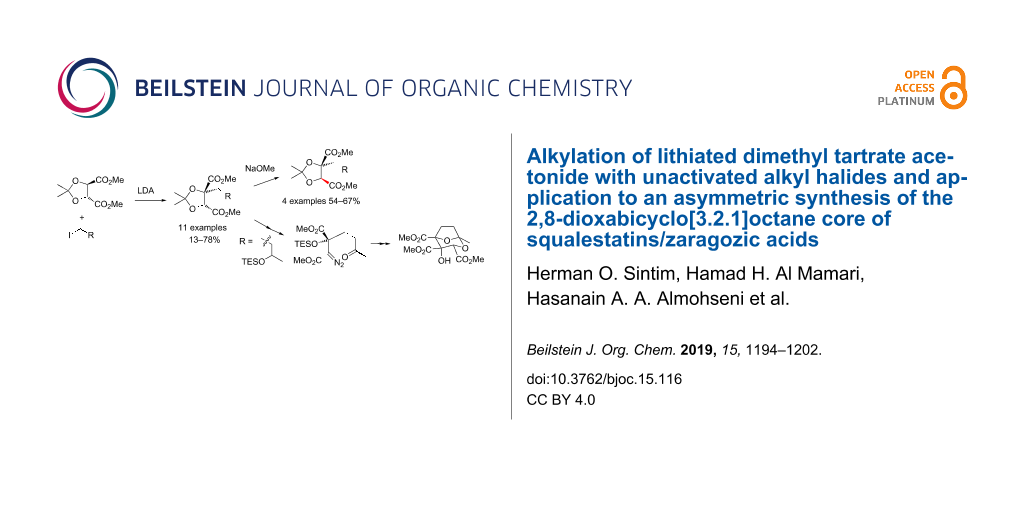
Abstract
(R,R)-Dimethyl tartrate acetonide 7 in THF/HMPA undergoes deprotonation with LDA and reaction at −78 °C during 12–72 h with a range of alkyl halides, including non-activated substrates, to give single diastereomers (at the acetonide) of monoalkylated tartrates 17, 24, 33a–f, 38a,b, 41 of R,R-configuration, i.e., a stereoretentive process (13–78% yields). Separable trans-dialkylated tartrates 34a–f can be co-produced in small amounts (9–14%) under these conditions, and likely arise from the achiral dienolate 36 of tartrate 7. Enolate oxidation and acetonide removal from γ-silyloxyalkyl iodide-derived alkylated tartrates 17 and 24 give ketones 21 and 26 and then Bamford–Stevens-derived diazoesters 23 and 27, respectively. Only triethylsilyl-protected diazoester 27 proved viable to deliver a diazoketone 28. The latter underwent stereoselective carbonyl ylide formation–cycloaddition with methyl glyoxylate and acid-catalysed rearrangement of the resulting cycloadduct 29, to give the 3,4,5-tricarboxylate-2,8-dioxabicyclo[3.2.1]octane core 31 of squalestatins/zaragozic acids. Furthermore, monoalkylated tartrates 33a,d,f, and 38a on reaction with NaOMe in MeOH at reflux favour (≈75:25) the cis-diester epimers epi-33a,d,f and epi-38a (54–67% isolated yields), possessing the R,S-configuration found in several monoalkylated tartaric acid motif-containing natural products.

Graphical Abstract
Introduction
Since their isolation was reported in the early 1990s [1,2], the squalestatins/zaragozic acids (e.g., squalestatin S1/zaragozic acid A (1), Figure 1) have been of enduring interest to synthetic chemists, due to a combination of a synthetically challenging densely functionalised 2,8-dioxabicyclo[3.2.1]octane core [3-6], combined with an increasing range of intriguing biological activities [7-11]. Here, we report in detail the evolution of chemistry that provides an asymmetric entry to the tricarboxylate core of these natural products, with particular focus on tartrate alkylation methodology to establish the fully-substituted C-5 stereocentre (squalestatin numbering).
![[1860-5397-15-116-1]](/bjoc/content/figures/1860-5397-15-116-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Squalestatin S1/zaragozic acid A (1) and DDSQ (2).
Figure 1: Squalestatin S1/zaragozic acid A (1) and DDSQ (2).
Our studies in this area have recently culminated in two communicated syntheses of 6,7-dideoxysqualestatin H5 (DDSQ (2), Figure 1) [12,13]. The centrepiece of both of these strategies is a rhodium(II)-catalysed tandem carbon ylide formation from a diazoketone 3 (Scheme 1) and stereoselective [3 + 2] cycloaddition with a glyoxylate (3 → 4 → 5) [14,15], followed by an acid-catalysed rearrangement to generate the desired dideoxysqualestatin core 6 with the requisite tricarboxylate functionality installed. While we had earlier established the viability of this approach in a racemic model study (X = H) [14], extension to an asymmetric variant of our aldol route (α-diazoacetate ester anion addition to an α-ketoester) to the cycloaddition substrate 3 (X = H) did not appear promising [16].
![[1860-5397-15-116-i1]](/bjoc/content/inline/1860-5397-15-116-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Carbonyl ylide cycloaddition–rearrangement to the squalestatin core [12,13].
Scheme 1: Carbonyl ylide cycloaddition–rearrangement to the squalestatin core [12,13].
An alternative and asymmetric route to such substrates, ultimately successful, built on stereoselective alkylation of enolates of tartrates (e.g., 7, Scheme 2) was originally reported by Seebach and co-workers for ‘activated’ (allylic, benzylic) alkyl halides [17-19]. If an alkylated tartrate 9 could be accessed from a silyloxy-substituted alkyl iodide 8 and subsequently oxidised (for example via a second tartrate enolate) with acetonide removal, this would give an α-ketoester 10. The latter should in principle be a progenitor to the desired α-diazo ester 3, following condensation with tosylhydrazide, then Bamford–Stevens-type base-induced sulfinate elimination [20] and oxidation of the secondary silyl ether.
![[1860-5397-15-116-i2]](/bjoc/content/inline/1860-5397-15-116-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Tartrate alkylation strategy to cycloaddition substrate.
Scheme 2: Tartrate alkylation strategy to cycloaddition substrate.
Results and Discussion
The general viability of the α-ketoester to α-diazoester functional group interconversion envisaged in Scheme 2 (10 → 3) was readily established on a simpler but closely structurally-related system (Scheme 3). Thus, the known Z-hydrazone 12, previously prepared by us from α-ketoester 11 in 75% yield [21], gave α-diazo ester 13 in 76% yield following reaction with NaOMe. Furthermore, our earlier racemic model study had established that deprotection and oxidation of a secondary silyl ether in the presence of α-diazo ester functionality was feasible, which constitutes precedent for the generation of the ketone functionality in 3 [14]. These observations led us to examine the possibility of substrate assembly through Seebach’s tartrate alkylation methodology.
![[1860-5397-15-116-i3]](/bjoc/content/inline/1860-5397-15-116-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Conversion of α-ketoester to α-diazoester.
Scheme 3: Conversion of α-ketoester to α-diazoester.
In 1981, Seebach and Naef communicated that (R,R)-tartrate acetonide 7 could be deprotonated and undergo stereoselective alkylation (≈80:20 dr) with reactive organohalides (Scheme 4) [17]. The process was valuable, because it allowed direct elaboration of a chiral pool building block that was readily available as either antipode [22,23], with the major alkylated diastereomer 15 being generated in 97:3 er [18]. The study was also notable in showing that the intermediate ester enolate 14 possessed sufficient stability not to undergo significant β-elimination under conditions of its generation and its alkylation: slow addition of pre-cooled LDA (−70 °C) to a mixture of the acetonide and electrophile in THF/HMPA at −78 °C, followed by slow warming to ≈−10 °C before work-up. Finally, the reaction displayed remarkable stereoselectivity, in that the electrophile was introduced on ostensibly the more hindered face of the enolate (that is, cis (“contrasteric”) [24] to the unenolised ester group). The former observation was rationalised due to the enolate π-system and potentially cleavable beta σ-C–O bond lying mutually orthogonal, while the latter was subsequently ascribed to alkylation occurring from an envelope conformation wherein the unenolised ester resided pseudoequatorial to avoid 1,3-steric interactions with a pseudoaxial methyl of the gem-dimethyl group [18,24]; it was proposed that the axial methyl group directed electrophile incorporation away from itself (Scheme 4).
![[1860-5397-15-116-i4]](/bjoc/content/inline/1860-5397-15-116-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Seebach’s tartrate alkylation and rationalisation of stereoselectivity [17-19].
Scheme 4: Seebach’s tartrate alkylation and rationalisation of stereoselectivity [17-19].
The fragile nature of the lithium ester enolate of dimethyl tartrate acetonide (to β-elimination with loss of acetone) was evident from Seebach’s work, which concluded that only especially reactive halides (methyl, benzylic, allylic) were feasible electrophiles; with iodoethane, 1-iodo-2-methylpropane and chloromethoxymethane no alkylation products were formed [17-19]. Given these rather discouraging observations in the context of our proposed chemistry (Scheme 2), we were pleased to find that initial studies with (R,R)-tartrate 7 and a 3-silyloxy-1-iodobutane 16 did generate alkylated tartrates (Scheme 5). Following Seebach’s protocol, with warming overnight to room temperature, gave a 50% yield (90% based on recovered iodide 16) of a separable 76:24 mixture of alkylated tartrates 17 and 18, respectively. The relative stereochemistry was assigned by analogy with Seebach’s findings for substituted tartrates: that the diastereomer with the ring methine cis to the ester group (i.e., 17) always displays the higher chemical shift (≈5 ppm vs ≈4.5 ppm in CDCl3) [18], and was further supported by 1D NOESY experiments on both alkylated tartrates 17 and 18. The use of DMPU as co-solvent [18,25] reversed the ratio, with the currently undesired diastereomer 18 becoming favoured (37:63, 17:18). However, with HMPA the proportion of 17 improved significantly (>90:10, 17:18) if the reaction was maintained at −78 °C for several hours before quenching at that temperature, giving isolated yields of 30–50% for 17. The absolute configuration of 17 was based on the chemical correlation studies of Seebach [17-19] and of Pan [26] for benzylations, and subsequently those of Nagano [27] and of Li [28] for allylations, i.e., stereoretentive (contrasteric) alkylation. As noted above, Seebach recorded 97:3 er (by NMR using a chiral shift reagent) for a benzylated tartrate [18]; in the present case, chiral HPLC comparison of 17 with the corresponding adduct from S,S-tartrate (ent)-7 indicated they were both of >98:2 enantiopurity in the tartrate portion.
![[1860-5397-15-116-i5]](/bjoc/content/inline/1860-5397-15-116-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Tartrate alkylation with a non-activated alkyl iodide.
Scheme 5: Tartrate alkylation with a non-activated alkyl iodide.
Before exploring the scope of the alkylation chemistry further, it was considered prudent for the proposed asymmetric approach to the dideoxysqualestatin core 6 (Scheme 1 and Scheme 2) to establish the viability of the rest of sequence outlined in Scheme 2 from an alkylated tartrate. While further C–C bond formation by enolate formation at the remaining methine on a monoalkylated tartrate acetonide had been reported by Molander and Harris [29], and by Kelly and co-workers [30]; the question whether such an enolate could be oxidised required investigation. Although the reaction of alkylated tartrate 17 with NaHMDS/2-(phenylsulfonyl)-3-phenyloxaziridine [31] gave an unidentifiable mixture, the use of LDA and MoOPH [32,33] at −78 °C followed by warming to −50 °C for 3 h gave the hydroxy acetonide 19a (Scheme 6) in 92% yield as a mixture of 4 diastereomers. Similarly, the enolate of the simpler propylated tartrate 33a [12] reacted with MoOPH to give the analogous hydroxy acetonide 19b in 96% yield (3:1 dr); if the MoOPH was added to the enolate which had been warmed to −40 °C, a more typical hydroxylation temperature, then a reduced yield of 19b was observed (53%). Indirect hydroxylation of the propylated tartrate enolate was also attempted using CBr4 (at −78 °C) as a more readily available/convenient electrophile, which also gave the hydroxy acetonide 19b presumably by way of hydrolysis on work-up of an intermediate bromo acetonide, albeit in significantly reduced yield (33%).
![[1860-5397-15-116-i6]](/bjoc/content/inline/1860-5397-15-116-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Alkylated tartrate to diazoester sequence.
Scheme 6: Alkylated tartrate to diazoester sequence.
In contrast to a simple hydroxy acetonide (formally derived from an α-hydroxy aldehyde and acetone) [34], hydroxy acetonide 19a was found stable to mild bases such as Et3N and iPr2NH, whereas the use of NaH or NaHMDS in THF both decomposed 19a into unidentifiable polar products. Attempted acid-induced loss of acetone with PTSA in MeOH, PPTS in refluxing MeOH, or 80% AcOH at reflux [27] all led to quantitative recovery of hydroxy acetonide 19a, whereas 5% aq HCl resulted in acetonide removal and concomitant desilylation. Initial Lewis acids screened either failed to react (PdCl2(MeCN)2), or led to complex mixtures (BF3, YbOTf, TBSOTf). More encouragingly, both AlCl3 and FeCl3 were found to cleave the acetonide 19a at rt, with the TBDPS group only being partially lost (≈15%) in both cases. AlCl3 was observed to deprotect the TBDPS ether more slowly than FeCl3, and adjustment of the reaction conditions with AlCl3 (to 2 equiv in CH2Cl2, −78 °C, followed by slow warming to −50 °C) cleanly provided α-ketoester 20 (Scheme 6). α-Ketoesters can be prone to hydrate easily (2D TLC analysis of 20 indicated decomposition); therefore, 20 and the derived tertiary TBS ether 21 were carried on directly to form hydrazone 22 (51% yield over 3 steps from hydroxy acetonide 19a). Unlike with hydrazone 12 (Scheme 3), application of NaOMe was not conducive to effective diazo formation from hydrazone 22, giving a mixture of unidentified products; however, hydrazone 22 was cleanly converted into α-diazo ester 23 (88%) using Et3N [35,36].
TBDPS protection for the secondary alcohol had originally been selected principally for its likely tolerance to potential (hydroxy) acetonide removal conditions, and with the possibility [37,38] of its selective deprotection in α-diazo ester 23 in the presence of the tertiary TBS ether. It was considered important that the tertiary alcohol remain masked during projected oxidation of the released secondary alcohol to give the ketone functionality in the cycloaddition substrate, as otherwise essentially irretrievable five-membered lactol formation would be expected [39]. Unfortunately, various reagents (TBAF/AcOH [40], NaH/HMPA [41], Bu4OH/DMF [40], NaOMe/MeOH) failed to selectively deprotect the secondary TBDPS ether in α-diazo ester 23 in the presence of the tertiary TBS ether. Reassessment of the protecting group strategy led us to TES protection at both alcohols, on the basis that this group should be robust enough to withstand the enolate manipulation chemistry, that desilylation of the secondary TES ether during acetonide removal could be restored in the subsequent tertiary alcohol silylation step, that selective 2° over 3° TES ether desilylation should be readily achievable using AcOH [14], and that the remaining tertiary TES ether should be potentially labile enough to be removed under typical transketalisation conditions (TFA/CH2Cl2/H2O (10:20:1), 40 °C, 48–68 h [14], cf, Scheme 1), thereby circumventing the separate prior desilylation step in our earlier racemic model study [14]. In the event, application of this TES protection approach did provide access to diazo alcohol 27 (Scheme 7). As anticipated from the above deprotection studies with TBDPS ether 23, 1% aq HCl removed both the acetonide and TES groups in hydroxy acetonide 25. Subsequent silylation using TESOTf gave the bis-TES ketone 26, which was not purified but taken on through diazo formation and desilylation to give diazo alcohol 27 (17% from 25). The efficiency of the sequence from hydroxy acetonide 25 to diazo alcohol 27 could be improved (to 37%) using ZnCl2 for the initial deprotection and TESCl in the silylation; the latter minimises formation of the undesired silylated six-membered lactol form of 26. The remaining steps to the model core 31 (Scheme 7) closely mirrored our previous racemic synthesis of 31 (from the corresponding 3° TBS ether) [14], and this strategy was subsequently also successfully applied in our most recent total synthesis of DDSQ (2) [13].
![[1860-5397-15-116-i7]](/bjoc/content/inline/1860-5397-15-116-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: TES protection approach to the squalestatin core.
Scheme 7: TES protection approach to the squalestatin core.
We now returned to study the Seebach alkylation chemistry in more detail. In 2008, Lipton and co-workers had observed that the cyclopentylidene derivative of diethyl tartrate reacted with LiHMDS in the presence of LiCl and MeI, to give exclusively the corresponding trans-monomethylated tartrate in 72% yield [42]; this compares with methylation of acetonide tartrate 7 by Seebach, which was reported to give an inseparable mixture of monomethylated product (86:14 dr), dimethylated, and recovered 7 (50%, 79:15:6, respectively) [17-19]. Application of Lipton's conditions to tartrate 7, cleanly gave the trans-monomethylated product 32, albeit in moderate yield (39%, Scheme 8); however, extension to a higher alkylating agent (PrI) was unsuccessful, returning only ≈25% of tartrate 7 in an impure state.
![[1860-5397-15-116-i8]](/bjoc/content/inline/1860-5397-15-116-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Tartrate acetonide methylation.
Scheme 8: Tartrate acetonide methylation.
In contrast to the unsuccessful propylation with LiHMDS/LiCl mentioned above, following our modified Seebach‘s protocol, propylation could be achieved to give propylated tartrate 33a [12] (Scheme 9), in 66% yield and 97:3 er by chiral HPLC, with the trans-dipropylated product 34a also being separately isolated, in 7% yield. Other primary ‘non-activated’ alkyl iodides also led to alkylated tartrates 33b,c and the corresponding dialkylated side-products 34b,c (Scheme 9). Under our modified alkylation conditions, ‘activated’ bromides (benzyl, allyl and prenyl), previously examined by Seebach [17-19], all gave the corresponding monoalkylated tartrates 33d–f also as single diastereomers (along with separable trans-dialkylated material 34d–f, Scheme 9). These latter results indicate that it is the modified reaction conditions, rather than the nature of the alkylating agent, which leads to the improved diastereoselectivity [43]. The trans stereochemistry assignment for the dialkylated products 34 follow from the observed equivalence of the acetonide methyl groups in all their proton and carbon NMR spectra.
![[1860-5397-15-116-i9]](/bjoc/content/inline/1860-5397-15-116-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Tartrate alkylation with various alkyl halides.
Scheme 9: Tartrate alkylation with various alkyl halides.
In Seebach's original studies, which established retention on tartrate alkylation for the major diastereomer (formed, as noted above, in 97:3 er), a further intriguing observation was made: that the minor diastereomer was obtained in virtually racemic form (determined by NMR using a chiral shift reagent) [18,19]. It was suggested that the latter principally arose from alkylation (then protonation, by unreacted 7) [19] of the (achiral) dienolate 36 (Scheme 10) of 7. In the current work, where the major difference is prolonged reaction time at low temperature and no warming before quenching, the minor diastereomer is not observed – but dialkylated byproduct 34 is. An attempt to propylate the monopropylated tartrate 33a under our conditions mainly returned starting 33a (68%), with only a trace of trans-dipropylated product 34a being isolated (2%). Seebach’s observations, together with ours, indicate that the dienolate 36 does form to some extent at −78 °C (Scheme 10) [44-46], and both it and the derived alkylated mono-anion rac-35 are quite reactive to alkylation, but protonation of the alkylated mono-anion rac-35 (by unreacted 7) does not occur at that temperature; also, once monoalkylation has occurred from the mono-enolate 14, then 33a is not readily deprotonated at −78 °C. Clearly, monoalkylated tartrate can be deprotonated and trapped with electrophiles if the system is warmed above −78 °C: to −30 °C by Molander and Harris [29], whereas −50 °C was sufficient for the enolate oxidation steps in Scheme 6 and Scheme 7. Indeed, propylation of the monopropylated tartrate 33a can be achieved in 34% yield, if the reaction mixture is warmed to −50 °C (Scheme 10). Chiral HPLC comparison of the trans-dipropylated material 34a obtained as the byproduct from the alkylation of tartrate acetonide 7, and from propylation of the monopropylated tartrate 33a, confirmed that the former was essentially racemic (52:48 er), while the latter was 98:2 er. Therefore, if one wishes to generate (R,R)- (or (S,S)-) C2-symmetric dialkylated tartrates [47-49], an important conclusion from the above observations and analysis is that monoalkylation should be carried out first, and the isolated monoalkylated material then separately subjected to a second alkylation, allowing warming to ≈−40 °C.
![[1860-5397-15-116-i10]](/bjoc/content/inline/1860-5397-15-116-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Rationalisation of dialkylation observations.
Scheme 10: Rationalisation of dialkylation observations.
Our first synthesis of 6,7-dideoxysqualestatin H5 (DDSQ (2), Figure 1), required extension of the above tartrate alkylation chemistry to a homoallylic halide as the electrophile (Scheme 11) [12]. An initial experiment with 4-bromobut-1-ene (37a) was not encouraging, delivering the homo-allylated product 38a in only 13% yield. It was suspected that competing elimination (to give butadiene) contributed to the low yield. Support for this was found with the real system, where the corresponding diene 39 was isolated in up to 36% yield. In our earlier studies, typically approximately equimolar quantities of tartrate and alkylating agent were used, but with the halide now being synthetically more valuable, efforts focused on conditions which gave the best yields using it as the limiting agent. A slight excess of tartrate relative to iodide produce the homo-allylated tartrate 38b in 34% yield, but this could be improved to 60–78% by using 100% excess of tartrate [12]. It was also noted that minimal contact time between the sensitive iodide 37b and HMPA at low temperature before slow addition of the pre-cooled LDA minimised diene formation, with the excess residual unreacted tartrate being most conveniently removed by distillation on large-scale. Our second synthesis of DDSQ (2) introduced the full side-chain through alkylation with iodide 40 to give the alkylated tartrate 41 in 71% yield, and used only a slight excess of tartrate 7 and LDA (1.2 and 1.5 equiv, respectively) for 72 h at −78 °C [13]. These examples demonstrate the viability of the tartrate alkylation chemistry with more complex and valuable electrophiles.
![[1860-5397-15-116-i11]](/bjoc/content/inline/1860-5397-15-116-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Tartrate alkylation chemistry with more complex alkyl iodides [12,13].
Scheme 11: Tartrate alkylation chemistry with more complex alkyl iodides [12,13].
While monoalkylated tartrate acetonides were applied in squalestatin syntheses as highlighted above, the monoalkylated tartaric acid motif is also directly present in several natural products, such as hydroxybenzyl-substituted piscidic acid (42) and congeners (fukiic and cimicifugic acids) [50], and the Cephalotaxus alkaloids isoharringtonine (43) and cephalezomine C (44) [51]. In these latter natural products, the monoalkylated tartaric acid residues typically possess 2R,3S stereochemistry (Figure 2); one exception is cephalezomine D [51], which is the 3R-epimer of cephalezomine C (44). Since the chiral (R,R- S,S-) tartrate acetonides undergo stereoretentive alkylation, then direct access to the stereochemistry present in the majority of these natural products is not possible. Nevertheless, a couple of isolated examples in the Chinese chemical literature from the late 1980s indicated that subsequent base-induced epimerisation of monoalkylated (p-(benzyloxy)benzylated and prenylated) tartrate acetonides favours R,S stereochemistry, providing access to piscidic acid (42) [26] and the tartrate residue of isoharringtonine (43) [28].
![[1860-5397-15-116-2]](/bjoc/content/figures/1860-5397-15-116-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Natural product examples containing the monoalkylated tartaric acid motif.
Figure 2: Natural product examples containing the monoalkylated tartaric acid motif.
We studied the previously reported epimerisation of prenylated tartrate 33f, on 0.5 mmol scale ([28] 5 mmol), using NaOMe (1 equiv) in MeOH (0.5 M) at room temperature for 12 h. Under these conditions we observed only partial epimerisation (33f/epi-33f 75:25, [[28] 12:88]), but a 25:75 ratio in favour of epi-33f could be achieved in MeOH (0.06 M) at reflux for 30 h (Scheme 12). That equilibrium had been reached was established by subjecting epi-33f to these latter reaction conditions, which returned the same 25:75 ratio of 33f/epi-33f. Other monoalkylated tartrates were found to give similar levels of epimerisation (Scheme 12), generating the chromatographically separable epimerised tartrates in 54–67% isolated yields.
![[1860-5397-15-116-i12]](/bjoc/content/inline/1860-5397-15-116-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Epimerisation of monoalkylated tartrates.
Scheme 12: Epimerisation of monoalkylated tartrates.
Conclusion
Contrary to the seminal observations of Seebach and Naef, we have demonstrated that lithiated dimethyl tartrate acetonide can undergo stereoretentive alkylation even with “non-activated” alkyl halides, in synthetically useful yields and high er. Optimal reaction conditions are prolonged reaction time at −78 °C, followed by quenching at that temperature; these conditions also avoid cogeneration of the (racemic) diastereomer side-product originally observed on warming. Essentially racemic dialkylated tartrate is observed as a minor side-product under our modified conditions. The reaction pathways that lead to these racemic products under the different reaction conditions have been rationalised. Application of this methodology to generate the 6,8-dioxabicyclo[3.2.1]octane core of the squalestatins/zaragozic acids in an asymmetric fashion, is shown to evolve from a carefully orchestrated sequence of oxidation and judicious protecting group manipulation. Base-induced epimerisation of the monoalkylated tartrates favours cis-disposition of the ester groups on the five-membered ring, thereby accessing the predominant stereochemistry found in several substituted tartaric acid-containing natural products. Our findings on tartrate alkylation with non-activated alkyl halides, on origins of side-products, and on conditions for epimerisation of the monoalkylated tartrates, significantly broaden the scope and understanding of Seebach’s alkylation chemistry.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterisation data and 1H and 13C NMR spectra for all new compounds. | ||
| Format: PDF | Size: 5.8 MB | Download |
Acknowledgements
We thank the University of Oxford, the Sultanate of Oman, the Higher Committee for Education Development in Iraq, and the EPSRC for studentship support (to H.O.S., H.H.A.M., H.A.A.A., and Y.F-H., respectively), and Dr. Anne Valade for the butylation result in Scheme 9.
References
-
Dawson, M. J.; Farthing, J. E.; Marshall, P. S.; Middleton, R. F.; O’Neill, M. J.; Shuttleworth, A.; Stylli, C.; Tait, R. M.; Taylor, P. M.; Wildman, H. G.; Buss, A. D.; Langley, D.; Hayes, M. V. J. Antibiot. 1992, 45, 639–647. doi:10.7164/antibiotics.45.639
Return to citation in text: [1] -
Dufresne, C.; Wilson, K. E.; Zink, D.; Smith, J.; Bergstrom, J. D.; Kurtz, M.; Rew, D.; Nallin, M.; Jenkins, R.; Bartizal, K.; Trainor, C.; Bills, G.; Meinz, M.; Huang, L.; Onishi, J.; Milligan, J.; Mojena, M.; Pelaez, F. Tetrahedron 1992, 48, 10221–10226. doi:10.1016/s0040-4020(01)88327-7
Return to citation in text: [1] -
Armstrong, A.; Blench, T. J. Tetrahedron 2002, 58, 9321–9349. doi:10.1016/s0040-4020(02)00993-6
Return to citation in text: [1] -
Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Johnson, J. S. J. Am. Chem. Soc. 2008, 130, 17281–17283. doi:10.1021/ja808347q
Return to citation in text: [1] -
Wang, Y.; Metz, P. Chem. – Eur. J. 2011, 17, 3335–3337. doi:10.1002/chem.201003399
Return to citation in text: [1] -
Kawamata, T.; Nagatomo, M.; Inoue, M. J. Am. Chem. Soc. 2017, 139, 1814–1817. doi:10.1021/jacs.6b13263
Return to citation in text: [1] -
Liu, C.-I.; Jeng, W.-Y.; Chang, W.-J.; Ko, T.-P.; Wang, A. H.-J. J. Biol. Chem. 2012, 287, 18750–18757. doi:10.1074/jbc.m112.351254
Return to citation in text: [1] -
Guan, Z.; Wen, R.; Lam, B. I. Compositions and Methods for the Diagnosis and Treatment of Dolichol Deficiency Related Disorders. PCT Int. Appl. WO2014138586 A1, Sept 12, 2014.
Return to citation in text: [1] -
Gabriel, H. B.; Silva, M. F.; Kimura, E. A.; Wunderlich, G.; Katzin, A. M.; Azevedo, M. F. Antimicrob. Agents Chemother. 2015, 59, 3180–3188. doi:10.1128/aac.04500-14
Return to citation in text: [1] -
Saito, K.; Shirasago, Y.; Suzuki, T.; Aizaki, H.; Hanada, K.; Wakita, T.; Nishijima, M.; Fukasawa, M. J. Virol. 2015, 89, 2220–2232. doi:10.1128/jvi.03385-14
Return to citation in text: [1] -
Lanterna, C.; Musumeci, A.; Raccosta, L.; Corna, G.; Moresco, M.; Maggioni, D.; Fontana, R.; Doglioni, C.; Bordignon, C.; Traversari, C.; Russo, V. Cancer Immunol. Immunother. 2016, 65, 1303–1315. doi:10.1007/s00262-016-1884-8
Return to citation in text: [1] -
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d
Return to citation in text: [1] [2] [3] [4] [5] -
Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Hodgson, D. M.; Labande, A. H.; Muthusamy, S. Org. React. 2013, 80, 133–496. doi:10.1002/0471264180.or080.02
Return to citation in text: [1] -
Wang, F.; Liu, X.; Zhang, Y.; Lin, L.; Feng, X. Chem. Commun. 2009, 7297–7299. doi:10.1039/b913520k
Return to citation in text: [1] -
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] -
Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Regitz, M.; Maas, G. The Bamford-Stevens Reaction. Diazo Compounds: Properties and Synthesis; Academic Press: London, 1986; pp 257–295. doi:10.1016/b978-0-12-585840-3.50014-3
Return to citation in text: [1] -
Fegheh-Hassanpour, Y.; Ebrahim, F.; Arif, T.; Sintim, H. O.; Claridge, T. D. W.; Amin, N. T.; Hodgson, D. M. Org. Biomol. Chem. 2018, 16, 2876–2884. doi:10.1039/c8ob00435h
Return to citation in text: [1] -
Gawronski, J.; Gawronska, K. Tartaric and Malic Acids in Synthesis; Wiley: New York, 1999.
Return to citation in text: [1] -
Ghosh, A. K.; Koltun, E. S.; Bilcer, G. Synthesis 2001, 1281–1301. doi:10.1055/s-2001-15217
Return to citation in text: [1] -
Ladner, W. Angew. Chem. 1982, 94, 459–460. doi:10.1002/ange.19820940623
Angew. Chem., Int. Ed. Engl. 1982, 21, 459–460. doi:10.1002/anie.198204492
Return to citation in text: [1] [2] -
Mukhopadhyay, T.; Seebach, D. Helv. Chim. Acta 1982, 65, 385–391. doi:10.1002/hlca.19820650141
Return to citation in text: [1] -
Nie, X.; Wang, Q.; Li, Y.; Pan, X. Gaodeng Xuexiao Huaxue Xuebao 1987, 8, 620–622.
Return to citation in text: [1] [2] -
Tokunaga, Y.; Nagano, H.; Shiota, M. J. Chem. Soc., Perkin Trans. 1 1986, 581–584. doi:10.1039/p19860000581
Return to citation in text: [1] [2] -
Zhang, G.-L.; Li, S.-B.; Li, Y.-L. Acta Chim. Sin. (Chin. Ed.) 1989, 47, 1087–1092.
Return to citation in text: [1] [2] [3] [4] -
Molander, G. A.; Harris, C. R. J. Am. Chem. Soc. 1996, 118, 4059–4071. doi:10.1021/ja952619k
Return to citation in text: [1] [2] -
Kelly, T. R.; Cai, X.; Tu, B.; Elliott, E. L.; Grossmann, G.; Laurent, P. Org. Lett. 2004, 6, 4953–4956. doi:10.1021/ol047922h
Return to citation in text: [1] -
Evans, D. A.; Trotter, B. W.; Barrow, J. C. Tetrahedron 1997, 53, 8779–8794. doi:10.1016/s0040-4020(97)90390-2
Return to citation in text: [1] -
Vedejs, E.; Engler, D. A.; Telschow, J. E. J. Org. Chem. 1978, 43, 188–196. doi:10.1021/jo00396a002
Return to citation in text: [1] -
Vedejs, E.; Larsen, S. Org. Synth., Coll. Vol. VII 1990, 277–282.
Return to citation in text: [1] -
Jarosz, S.; Skóra, S.; Szewczyk, K. Tetrahedron: Asymmetry 2000, 11, 1997–2006. doi:10.1016/s0957-4166(00)00135-x
Return to citation in text: [1] -
House, H. O.; Blankley, C. J. J. Org. Chem. 1968, 33, 53–60. doi:10.1021/jo01265a011
Return to citation in text: [1] -
Blankley, C. J.; Sauter, F. J.; House, H. O. Org. Synth., Coll. Vol. V 1973, 258–263.
Return to citation in text: [1] -
Crouch, R. D. Tetrahedron 2013, 69, 2383–2417. doi:10.1016/j.tet.2013.01.017
Return to citation in text: [1] -
Wuts, P. G. M., Ed. Greene's Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2014; pp 257–262. doi:10.1002/9781118905074
Return to citation in text: [1] -
Villalonga–Barber, C. Synthetic studies towards the zaragozic acids. Ph.D. Thesis, University of Oxford, Oxford, U.K., 2001.
Return to citation in text: [1] -
Higashibayashi, S.; Shinko, K.; Ishizu, T.; Hashimoto, K.; Shirahama, H.; Nakata, M. Synlett 2000, 1306–1308. doi:10.1055/s-2000-7158
Return to citation in text: [1] [2] -
Shekhani, M. S.; Khan, K. M.; Mahmood, K.; Mozzam Shah, P.; Malik, S. Tetrahedron Lett. 1990, 31, 1669–1670. doi:10.1016/s0040-4039(00)88849-8
Return to citation in text: [1] -
Ramamoorthy, G.; Acevedo, C. M.; Alvira, E.; Lipton, M. A. Tetrahedron: Asymmetry 2008, 19, 2546–2554. doi:10.1016/j.tetasy.2008.09.031
Return to citation in text: [1] -
Crich, D.; Hao, X. J. Org. Chem. 1999, 64, 4016–4024. doi:10.1021/jo990046e
Return to citation in text: [1] -
Barros, M. T.; Burke, A. J.; Maycock, C. D. Tetrahedron Lett. 1999, 40, 1583–1586. doi:10.1016/s0040-4039(98)02650-1
Return to citation in text: [1] -
Barros, M. T.; Burke, A. J.; Lou, J.-D.; Maycock, C. D.; Wahnon, J. R. J. Org. Chem. 2004, 69, 7847–7850. doi:10.1021/jo049027h
Return to citation in text: [1] -
Burke, A. J.; Maycock, C. D.; Ventura, M. R. Org. Biomol. Chem. 2006, 4, 2361–2363. doi:10.1039/b606362b
Return to citation in text: [1] -
Zhu, J.; Yuan, Y.; Wang, S.; Yao, Z.-J. ACS Omega 2017, 2, 4665–4677. doi:10.1021/acsomega.7b00749
Return to citation in text: [1] -
Wang, N.; Song, J.; Jang, K. H.; Lee, H.-S.; Li, X.; Oh, K.-B.; Shin, J. J. Nat. Prod. 2008, 71, 551–557. doi:10.1021/np0780147
Return to citation in text: [1] -
Balansa, W.; Islam, R.; Fontaine, F.; Piggott, A. M.; Zhang, H.; Webb, T. I.; Gilbert, D. F.; Lynch, J. W.; Capon, R. J. Bioorg. Med. Chem. 2010, 18, 2912–2919. doi:10.1016/j.bmc.2010.03.002
Return to citation in text: [1] -
Miranda, V.; Maycock, C. D.; Ventura, M. R. Eur. J. Org. Chem. 2015, 7529–7533. doi:10.1002/ejoc.201501002
Return to citation in text: [1] -
Morita, H.; Arisaka, M.; Yoshida, N.; Kobayashi, J. Tetrahedron 2000, 56, 2929–2934. doi:10.1016/s0040-4020(00)00091-0
Return to citation in text: [1] [2]
| 30. | Kelly, T. R.; Cai, X.; Tu, B.; Elliott, E. L.; Grossmann, G.; Laurent, P. Org. Lett. 2004, 6, 4953–4956. doi:10.1021/ol047922h |
| 31. | Evans, D. A.; Trotter, B. W.; Barrow, J. C. Tetrahedron 1997, 53, 8779–8794. doi:10.1016/s0040-4020(97)90390-2 |
| 32. | Vedejs, E.; Engler, D. A.; Telschow, J. E. J. Org. Chem. 1978, 43, 188–196. doi:10.1021/jo00396a002 |
| 33. | Vedejs, E.; Larsen, S. Org. Synth., Coll. Vol. VII 1990, 277–282. |
| 40. | Higashibayashi, S.; Shinko, K.; Ishizu, T.; Hashimoto, K.; Shirahama, H.; Nakata, M. Synlett 2000, 1306–1308. doi:10.1055/s-2000-7158 |
| 41. | Shekhani, M. S.; Khan, K. M.; Mahmood, K.; Mozzam Shah, P.; Malik, S. Tetrahedron Lett. 1990, 31, 1669–1670. doi:10.1016/s0040-4039(00)88849-8 |
| 37. | Crouch, R. D. Tetrahedron 2013, 69, 2383–2417. doi:10.1016/j.tet.2013.01.017 |
| 38. | Wuts, P. G. M., Ed. Greene's Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2014; pp 257–262. doi:10.1002/9781118905074 |
| 39. | Villalonga–Barber, C. Synthetic studies towards the zaragozic acids. Ph.D. Thesis, University of Oxford, Oxford, U.K., 2001. |
| 27. | Tokunaga, Y.; Nagano, H.; Shiota, M. J. Chem. Soc., Perkin Trans. 1 1986, 581–584. doi:10.1039/p19860000581 |
| 35. | House, H. O.; Blankley, C. J. J. Org. Chem. 1968, 33, 53–60. doi:10.1021/jo01265a011 |
| 36. | Blankley, C. J.; Sauter, F. J.; House, H. O. Org. Synth., Coll. Vol. V 1973, 258–263. |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 34. | Jarosz, S.; Skóra, S.; Szewczyk, K. Tetrahedron: Asymmetry 2000, 11, 1997–2006. doi:10.1016/s0957-4166(00)00135-x |
| 40. | Higashibayashi, S.; Shinko, K.; Ishizu, T.; Hashimoto, K.; Shirahama, H.; Nakata, M. Synlett 2000, 1306–1308. doi:10.1055/s-2000-7158 |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 13. | Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d |
| 42. | Ramamoorthy, G.; Acevedo, C. M.; Alvira, E.; Lipton, M. A. Tetrahedron: Asymmetry 2008, 19, 2546–2554. doi:10.1016/j.tetasy.2008.09.031 |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 44. | Barros, M. T.; Burke, A. J.; Maycock, C. D. Tetrahedron Lett. 1999, 40, 1583–1586. doi:10.1016/s0040-4039(98)02650-1 |
| 45. | Barros, M. T.; Burke, A. J.; Lou, J.-D.; Maycock, C. D.; Wahnon, J. R. J. Org. Chem. 2004, 69, 7847–7850. doi:10.1021/jo049027h |
| 46. | Burke, A. J.; Maycock, C. D.; Ventura, M. R. Org. Biomol. Chem. 2006, 4, 2361–2363. doi:10.1039/b606362b |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 1. | Dawson, M. J.; Farthing, J. E.; Marshall, P. S.; Middleton, R. F.; O’Neill, M. J.; Shuttleworth, A.; Stylli, C.; Tait, R. M.; Taylor, P. M.; Wildman, H. G.; Buss, A. D.; Langley, D.; Hayes, M. V. J. Antibiot. 1992, 45, 639–647. doi:10.7164/antibiotics.45.639 |
| 2. | Dufresne, C.; Wilson, K. E.; Zink, D.; Smith, J.; Bergstrom, J. D.; Kurtz, M.; Rew, D.; Nallin, M.; Jenkins, R.; Bartizal, K.; Trainor, C.; Bills, G.; Meinz, M.; Huang, L.; Onishi, J.; Milligan, J.; Mojena, M.; Pelaez, F. Tetrahedron 1992, 48, 10221–10226. doi:10.1016/s0040-4020(01)88327-7 |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 15. | Hodgson, D. M.; Labande, A. H.; Muthusamy, S. Org. React. 2013, 80, 133–496. doi:10.1002/0471264180.or080.02 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 50. | Miranda, V.; Maycock, C. D.; Ventura, M. R. Eur. J. Org. Chem. 2015, 7529–7533. doi:10.1002/ejoc.201501002 |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 13. | Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d |
| 24. |
Ladner, W. Angew. Chem. 1982, 94, 459–460. doi:10.1002/ange.19820940623
Angew. Chem., Int. Ed. Engl. 1982, 21, 459–460. doi:10.1002/anie.198204492 |
| 7. | Liu, C.-I.; Jeng, W.-Y.; Chang, W.-J.; Ko, T.-P.; Wang, A. H.-J. J. Biol. Chem. 2012, 287, 18750–18757. doi:10.1074/jbc.m112.351254 |
| 8. | Guan, Z.; Wen, R.; Lam, B. I. Compositions and Methods for the Diagnosis and Treatment of Dolichol Deficiency Related Disorders. PCT Int. Appl. WO2014138586 A1, Sept 12, 2014. |
| 9. | Gabriel, H. B.; Silva, M. F.; Kimura, E. A.; Wunderlich, G.; Katzin, A. M.; Azevedo, M. F. Antimicrob. Agents Chemother. 2015, 59, 3180–3188. doi:10.1128/aac.04500-14 |
| 10. | Saito, K.; Shirasago, Y.; Suzuki, T.; Aizaki, H.; Hanada, K.; Wakita, T.; Nishijima, M.; Fukasawa, M. J. Virol. 2015, 89, 2220–2232. doi:10.1128/jvi.03385-14 |
| 11. | Lanterna, C.; Musumeci, A.; Raccosta, L.; Corna, G.; Moresco, M.; Maggioni, D.; Fontana, R.; Doglioni, C.; Bordignon, C.; Traversari, C.; Russo, V. Cancer Immunol. Immunother. 2016, 65, 1303–1315. doi:10.1007/s00262-016-1884-8 |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 13. | Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d |
| 3. | Armstrong, A.; Blench, T. J. Tetrahedron 2002, 58, 9321–9349. doi:10.1016/s0040-4020(02)00993-6 |
| 4. | Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Johnson, J. S. J. Am. Chem. Soc. 2008, 130, 17281–17283. doi:10.1021/ja808347q |
| 5. | Wang, Y.; Metz, P. Chem. – Eur. J. 2011, 17, 3335–3337. doi:10.1002/chem.201003399 |
| 6. | Kawamata, T.; Nagatomo, M.; Inoue, M. J. Am. Chem. Soc. 2017, 139, 1814–1817. doi:10.1021/jacs.6b13263 |
| 22. | Gawronski, J.; Gawronska, K. Tartaric and Malic Acids in Synthesis; Wiley: New York, 1999. |
| 23. | Ghosh, A. K.; Koltun, E. S.; Bilcer, G. Synthesis 2001, 1281–1301. doi:10.1055/s-2001-15217 |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 13. | Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 21. | Fegheh-Hassanpour, Y.; Ebrahim, F.; Arif, T.; Sintim, H. O.; Claridge, T. D. W.; Amin, N. T.; Hodgson, D. M. Org. Biomol. Chem. 2018, 16, 2876–2884. doi:10.1039/c8ob00435h |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 13. | Almohseni, H. A. A.; Al Mamari, H. H.; Valade, A.; Sintim, H. O.; Hodgson, D. M. Chem. Commun. 2018, 54, 5354–5356. doi:10.1039/c8cc02690d |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 12. |
Fegheh-Hassanpour, Y.; Arif, T.; Sintim, H. O.; Al Mamari, H. H.; Hodgson, D. M. Org. Lett. 2017, 19, 3540–3543. doi:10.1021/acs.orglett.7b01513
Corrigendum: Org. Lett. 2018, 20, 5528. doi:10.1021/acs.orglett.8b02330 |
| 16. | Wang, F.; Liu, X.; Zhang, Y.; Lin, L.; Feng, X. Chem. Commun. 2009, 7297–7299. doi:10.1039/b913520k |
| 29. | Molander, G. A.; Harris, C. R. J. Am. Chem. Soc. 1996, 118, 4059–4071. doi:10.1021/ja952619k |
| 14. | Hodgson, D. M.; Villalonga-Barber, C.; Goodman, J. M.; Pellegrinet, S. C. Org. Biomol. Chem. 2010, 8, 3975–3984. doi:10.1039/c004496b |
| 20. | Regitz, M.; Maas, G. The Bamford-Stevens Reaction. Diazo Compounds: Properties and Synthesis; Academic Press: London, 1986; pp 257–295. doi:10.1016/b978-0-12-585840-3.50014-3 |
| 47. | Zhu, J.; Yuan, Y.; Wang, S.; Yao, Z.-J. ACS Omega 2017, 2, 4665–4677. doi:10.1021/acsomega.7b00749 |
| 48. | Wang, N.; Song, J.; Jang, K. H.; Lee, H.-S.; Li, X.; Oh, K.-B.; Shin, J. J. Nat. Prod. 2008, 71, 551–557. doi:10.1021/np0780147 |
| 49. | Balansa, W.; Islam, R.; Fontaine, F.; Piggott, A. M.; Zhang, H.; Webb, T. I.; Gilbert, D. F.; Lynch, J. W.; Capon, R. J. Bioorg. Med. Chem. 2010, 18, 2912–2919. doi:10.1016/j.bmc.2010.03.002 |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 24. |
Ladner, W. Angew. Chem. 1982, 94, 459–460. doi:10.1002/ange.19820940623
Angew. Chem., Int. Ed. Engl. 1982, 21, 459–460. doi:10.1002/anie.198204492 |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 26. | Nie, X.; Wang, Q.; Li, Y.; Pan, X. Gaodeng Xuexiao Huaxue Xuebao 1987, 8, 620–622. |
| 28. | Zhang, G.-L.; Li, S.-B.; Li, Y.-L. Acta Chim. Sin. (Chin. Ed.) 1989, 47, 1087–1092. |
| 51. | Morita, H.; Arisaka, M.; Yoshida, N.; Kobayashi, J. Tetrahedron 2000, 56, 2929–2934. doi:10.1016/s0040-4020(00)00091-0 |
| 51. | Morita, H.; Arisaka, M.; Yoshida, N.; Kobayashi, J. Tetrahedron 2000, 56, 2929–2934. doi:10.1016/s0040-4020(00)00091-0 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 29. | Molander, G. A.; Harris, C. R. J. Am. Chem. Soc. 1996, 118, 4059–4071. doi:10.1021/ja952619k |
| 27. | Tokunaga, Y.; Nagano, H.; Shiota, M. J. Chem. Soc., Perkin Trans. 1 1986, 581–584. doi:10.1039/p19860000581 |
| 28. | Zhang, G.-L.; Li, S.-B.; Li, Y.-L. Acta Chim. Sin. (Chin. Ed.) 1989, 47, 1087–1092. |
| 17. |
Naef, R.; Seebach, D. Angew. Chem. 1981, 93, 1113. doi:10.1002/ange.19810931239
Angew. Chem., Int. Ed. Engl. 1981, 20, 1030–1031. doi:10.1002/anie.198110301 |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 19. | Naef, R. Chiral enolates, Dissertation No. 7442, ETH Zürich, Switzerland, 1983. |
| 26. | Nie, X.; Wang, Q.; Li, Y.; Pan, X. Gaodeng Xuexiao Huaxue Xuebao 1987, 8, 620–622. |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 28. | Zhang, G.-L.; Li, S.-B.; Li, Y.-L. Acta Chim. Sin. (Chin. Ed.) 1989, 47, 1087–1092. |
| 18. | Seebach, D.; Aebi, J. D.; Gander-Coquoz, M.; Naef, R. Helv. Chim. Acta 1987, 70, 1194–1216. doi:10.1002/hlca.19870700426 |
| 25. | Mukhopadhyay, T.; Seebach, D. Helv. Chim. Acta 1982, 65, 385–391. doi:10.1002/hlca.19820650141 |
| 28. | Zhang, G.-L.; Li, S.-B.; Li, Y.-L. Acta Chim. Sin. (Chin. Ed.) 1989, 47, 1087–1092. |
© 2019 Sintim et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)