
Return to citation in text:
[1]
Wu, X.; Schmidt, R. R. Eur. J. Org. Chem.2004, 2826. doi:10.1002/ejoc.200400111
Return to citation in text:
[1]
Mezzato, S.; Schaffrath, M.; Unverzagt, C. Angew. Chem., Int. Ed.2005,44, 1650. doi:10.1002/anie.200461125
Return to citation in text:
[1]
de Paz, J. L.; Kayser, M. M.; Macchione, G.; Nieto, P. M. Carbohydr. Res.2010,345, 565. doi:10.1016/j.carres.2009.12.021
Return to citation in text:
[1]
Seeberger, P. H., Ed. Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries; John Wiley & Sons Inc.: New York, 2001. doi:10.1002/0471220442
Werz, D. B.; Castagner, B.; Seeberger, P. H. J. Am. Chem. Soc.2007,129, 2770. doi:10.1021/ja069218x
10.
Codée, J. D. C.; Kröck, L.; Castagner, B.; Seeberger, P. H. Chem.–Eur. J.2008,14, 3987. doi:10.1002/chem.200701864
11.
Bindschädler, P. Different aspects of automeated heparin synthesis: de novo synthesis of building blocks, a new linker, and synthesis of heparin oligosaccharides. Ph.D. Thesis, Eidgenössische Technische Hochschule Zürich, Switzerland, 2008.


![[1860-5397-8-232-1]](/bjoc/content/figures/1860-5397-8-232-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-232-i1]](/bjoc/content/inline/1860-5397-8-232-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-232-i2]](/bjoc/content/inline/1860-5397-8-232-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-232-i3]](/bjoc/content/inline/1860-5397-8-232-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-232-i4]](/bjoc/content/inline/1860-5397-8-232-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)