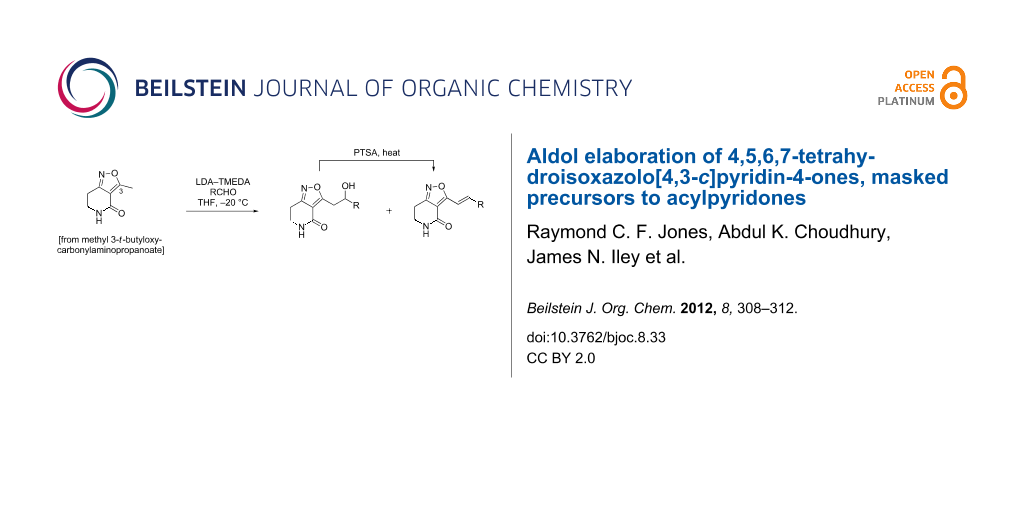
Abstract
A core 4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridine-4-one scaffold is elaborated at C-3(Me) by base-mediated aldol condensation to give new 3-alkenyl-4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridine-4-ones, which are masked forms related to the acylpyridone natural products.

Graphical Abstract
Introduction
The 3-acyl-4-hydroxypyridin-2-one moiety 1 (Figure 1) is the common structural unit of a family of natural products with a range of interesting biological activities [1]. Examples are the pigments tenellin (2a) and bassianin (2b) from insect pathogenic fungus Beauveria bassiana [2,3], pyridovericin (2c) [4] (a tyrosine kinase inhibitor) and the elfamycin antibiotics [5]. Interest has also been stimulated in these metabolites by the use of the entomopathogenic fungi such as Cordyceps sp., many of which contain pyridone metabolites, in traditional Chinese medicine to strengthen the immune system and improve cognitive function. Farinosone A (2d) from Paecilomyces farinosus, for example, induces and enhances neurite outgrowth in the PC-12 cell line, although it is not clear whether the pyridones in general display neuritogenic properties [6]. The biosynthesis of tenellin and bassianin in Beauveria bassiana has recently been studied in detail by using genetic techniques, and been shown to involve conversion from an acyltetramic acid by oxidative ring expansion [7,8].
![[1860-5397-8-33-1]](/bjoc/content/figures/1860-5397-8-33-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The 3-acyl-4-hydroxypyridin-2-one motif, example natural products and the isoxazolopyridone scaffolds.
Figure 1: The 3-acyl-4-hydroxypyridin-2-one motif, example natural products and the isoxazolopyridone scaffol...
During a programme of synthesis towards metabolites containing the enolised heterocyclic tricarbonyl motif 3 [9-16], we have reported nitrile oxide dipolar-cycloaddition strategies to access the isoxazolo[4,3-c]pyridin-4-one 4 as 2nd-generation masked nonpolar scaffolds for the 3-acyl-4-hydroxypyridin-2-one nucleus [12]; our 1st-generation approach had employed the [4,5-c] isomer 5 [13-15]. We have recently reported on elaboration of isoxazolopyridone 4 at C-3(Me) and C-7 towards natural products and analogues [16]. An intermediate en route to scaffold 4 is the 4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridin-4-one 6 [12,16], and the availability of this compound, combined with an interest in the biological potential of the acylpyridones and their dihydro derivatives (which are also ring homologues of bioactive 3-acyltetramic acid metabolites), led us to explore elaboration of this 6,7-dihydro-scaffold at C-3(Me) to produce masked forms of the acyldihydropyridones. We report here on these findings.
Results and Discussion
Our approach to the elaboration of the tetrahydroisoxazolopyridone 6 was to use the direct deprotonation strategy employed with the corresponding dehydro derivative 4 [16]. It is well-precedented that 3,5-disubstituted isoxazoles undergo lateral deprotonation–metalation preferentially at the C-5 substituent [17,18] (isoxazole numbering), which corresponds to the C-3 substituent in isoxazolopyridine 6. We planned to undertake aldol-type reactions to lead to 3-alkenylisoxazolopyridones as masked forms of the corresponding acylpyridones, in which 3-alkenoyl substituents feature prominently, see Figure 1. Thus the 3-methyl compound 6 was treated with LDA–TMEDA (2.1 mol equiv of each; THF, −20 °C ice–salt bath) before addition of benzaldehyde (3.5 mol equiv; THF, −20 °C). After 1 hour at −20 °C the mixture was allowed to warm to 20 °C and stirred for 4 days. After this time, we were pleased to be able to isolate the condensation product, 3-(2-phenylethenyl)tetrahydroisoxazolopyridone 8a (53%) rather than the presumed intermediate 3-(2-hydroxy-2-phenylethyl) compound 7a (Scheme 1), along with unchanged starting material 6 (17%). 1H NMR studies indicated the newly formed alkene bond to have the expected E-configuration, with 3JCH=CH = 16.5 Hz. The structure of the 3-alkenyl compound 8a was confirmed by an X-ray crystallographic analysis (Figure 2, Supporting Information File 1 for crystal data) [19].
![[1860-5397-8-33-i1]](/bjoc/content/inline/1860-5397-8-33-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Aldol reactions of tetrahydroisoxazolopyridone 6. Reagents: (i) LDA–TMEDA, RCHO, THF, −20 °C; (ii) toluene, reflux, PTSA.
Scheme 1: Aldol reactions of tetrahydroisoxazolopyridone 6. Reagents: (i) LDA–TMEDA, RCHO, THF, −20 °C; (ii) ...
![[1860-5397-8-33-2]](/bjoc/content/figures/1860-5397-8-33-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of 3-(2-phenylethenyl)-4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridin-4-one (8a) [19].
Figure 2: X-ray crystal structure of 3-(2-phenylethenyl)-4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridin-4-one (8a) ...
By using the same protocol, the reaction was repeated with 4-nitrobenzaldehyde to afford 8b (52%). 4-Bromobenzaldehyde gave condensation product 8c (15%) with hydroxy-adduct 7c as the major product (35%); 2,4-dichlorobenzaldehyde similarly afforded 8d (22%) and 7d (51%), and the nonenolisable 2,2-dimethylpropanal led to 8e in low yield (4%) along with adduct 7e (55%). 3-Phenylpropenal gave condensation product 8f but in a poor yield (12%) along with recovered starting material 6 (14%). These results are summarized in Table 1. In all cases the new alkene bonds have E-configuration (3JCH=CH = 16.5–16.8 Hz). The less electrophilic 2,4-dimethoxy- and 4-dimethylaminobenzaldehydes did not react with the C-3(Me) anion. The hydroxy adducts 7c–e could be made to undergo elimination under Dean–Stark conditions of water removal (toluene, reflux, PTSA in 2 portions; for quantities see Supporting Information File 1) to deliver more of the condensation products 8c–e (41, 38, 44%, respectively, Table 1, see Supporting Information File 1 for full experimental and spectroscopic data).
Table 1: Preparation of 3-alkenylisoxazolopyridones 8, hydroxy adducts 7, and dehydration of adducts 7.
| R | Aldol reaction products 7 and 8a | Dehydration of adducts 7b | |
|---|---|---|---|
| 3-Alkenyl compounds 8 (yield %) | Hydroxy adducts 7 (yield %) | 3-Alkenyl compounds 8 (yield %) | |
| Ph | 8a (53%) | 7a (–) | – |
| 4-O2NC6H4 | 8b (52%) | 7b (–) | – |
| 4-BrC6H4 | 8c (15%) | 7c (35%) | 8c (41%) |
| 2,4-Cl2C6H3 | 8d (22%) | 7d (51%) | 8d (38%) |
| CMe3 | 8e (4%) | 7e (55%) | 8e (44%) |
| CH=CHPh | 8f (12%) | 7f (–) | – |
aReagents: LDA–TMEDA, RCHO, THF, −20 °C; bReagents: toluene, reflux, PTSA.
As an alternative to the LDA protocol described above, the tetrahydroisoxazolopyridone 6 was deprotonated, by using BuLi (2.3 mol equiv; THF–hexanes, −78 °C), followed by addition of benzaldehyde (1.5 mol equiv) to afford the hydroxy adduct 7a (70%). Dehydration was accomplished under Dean–Stark conditions (toluene, reflux, PTSA, 1.3 mol equiv) to give the 3-alkenyl compound 8a (63%). This revised protocol has not yet been extended to the reaction of pyridone 6 with other aldehydes, but has been routinely employed for elaboration of the dehydro version 4 [16].
The acylpyridone antifungal natural product ilicicolin H (9) (Figure 3) [20,21] along with fischerin, apiosporamide and YM-215343, displays a 3-decalinoyl-4-hydroxypyridin-2-one skeleton wherein the decalin unit may arise biosynthetically from a Diels–Alder reaction within the polyketide-derived side chain [1,22]. As a further demonstration of the aldol methodology, we proposed to construct the precursor triene 11, which would facilitate a biomimetic cycloaddition approach to a benzo-fused decalin-substituted isoxazolopyridone, such as 10 (Scheme 2).
![[1860-5397-8-33-3]](/bjoc/content/figures/1860-5397-8-33-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Ilicicolin H as an example of a 3-decalinoyl-4-hydroxypyridin-2-one metabolite.
Figure 3: Ilicicolin H as an example of a 3-decalinoyl-4-hydroxypyridin-2-one metabolite.
![[1860-5397-8-33-i2]](/bjoc/content/inline/1860-5397-8-33-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of a model 3-decalinyl-4,5,6,7-tetrahydroisoxazolopyridone 10.
Scheme 2: Retrosynthetic analysis of a model 3-decalinyl-4,5,6,7-tetrahydroisoxazolopyridone 10.
The aldehyde 12 required for the 3-alkenyl side chain of triene 11 was prepared in four steps by the route shown in Scheme 3, involving palladium-catalysed decarboxylative ring opening of cyclic carbonates [23]. Thus the enolate of α-tetralone was added to propenal in an aldol addition (LDA, THF, −78 °C; 86%). The second step involved the reduction of the intermediate β-hydroxyketone 13, (LiAlH4, THF) to give the 1,3-diol 14 in 75% yield. This was followed by synthesis of the cyclic carbonate 15 by treating the 1,3-diol 14 with methyl chloroformate (Et3N, CH2Cl2; 74%). In the final step, carbonate 15 was treated with palladium dibenzylideneacetone in dry MeCN at room temperature to afford the desired aldehyde 12 in 65% yield. Reaction of this aldehyde (10 mol equiv) with tetrahydroisoxazolopyridone 6 by using BuLi as a base (4.8 mol equiv; THF–hexanes, −78 °C) led to the aldol-type adduct 16 in 53% yield with some of the dehydrated condensation product also isolated as a minor product. This dehydration product, 3-alkenylisoxazolopyridone 11, was prepared from the hydroxy adduct 16 by heating under a Dean–Stark water separator (toluene, reflux, PTSA, 1.5 mol equiv; 78%).
![[1860-5397-8-33-i3]](/bjoc/content/inline/1860-5397-8-33-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of triene 11. Reagents: (i), LDA, propenal, THF, −78 °C; (ii), LiAlH4, THF, 20 °C (75%); (iii), MeOCOCl, Et3N, CH2Cl2 (74%); (iv), Pd2(dba)3·CHCl3, MeCN, 20 °C (65%); (v), BuLi, isoxazolopyridone 6, THF–hexanes, −78 °C (53%); (vi), toluene reflux, PTSA (78%).
Scheme 3: Synthesis of triene 11. Reagents: (i), LDA, propenal, THF, −78 °C; (ii), LiAlH4, THF, 20 °C (75%); ...
Conclusion
We have thus developed suitable protocols for C-3 elaboration of the 3-methyl-4,5,6,7-tetrahydroisoxazolo[4,3-c]pyridin-4-one (6) to give 3-alkenyl derivatives 8 and 11. These are masked forms of the corresponding 3-acyl-4-hydroxy-1,2,5,6-tetrahydropyridin-2-ones, which are of interest as dihydro relatives of the bioactive acylpyridone natural products and homologues of acyltetramic acids. Reductive methods to reveal the cyclic tricarbonyl moiety in such compounds have been reported by us previously [9-16].
Supporting Information
Supporting information features full experimental and spectroscopic details for alkenylisoxazolopyridones 8a–f, hydroxy adducts 7c–e, compounds 11–16 and crystallographic data for 8a (Figure 2).
| Supporting Information File 1: Experimental and spectroscopic details. | ||
| Format: PDF | Size: 210.1 KB | Download |
Acknowledgements
We thank the Open University (G. L.) and Loughborough University (A. K. C. & T. A. P.) for studentship support, Syngenta for additional support (A. K. C.), Dr. N. B. Carter for helpful discussions, and the EPSRC Mass Spectrometry Service Centre (Swansea) for some high-resolution MS data.
References
-
Jessen, H. J.; Gademann, K. Nat. Prod. Rep. 2010, 27, 1168–1185. doi:10.1039/b911516c
See for a review.
Return to citation in text: [1] [2] -
El Basyouni, S. H.; Brewer, D.; Vining, L. C. Can. J. Bot. 1968, 46, 441–448. doi:10.1139/b68-067
Return to citation in text: [1] -
Wat, C.-K.; McInnes, A. G.; Smith, D. G.; Wright, J. L. C.; Vining, L. C. Can. J. Chem. 1977, 55, 4090–4098. doi:10.1139/v77-580
Return to citation in text: [1] -
Takahashi, S.; Uchida, K.; Kakinuma, N.; Hashimoto, R.; Yanagisawa, T.; Nakagawa, A. J. Antibiot. 1998, 51, 1051–1054.
Return to citation in text: [1] -
Dolle, R. E.; Nicolaou, K. C. J. Am. Chem. Soc. 1985, 107, 1691–1694. doi:10.1021/ja00292a038
See for leading references.
Return to citation in text: [1] -
Cheng, Y.; Schneider, B.; Riese, U.; Schubert, B.; Li, Z.; Hamburger, M. J. Nat. Prod. 2004, 67, 1854–1858. doi:10.1021/np049761w
Return to citation in text: [1] -
Halo, L. M.; Marshall, J. W.; Yakasai, A. A.; Song, Z.; Butts, C. P.; Crump, M. P.; Heneghan, M.; Bailey, A. M.; Simpson, T. J.; Lazarus, C. M.; Cox, R. J. ChemBioChem 2008, 9, 585–594. doi:10.1002/cbic.200700390
Return to citation in text: [1] -
Halo, L. M.; Heneghan, M. N.; Yakasai, A. A.; Song, Z.; Williams, K.; Bailey, A. M.; Cox, R. J.; Lazarus, C. M.; Simpson, T. J. J. Am. Chem. Soc. 2008, 130, 17988–17996. doi:10.1021/ja807052c
Return to citation in text: [1] -
Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J. Synlett 1999, 873–876. doi:10.1055/s-1999-3091
See for 3-acyltetramic acids.
Return to citation in text: [1] [2] -
Jones, R. C. F.; Pillainayagam, T. A. Synlett 2004, 2815–2817. doi:10.1055/s-2004-835640
See for 3-acyltetramic acids.
Return to citation in text: [1] [2] -
Law, C. C. M. Ph.D. Thesis, Loughborough University, U.K., 2008.
See for 3-acyltetramic acids.
Return to citation in text: [1] [2] -
Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J.; Patel, P. Tetrahedron Lett. 1999, 40, 4085–4088. doi:10.1016/S0040-4039(99)00655-3
Return to citation in text: [1] [2] [3] [4] -
Jones, R. C. F.; Duller, K. A. M.; Vulto, S. I. E. J. Chem. Soc., Perkin Trans. 1 1998, 411–416. doi:10.1039/a707282a
See for our 1st generation approach.
Return to citation in text: [1] [2] [3] -
Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/A900120D
See for our 1st generation approach.
Return to citation in text: [1] [2] [3] -
Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, viii, 34–39.
See for our 1st generation approach.
Return to citation in text: [1] [2] [3] -
Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lang, S. A., Jr.; Lin, Y.-I. Isoxazoles and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry I; Katritzxy, A. R.; Rees, C. W., Eds.; Elsevier: Oxford, 1984; Vol. 6, Part 4B, pp 48–50.
Return to citation in text: [1] -
Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, 2008; Vol. 4, pp 404–406.
Return to citation in text: [1] -
Complete crystallographic data (excluding structure factors) have been deposited at the Cambridge Crystallographic Data Centre under the number CCDC 842696. These data can be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1RZ, England, or via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] [2] -
Matsumoto, M.; Minato, H. Tetrahedron Lett. 1976, 17, 3827–3830. doi:10.1016/S0040-4039(00)93121-6
Return to citation in text: [1] -
Tanabe, M.; Urano, S. Tetrahedron 1983, 39, 3569–3574. doi:10.1016/S0040-4020(01)88667-1
Return to citation in text: [1] -
Williams, D. R.; Bremmer, M. L.; Brown, D. L.; D’Antuono, J. J. Org. Chem. 1985, 50, 2807–2809. doi:10.1021/jo00215a053
See for a synthesis of ilicicolin J using a Diels–Alder approach.
Return to citation in text: [1] -
Harayama, H.; Kuroki, T.; Kimura, M.; Tanaka, S.; Tamaru, Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 2352–2354. doi:10.1002/anie.199723521
Return to citation in text: [1]
| 23. | Harayama, H.; Kuroki, T.; Kimura, M.; Tanaka, S.; Tamaru, Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 2352–2354. doi:10.1002/anie.199723521 |
| 20. | Matsumoto, M.; Minato, H. Tetrahedron Lett. 1976, 17, 3827–3830. doi:10.1016/S0040-4039(00)93121-6 |
| 21. | Tanabe, M.; Urano, S. Tetrahedron 1983, 39, 3569–3574. doi:10.1016/S0040-4020(01)88667-1 |
| 1. |
Jessen, H. J.; Gademann, K. Nat. Prod. Rep. 2010, 27, 1168–1185. doi:10.1039/b911516c
See for a review. |
| 22. |
Williams, D. R.; Bremmer, M. L.; Brown, D. L.; D’Antuono, J. J. Org. Chem. 1985, 50, 2807–2809. doi:10.1021/jo00215a053
See for a synthesis of ilicicolin J using a Diels–Alder approach. |
| 1. |
Jessen, H. J.; Gademann, K. Nat. Prod. Rep. 2010, 27, 1168–1185. doi:10.1039/b911516c
See for a review. |
| 6. | Cheng, Y.; Schneider, B.; Riese, U.; Schubert, B.; Li, Z.; Hamburger, M. J. Nat. Prod. 2004, 67, 1854–1858. doi:10.1021/np049761w |
| 19. | Complete crystallographic data (excluding structure factors) have been deposited at the Cambridge Crystallographic Data Centre under the number CCDC 842696. These data can be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1RZ, England, or via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 5. |
Dolle, R. E.; Nicolaou, K. C. J. Am. Chem. Soc. 1985, 107, 1691–1694. doi:10.1021/ja00292a038
See for leading references. |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
| 4. | Takahashi, S.; Uchida, K.; Kakinuma, N.; Hashimoto, R.; Yanagisawa, T.; Nakagawa, A. J. Antibiot. 1998, 51, 1051–1054. |
| 17. | Lang, S. A., Jr.; Lin, Y.-I. Isoxazoles and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry I; Katritzxy, A. R.; Rees, C. W., Eds.; Elsevier: Oxford, 1984; Vol. 6, Part 4B, pp 48–50. |
| 18. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, 2008; Vol. 4, pp 404–406. |
| 2. | El Basyouni, S. H.; Brewer, D.; Vining, L. C. Can. J. Bot. 1968, 46, 441–448. doi:10.1139/b68-067 |
| 3. | Wat, C.-K.; McInnes, A. G.; Smith, D. G.; Wright, J. L. C.; Vining, L. C. Can. J. Chem. 1977, 55, 4090–4098. doi:10.1139/v77-580 |
| 19. | Complete crystallographic data (excluding structure factors) have been deposited at the Cambridge Crystallographic Data Centre under the number CCDC 842696. These data can be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1RZ, England, or via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 13. |
Jones, R. C. F.; Duller, K. A. M.; Vulto, S. I. E. J. Chem. Soc., Perkin Trans. 1 1998, 411–416. doi:10.1039/a707282a
See for our 1st generation approach. |
| 14. |
Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/A900120D
See for our 1st generation approach. |
| 15. |
Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, viii, 34–39.
See for our 1st generation approach. |
| 12. | Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J.; Patel, P. Tetrahedron Lett. 1999, 40, 4085–4088. doi:10.1016/S0040-4039(99)00655-3 |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
| 12. | Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J.; Patel, P. Tetrahedron Lett. 1999, 40, 4085–4088. doi:10.1016/S0040-4039(99)00655-3 |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
| 9. |
Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J. Synlett 1999, 873–876. doi:10.1055/s-1999-3091
See for 3-acyltetramic acids. |
| 10. |
Jones, R. C. F.; Pillainayagam, T. A. Synlett 2004, 2815–2817. doi:10.1055/s-2004-835640
See for 3-acyltetramic acids. |
| 11. |
Law, C. C. M. Ph.D. Thesis, Loughborough University, U.K., 2008.
See for 3-acyltetramic acids. |
| 12. | Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J.; Patel, P. Tetrahedron Lett. 1999, 40, 4085–4088. doi:10.1016/S0040-4039(99)00655-3 |
| 13. |
Jones, R. C. F.; Duller, K. A. M.; Vulto, S. I. E. J. Chem. Soc., Perkin Trans. 1 1998, 411–416. doi:10.1039/a707282a
See for our 1st generation approach. |
| 14. |
Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/A900120D
See for our 1st generation approach. |
| 15. |
Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, viii, 34–39.
See for our 1st generation approach. |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
| 9. |
Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J. Synlett 1999, 873–876. doi:10.1055/s-1999-3091
See for 3-acyltetramic acids. |
| 10. |
Jones, R. C. F.; Pillainayagam, T. A. Synlett 2004, 2815–2817. doi:10.1055/s-2004-835640
See for 3-acyltetramic acids. |
| 11. |
Law, C. C. M. Ph.D. Thesis, Loughborough University, U.K., 2008.
See for 3-acyltetramic acids. |
| 12. | Jones, R. C. F.; Dawson, C. E.; O’Mahony, M. J.; Patel, P. Tetrahedron Lett. 1999, 40, 4085–4088. doi:10.1016/S0040-4039(99)00655-3 |
| 13. |
Jones, R. C. F.; Duller, K. A. M.; Vulto, S. I. E. J. Chem. Soc., Perkin Trans. 1 1998, 411–416. doi:10.1039/a707282a
See for our 1st generation approach. |
| 14. |
Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/A900120D
See for our 1st generation approach. |
| 15. |
Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, viii, 34–39.
See for our 1st generation approach. |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
| 7. | Halo, L. M.; Marshall, J. W.; Yakasai, A. A.; Song, Z.; Butts, C. P.; Crump, M. P.; Heneghan, M.; Bailey, A. M.; Simpson, T. J.; Lazarus, C. M.; Cox, R. J. ChemBioChem 2008, 9, 585–594. doi:10.1002/cbic.200700390 |
| 8. | Halo, L. M.; Heneghan, M. N.; Yakasai, A. A.; Song, Z.; Williams, K.; Bailey, A. M.; Cox, R. J.; Lazarus, C. M.; Simpson, T. J. J. Am. Chem. Soc. 2008, 130, 17988–17996. doi:10.1021/ja807052c |
| 16. | Jones, R. C. F.; Choudhury, A. K.; Iley, J. N.; Loizou, G.; Lumley, C.; McKee, V. Synlett 2010, 654–658. doi:10.1055/s-0029-1219341 |
© 2012 Jones et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)