Abstract
Multicomponent reactions involving polyfunctional 4-amino-5-carboxamido-1,2,3-triazole and cyclic carbonyl-containing CH-acids were studied under conventional thermal heating, microwave and ultrasonic irradiation. The features of the reactions studied were discussed and the optimized procedures for the synthesis of final triazolopyrimidines were elaborated. In contrast to the similar MCRs of numerous other aminoazoles, a change of direction of the heterocyclizations in the case of 4-amino-5-carboxamido-1,2,3-triazole was not observed when microwave or thermal heating was substituted by ultrasonication at ambient temperature.

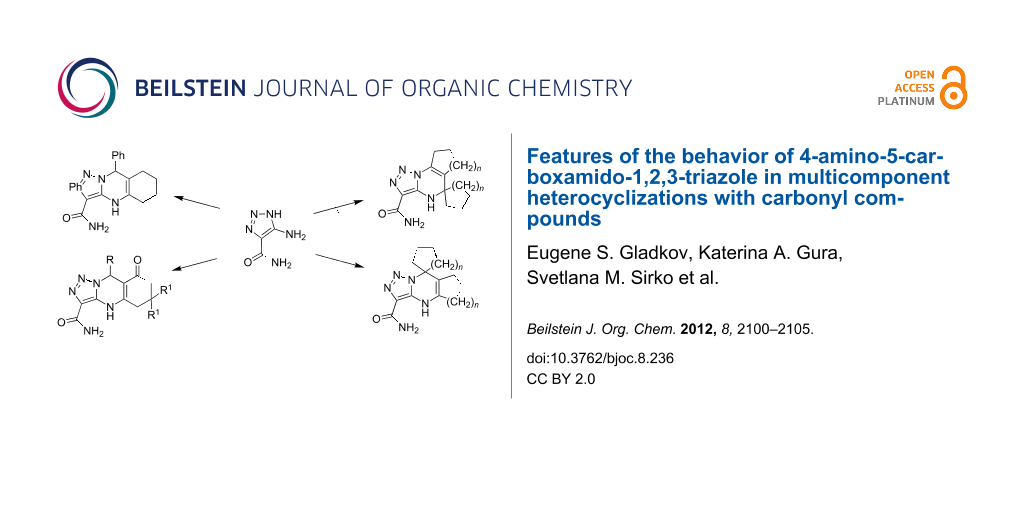
Graphical Abstract
Introduction
Multicomponent reactions (MCRs) [1-4] involving polyfunctional aminazoles as a key reagent are challenging objectives in the modern chemistry of heterocyclic compounds dealing with the synthesis of partially hydrogenated azoloazines [5-8]. The possibility of realization of several independent sequences of two-component stages of MCRs and the presence of alternative nucleophilic reaction centers in an aminazole molecule often allows the synthesis of different final heterocyclic systems from the same building blocks. However, one of the main problems preventing efficient application of polyfunctional starting materials in MCRs is the existence of simultaneous competing reactions leading to complicated mixtures of several compounds. Thereby, the development of a strategy for the control of the MCR direction and the elaboration of synthetic methods allowing tuning of their selectivity are the current objectives that have attracted our attention and efforts over the past few years [5-8].
In some of our recent publications it was demonstrated that the direction of MCRs involving aminazoles, carbonyls and active methylene compounds can be effectively controlled by variation of such reaction parameters as temperature, solvent type, catalytic system, method of activation, etc. [9-17]. Changing these parameters gives us the opportunity to synthesize different types of heterocyclic systems, with a high level of selectivity, from the same starting materials. For example, MCRs or 5-aminopyrazoles containing several nonequivalent nucleophilic reaction centers with aromatic aldehydes and derivatives of 1,3-cyclohexanedione [10,11] or pyruvic acids [12-14] can be selectively directed to the formation of one of the compounds I–III or IV–VI (Figure 1). Such an approach is in good correspondence with the “ideal synthesis” concept [18,19] and allows significant increasing of the molecular diversity of target azoloazine systems.
![[1860-5397-8-236-1]](/bjoc/content/figures/1860-5397-8-236-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some MCRs of aminoazoles with controlled switching of the direction.
Figure 1: Some MCRs of aminoazoles with controlled switching of the direction.
Among the polyfunctional aminoazoles that have been studied in heterocyclization reactions [5-8,20], derivatives of 4-amino-1,2,3-triazole have been less well investigated. There are only a few examples of their heterocyclizations with β-diketones [21], N-cyanomethaneimidates [22], isocyanates [23], chalcones [24] and derivatives of carboxylic acids [25,26]. The only MCR involving this type of aminoazole was described in our previous publication [27].
In the case of 4-amino-5-carboxamido-1,2,3-triazole, heterocyclizations can proceed in two main ways: “classical” for α-aminoazole direction with participation of the NH2-group and endocyclic NH (Figure 2, compound VII or its position isomer VIII), or an alternative pathway involving the NH2-group and carboxamide fragment [25,26] (compound IX).
![[1860-5397-8-236-2]](/bjoc/content/figures/1860-5397-8-236-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Some possible products of MCRs involving 4-amino-5-carboxamido-1,2,3-triazole and carbonyl compounds.
Figure 2: Some possible products of MCRs involving 4-amino-5-carboxamido-1,2,3-triazole and carbonyl compound...
Thus, there are several alternative directions for reactions between 4-amino-5-carboxamido-1,2,3-triazole, carbonyls and active methylene compounds. In the present article we disclose our recent results of a study of such MCRs and analyze the influence of some of the reaction parameters on their development and outcome.
Results and Discussion
It was found that reaction between one equivalent of aminotriazole 1 and two equivalents of cycloalkanones 2a,b (n = 1–2) in boiling dry ethanol for 3 h (Method A) proceeded as ABB’-type [28] MCR with chemodifferentiation of cyclic ketone and yielded spiroheterocycles 4a,b (31–37%, Scheme 1, Table 1) as the sole reaction products. However, better yields and purity of compounds 4a,b were observed when the starting materials 1 and 2a,b were treated in methanol under microwave (MW) irradiation at 120 °C for 30 min (Method B). At the same time, the reaction involving cycloheptanone (n = 3) did not allow the synthesis of compound 4c, which was not found in the reaction mixture even in trace amounts by TLC, while aminotriazole 1 was isolated unchanged from the reaction mixture.
![[1860-5397-8-236-i1]](/bjoc/content/inline/1860-5397-8-236-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Multicomponent and sequential reactions of 4-amino-5-carboxamido-1,2,3-triazole with cyclic ketones.
Scheme 1: Multicomponent and sequential reactions of 4-amino-5-carboxamido-1,2,3-triazole with cyclic ketones....
Table 1: Multicomponent synthesis of 4,5,6,7,8,9-hexahydro[1,2,3]triazolo[5,1-b]quinazoline-3-carboxamides 4a–c, 7 and 9a–j.
| Entry | Aldehyde | Ketone | Product | Methoda | Yield, % | ||
|---|---|---|---|---|---|---|---|
| No | R | No | R1 | ||||
| 1 | – | – | 5a | – | 4a | A (D) | 31 (67) |
| 2 | – | – | 5b | – | 4b | A (B,C,D) | 37 (45,27,81) |
| 3 | – | – | 5c | – | 4c | D | 45 |
| 4 | 6a | C6H5 | 2b | – | 7 | A (B,C,D) | 21 (41,60,71) |
| 5 | 6a | C6H5 | 8a | H | 9a | A (B,C) | 35 (80,61) |
| 6 | 6b | 4-CH3OC6H4 | 8a | H | 9b | B (C) | 90 (43) |
| 7 | 6c | 2-CH3OC6H4 | 8a | H | 9c | B (C) | 55 (51) |
| 8 | 6d | 4-BrC6H4 | 8a | H | 9d | B (C) | 70 (75) |
| 9 | 6e | 4-ClC6H4 | 8a | H | 9e | B (C) | 61 (69) |
| 10 | 6a | C6H5 | 8b | CH3 | 9f | A (B,C) | 37 (52,78) |
| 11 | 6b | 4-CH3OC6H4 | 8b | CH3 | 9g | B (C) | 66 (86) |
| 12 | 6c | 2-CH3OC6H4 | 8b | CH3 | 9h | B (C) | 51 (88) |
| 13 | 6d | 4-BrC6H4 | 8b | CH3 | 9i | B (C) | 90 (91) |
| 14 | 6e | 4-ClC6H4 | 8b | CH3 | 9j | B (C) | 85 (77) |
aMethod A: three-component reaction, EtOH, Δ; Method B: three-component reaction, MeOH, MW; Method C: three-component reaction, HOAc, ultrasonification (US); Method D: sequential reaction, MeOH, MW.
Positional-isomeric compounds 3, whose formation had been expected according to the data published earlier for the similar reactions of 3-amino-1,2,4-triazole [29], was not isolated or even chromatographically detected in either of the procedures applied.
It is worth noting that in our previous works [9-17] we also showed a possibility to change the direction of similar MCRs by replacement of conventional thermal or MW heating with room temperature ultrasonication. However, treatment of the starting compounds 1 and 2a,b under ultrasonication at ambient temperature in acetic acid, which was selected instead of methanol due to solubility problems, also yielded only heterocycles 4a,b (Method C). The yields and purity of the compounds obtained under ultrasonication were slightly lower than in the case of Methods A and B.
Spirocompound 4c was obtained by linear reaction via preliminary synthesis and isolation of cyclic α,β-unsaturated ketone 5c and its further treatment with 4-amino-5-carboxamido-1,2,3-triazole (1) in acetic acid under ultrasonication at ambient temperature, in boiling dry ethanol or under MW heating in methanol. The same procedure also allowed us to obtain compounds 5a,b. The best results in this case were observed when the microwave-assisted procedure (Method D) was applied.
It should be noted that for similar treatments involving 3-amino-1,2,4-triazole [7,29,30] the structures of the final products were different depending on whether a multicomponent or sequential procedure was used, while in our case both procedures gave the same compounds.
ABC-type MCR of aminoazole 1, cyclohexaneone 2b and benzaldehyde 6a was carried out in dry ethanol by conventional heating under reflux (Method A), in methanol under microwave irradiation at 120 °C (Method B), or by ultrasonication at ambient temperature in acetic acid (Method C). In all these cases triazolopyrimidine 7 was isolated as the sole reaction product (Scheme 2, Table 1). In most cases the best results from the viewpoint of yields and purity of the target compounds were observed when ultrasound-assisted method C was applied.
![[1860-5397-8-236-i2]](/bjoc/content/inline/1860-5397-8-236-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Multicomponent and sequential reactions of 4-amino-5-carboxamido-1,2,3-triazole with aldehydes and cyclic ketones.
Scheme 2: Multicomponent and sequential reactions of 4-amino-5-carboxamido-1,2,3-triazole with aldehydes and ...
Sequential reaction via synthesis of arylidenecyclohexanone 10 also gave as sole reaction product compound 7 under all the methods of activation applied. However, microwave heating (Method D) was the procedure of choice in this case as well.
Multicomponent heterocyclizations of aminotriazole 1 with aromatic aldehydes 6a–e and cyclohexanediones 8a,b carried out according to Method A, B or C yielded tricyclic compounds 9a–j in 51–91% (Scheme 2, Table 1). The most efficient synthetic procedure was the treatment of the starting materials in methanol under ultrasonication at ambient temperature. Very similar results were observed for MW-assisted syntheses.
The structures of the compounds synthesized were established by MS and NMR spectroscopy in combination with elemental analyses. However, MS data obtained for the reaction product isolated from the MCR between aminoazole 1 and cycloalkanone 2 may correspond to both structure 3 and 4. Moreover, 1H and 13C NMR spectra of these compounds should contain very similar signals, and we thus used the position of the signal of pyrimidine NH in the 1H NMR spectra, which strongly depends on type of the structure: for angular compounds such as 3 its signal appears at 5–6 ppm while for heterocycles 4 it should be at 7–8 ppm [8,29,31,32], as was observed in our case. Additionally, the HMBC spectrum of the heterocycles obtained does not contain cross-peaks between pyrimidine NH and spiro-carbon, which is also in good correlation with structure 4. Analogous elucidations allowed the structures of compounds 7 and 9 to be established.
Conclusion
In summary, two types of heterocyclization reactions involving 4-amino-5-carboxamido-1,2,3-triazole and cyclic ketones were studied under conventional thermal heating, and microwave and ultrasonic irradiation. ABB’-type MCR proceeding with chemodifferentiation of cyclopentanone or cyclohexanone molecules yields 4,5,6,7-tetrahydrospiro{cyclopenta[d][1,2,3]triazolo[1,5-a]pyrimidine-8,1'-cyclopentane}-3-carboxamide (4a) or 5,6,7,8-tetrahydro-4H-spiro{[1,2,3]triazolo[5,1-b]quinazoline-9,1'-cyclohexane}-3-carboxamide (4b) under all the conditions tested. The best results were observed in the case of microwave-assisted synthesis in methanol at 120 °C. Spirocompound 4c may be obtained by a sequential procedure via the preliminary synthesis of 2-cycloheptylidenecycloheptanone but not by a three-component reaction involving cycloheptanone. ABC-type MCR between 4-amino-5-carboxamido-1,2,3-triazole, aldehydes and cyclohexanone or 1,3-cyclohexandiones leads to the formation of 4,5,6,7,8,9-hexahydro[1,2,3]triazolo[5,1-b]quinazoline-3-carboxamides 7 or 9 under all the reactions conditions studied; however, the ultrasound-assisted procedure is the method of choice for this heterocyclization.
For all the MCRs studied, changes in the directions of the heterocyclizations were not observed when microwave or thermal heating was substituted by ultrasonication at ambient temperature or when the three-component reaction was replaced by a sequential protocol via the preliminary synthesis of cyclic α,β-unsaturated ketones.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data for all new compounds. | ||
| Format: PDF | Size: 127.0 KB | Download |
References
-
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Biggs-Houck, J. E.; Younai, A.; Shaw, J. T. Curr. Opin. Chem. Biol. 2010, 14, 371. doi:10.1016/j.cbpa.2010.03.003
Return to citation in text: [1] -
Ganem, B. Acc. Chem. Res. 2009, 42, 463. doi:10.1021/ar800214s
Return to citation in text: [1] -
Isambert, N.; Lavilla, R. Chem.–Eur. J. 2008, 14, 8444. doi:10.1002/chem.200800473
Return to citation in text: [1] -
Chebanov, V. A.; Gura, K. A.; Desenko, S. M. Top. Heterocycl. Chem. 2010, 23, 41. doi:10.1007/7081_2009_21
Return to citation in text: [1] [2] [3] -
Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer: Meppel, 2008.
Return to citation in text: [1] [2] [3] -
Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2012, 48, 566. doi:10.1007/s10593-012-1030-2
Return to citation in text: [1] [2] [3] [4] -
Sedash, Y. V.; Gorobets, N. Yu.; Chebanov, V. A.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. RSC Adv. 2012, 2, 6719. doi:10.1039/c2ra20195j
Return to citation in text: [1] [2] [3] [4] -
Chebanov, V. A.; Sakhno, Ya. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229. doi:10.1016/j.tet.2006.11.048
Return to citation in text: [1] [2] -
Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l
Return to citation in text: [1] [2] [3] -
Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c
Return to citation in text: [1] [2] [3] -
Sakhno, Ya. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041. doi:10.1016/j.tet.2008.09.089
Return to citation in text: [1] [2] [3] -
Muravyova, E. A.; Shishkina, S. V.; Musatov, V. I.; Knyazeva, I. V.; Shishkin, O. V.; Desenko, S. M.; Chebanov, V. A. Synthesis 2009, 1375. doi:10.1055/s-0028-1088024
Return to citation in text: [1] [2] [3] -
Sakhno, Ya. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523. doi:10.1007/s11030-010-9226-9
Return to citation in text: [1] [2] [3] -
Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Chebanov, V. A. Synthesis 2011, 1120. doi:10.1055/s-0030-1258468
Return to citation in text: [1] [2] -
Muravyova, E. A.; Desenko, S. M.; Rudenko, R. V.; Shishkina, S. V.; Shishkin, O. V.; Sen’ko, Y. V.; Vashchenko, E. V.; Chebanov, V. A. Tetrahedron 2011, 67, 9389. doi:10.1016/j.tet.2011.09.138
Return to citation in text: [1] [2] -
Chebanov, V. A.; Saraev, V. E.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Desenko, S. M. Eur. J. Org. Chem. 2012, 5515. doi:10.1002/ejoc.201200669
Return to citation in text: [1] [2] -
Dömling, A.; Ugi, I. Angew. Chem. Int. Ed. 2000, 39, 3168. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] -
Wender, P. A.; Handy, S. T.; Wright, D. L. Chem. Ind. (London) 1997, 765.
Return to citation in text: [1] -
Chebanov, V. A.; Desenko, S. M. Curr. Org. Chem. 2006, 10, 297. doi:10.2174/138527206775473904
Return to citation in text: [1] -
Okabe, T.; Bhooshan, B.; Novinson, T.; Hillyard, I. W.; Garner, G. E.; Robins, R. K. J. Heterocycl. Chem. 1983, 20, 735. doi:10.1002/jhet.5570200345
Return to citation in text: [1] -
Oakes, F. T.; Leonard, N. J. J. Org. Chem. 1985, 50, 4986. doi:10.1021/jo00224a074
Return to citation in text: [1] -
Ege, G.; Gilbert, K.; Maurer, K. Chem. Ber. 1987, 120, 1375. doi:10.1002/cber.19871200816
Return to citation in text: [1] -
Desenko, S. M.; Gladkov, E. S.; Shishkina, S. V.; Shishkin, O. V.; Komykhov, S. A.; Orlov, V. D.; Meier, H. J. Heterocycl. Chem. 2006, 43, 1563. doi:10.1002/jhet.5570430620
Return to citation in text: [1] -
Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Lucacchini, A. Farmaco 1989, 44, 843.
Return to citation in text: [1] [2] -
Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Tonetti, I.; Lucacchini, A. Farmaco 1990, 45, 979.
Return to citation in text: [1] [2] -
Gladkov, E. S.; Sirko, S. N.; Shishkina, S. V.; Shishkin, O. V.; Knyazeva, I. V.; Desenko, S. M.; Chebanov, V. A. Monatsh. Chem. 2010, 141, 773. doi:10.1007/s00706-010-0326-0
Return to citation in text: [1] -
Tejedor, D.; García-Tellado, F. Chem. Soc. Rev. 2007, 36, 484. doi:10.1039/b608164a
Return to citation in text: [1] -
Desenko, S. M.; Orlov, V. D.; Éstrada, K. Chem. Hetetocycl. Compd. 1990, 26, 839. doi:10.1007/BF00509729
Return to citation in text: [1] [2] [3] -
Chernyshev, V. M.; Khoroshkin, D. A.; Sokolov, A. N.; Taranushich, V. A.; Gladkov, E. S.; Shishkina, S. V.; Shishkin, O. V.; Desenko, S. M. J. Heterocycl. Chem. 2008, 45, 1419. doi:10.1002/jhet.5570450527
Return to citation in text: [1] -
Lipson, V. V.; Desenko, S. M.; Shirobokova, M. G.; Borodina, V. V. Chem. Heterocycl. Compd. 2003, 39, 1213. doi:10.1023/B:COHC.0000008269.69460.ac
Return to citation in text: [1] -
Desenko, S. M.; Orlov, V. D.; Getmanskii, N. V.; Shishkin, O. V.; Lindeman, S. V.; Struchkov, Yu. T. Chem. Heterocycl. Compd. 1993, 29, 406. doi:10.1007/BF00529878
Return to citation in text: [1]
| 7. | Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2012, 48, 566. doi:10.1007/s10593-012-1030-2 |
| 29. | Desenko, S. M.; Orlov, V. D.; Éstrada, K. Chem. Hetetocycl. Compd. 1990, 26, 839. doi:10.1007/BF00509729 |
| 30. | Chernyshev, V. M.; Khoroshkin, D. A.; Sokolov, A. N.; Taranushich, V. A.; Gladkov, E. S.; Shishkina, S. V.; Shishkin, O. V.; Desenko, S. M. J. Heterocycl. Chem. 2008, 45, 1419. doi:10.1002/jhet.5570450527 |
| 29. | Desenko, S. M.; Orlov, V. D.; Éstrada, K. Chem. Hetetocycl. Compd. 1990, 26, 839. doi:10.1007/BF00509729 |
| 9. | Chebanov, V. A.; Sakhno, Ya. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229. doi:10.1016/j.tet.2006.11.048 |
| 10. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l |
| 11. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 12. | Sakhno, Ya. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041. doi:10.1016/j.tet.2008.09.089 |
| 13. | Muravyova, E. A.; Shishkina, S. V.; Musatov, V. I.; Knyazeva, I. V.; Shishkin, O. V.; Desenko, S. M.; Chebanov, V. A. Synthesis 2009, 1375. doi:10.1055/s-0028-1088024 |
| 14. | Sakhno, Ya. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523. doi:10.1007/s11030-010-9226-9 |
| 15. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Chebanov, V. A. Synthesis 2011, 1120. doi:10.1055/s-0030-1258468 |
| 16. | Muravyova, E. A.; Desenko, S. M.; Rudenko, R. V.; Shishkina, S. V.; Shishkin, O. V.; Sen’ko, Y. V.; Vashchenko, E. V.; Chebanov, V. A. Tetrahedron 2011, 67, 9389. doi:10.1016/j.tet.2011.09.138 |
| 17. | Chebanov, V. A.; Saraev, V. E.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Desenko, S. M. Eur. J. Org. Chem. 2012, 5515. doi:10.1002/ejoc.201200669 |
| 1. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. |
| 2. | Biggs-Houck, J. E.; Younai, A.; Shaw, J. T. Curr. Opin. Chem. Biol. 2010, 14, 371. doi:10.1016/j.cbpa.2010.03.003 |
| 3. | Ganem, B. Acc. Chem. Res. 2009, 42, 463. doi:10.1021/ar800214s |
| 4. | Isambert, N.; Lavilla, R. Chem.–Eur. J. 2008, 14, 8444. doi:10.1002/chem.200800473 |
| 10. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l |
| 11. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 25. | Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Lucacchini, A. Farmaco 1989, 44, 843. |
| 26. | Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Tonetti, I.; Lucacchini, A. Farmaco 1990, 45, 979. |
| 9. | Chebanov, V. A.; Sakhno, Ya. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229. doi:10.1016/j.tet.2006.11.048 |
| 10. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l |
| 11. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 12. | Sakhno, Ya. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041. doi:10.1016/j.tet.2008.09.089 |
| 13. | Muravyova, E. A.; Shishkina, S. V.; Musatov, V. I.; Knyazeva, I. V.; Shishkin, O. V.; Desenko, S. M.; Chebanov, V. A. Synthesis 2009, 1375. doi:10.1055/s-0028-1088024 |
| 14. | Sakhno, Ya. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523. doi:10.1007/s11030-010-9226-9 |
| 15. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Chebanov, V. A. Synthesis 2011, 1120. doi:10.1055/s-0030-1258468 |
| 16. | Muravyova, E. A.; Desenko, S. M.; Rudenko, R. V.; Shishkina, S. V.; Shishkin, O. V.; Sen’ko, Y. V.; Vashchenko, E. V.; Chebanov, V. A. Tetrahedron 2011, 67, 9389. doi:10.1016/j.tet.2011.09.138 |
| 17. | Chebanov, V. A.; Saraev, V. E.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Desenko, S. M. Eur. J. Org. Chem. 2012, 5515. doi:10.1002/ejoc.201200669 |
| 28. | Tejedor, D.; García-Tellado, F. Chem. Soc. Rev. 2007, 36, 484. doi:10.1039/b608164a |
| 5. | Chebanov, V. A.; Gura, K. A.; Desenko, S. M. Top. Heterocycl. Chem. 2010, 23, 41. doi:10.1007/7081_2009_21 |
| 6. | Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer: Meppel, 2008. |
| 7. | Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2012, 48, 566. doi:10.1007/s10593-012-1030-2 |
| 8. | Sedash, Y. V.; Gorobets, N. Yu.; Chebanov, V. A.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. RSC Adv. 2012, 2, 6719. doi:10.1039/c2ra20195j |
| 25. | Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Lucacchini, A. Farmaco 1989, 44, 843. |
| 26. | Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Tonetti, I.; Lucacchini, A. Farmaco 1990, 45, 979. |
| 5. | Chebanov, V. A.; Gura, K. A.; Desenko, S. M. Top. Heterocycl. Chem. 2010, 23, 41. doi:10.1007/7081_2009_21 |
| 6. | Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer: Meppel, 2008. |
| 7. | Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2012, 48, 566. doi:10.1007/s10593-012-1030-2 |
| 8. | Sedash, Y. V.; Gorobets, N. Yu.; Chebanov, V. A.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. RSC Adv. 2012, 2, 6719. doi:10.1039/c2ra20195j |
| 27. | Gladkov, E. S.; Sirko, S. N.; Shishkina, S. V.; Shishkin, O. V.; Knyazeva, I. V.; Desenko, S. M.; Chebanov, V. A. Monatsh. Chem. 2010, 141, 773. doi:10.1007/s00706-010-0326-0 |
| 21. | Okabe, T.; Bhooshan, B.; Novinson, T.; Hillyard, I. W.; Garner, G. E.; Robins, R. K. J. Heterocycl. Chem. 1983, 20, 735. doi:10.1002/jhet.5570200345 |
| 23. | Ege, G.; Gilbert, K.; Maurer, K. Chem. Ber. 1987, 120, 1375. doi:10.1002/cber.19871200816 |
| 5. | Chebanov, V. A.; Gura, K. A.; Desenko, S. M. Top. Heterocycl. Chem. 2010, 23, 41. doi:10.1007/7081_2009_21 |
| 6. | Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer: Meppel, 2008. |
| 7. | Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2012, 48, 566. doi:10.1007/s10593-012-1030-2 |
| 8. | Sedash, Y. V.; Gorobets, N. Yu.; Chebanov, V. A.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. RSC Adv. 2012, 2, 6719. doi:10.1039/c2ra20195j |
| 20. | Chebanov, V. A.; Desenko, S. M. Curr. Org. Chem. 2006, 10, 297. doi:10.2174/138527206775473904 |
| 24. | Desenko, S. M.; Gladkov, E. S.; Shishkina, S. V.; Shishkin, O. V.; Komykhov, S. A.; Orlov, V. D.; Meier, H. J. Heterocycl. Chem. 2006, 43, 1563. doi:10.1002/jhet.5570430620 |
| 18. | Dömling, A.; Ugi, I. Angew. Chem. Int. Ed. 2000, 39, 3168. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 19. | Wender, P. A.; Handy, S. T.; Wright, D. L. Chem. Ind. (London) 1997, 765. |
| 8. | Sedash, Y. V.; Gorobets, N. Yu.; Chebanov, V. A.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. RSC Adv. 2012, 2, 6719. doi:10.1039/c2ra20195j |
| 29. | Desenko, S. M.; Orlov, V. D.; Éstrada, K. Chem. Hetetocycl. Compd. 1990, 26, 839. doi:10.1007/BF00509729 |
| 31. | Lipson, V. V.; Desenko, S. M.; Shirobokova, M. G.; Borodina, V. V. Chem. Heterocycl. Compd. 2003, 39, 1213. doi:10.1023/B:COHC.0000008269.69460.ac |
| 32. | Desenko, S. M.; Orlov, V. D.; Getmanskii, N. V.; Shishkin, O. V.; Lindeman, S. V.; Struchkov, Yu. T. Chem. Heterocycl. Compd. 1993, 29, 406. doi:10.1007/BF00529878 |
| 12. | Sakhno, Ya. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041. doi:10.1016/j.tet.2008.09.089 |
| 13. | Muravyova, E. A.; Shishkina, S. V.; Musatov, V. I.; Knyazeva, I. V.; Shishkin, O. V.; Desenko, S. M.; Chebanov, V. A. Synthesis 2009, 1375. doi:10.1055/s-0028-1088024 |
| 14. | Sakhno, Ya. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523. doi:10.1007/s11030-010-9226-9 |
| 22. | Oakes, F. T.; Leonard, N. J. J. Org. Chem. 1985, 50, 4986. doi:10.1021/jo00224a074 |
© 2012 Gladkov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)