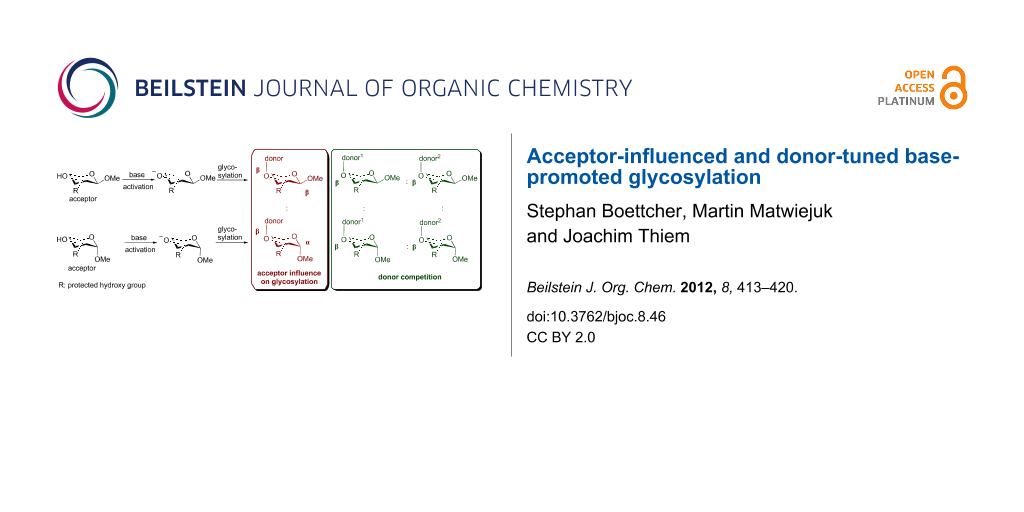
Abstract
Base-promoted glycosylation is a recently established stereoselective and regioselective approach for the assembly of di- and oligosaccharides by using partially protected acceptors and glycosyl halide donors. Initial studies were performed on partially methylated acceptor and donor moieties as a model system in order to analyze the key principles of oxyanion reactivities. In this work, extended studies on base-promoted glycosylation are presented by using benzyl protective groups in view of preparative applications. Emphases are placed on the influence of the acceptor anomeric configuration and donor reactivities.

Graphical Abstract
Introduction
For the assembly of oligosaccharides the complex and challenging control of regio- and stereochemistry has to be solved. Various contributions have facilitated enormously the access to complex oligosaccharide structures so far; however, regioselective and stereoselective glycosylations remain difficult and alternative concepts are welcome, and should be considered and studied.
Recently regioselective and β-stereospecific glycosylations employing saccharide oxyanions have been presented. Initial studies on base-promoted glycosylations were elaborated on partially methylated glucopyranosides as model systems. This systematic approach allowed determination of the preferred positions for glycosylation and the establishment of an oxyanion reactivity scale. In addition to studies with different base promoters and donors, the first successful conversions with benzylated acceptor and donor moieties were reported [1-3].
This novel glycosylation method could substantially reduce the need to apply protecting-group chemistry. Also, the β-specificity and base-promoted activation without any heavy-metal salt or Lewis acid are useful for synthetic applications. To develop the synthetic potential and evolve this method into an applicable alternative pathway towards di-, tri- or oligosaccharides, more research needs to be done.
So far, glycosylation reactions have been conducted by employing methyl α-glycopyranoside acceptors. Therefore, it was of particular interest to test the base-promoted glycosylation methodology with methyl β-glycopyranosides. Hence monobenzylated methyl α- and β-D-glycopyranosides were synthesized, then fucosylated by employing the base-promoted glycosylation methodology, and analysed with respect to the absolute and relative yields.
In addition, donor reactivity in base-promoted glycosylation reactions was investigated by employing partially benzylated methyl α- and β-D-glycopyranosides and donor halide mixtures.
Results and Discussion
To answer the questions above, monobenzylated methyl α- and β-D-gluco- and galactopyranosides 1–6 were selected as acceptor moieties (Figure 1) and synthesized by employing standard protecting-group chemistry. After base activation of the acceptor hydroxy groups, glycosylation was performed with the perbenzylated glycopyranosyl chlorides 7–9 (Figure 2).
![[1860-5397-8-46-1]](/bjoc/content/figures/1860-5397-8-46-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Monobenzylated methyl α- and β-D-gluco- and galactopyranoside acceptors 1–6.
Figure 1: Monobenzylated methyl α- and β-D-gluco- and galactopyranoside acceptors 1–6.
![[1860-5397-8-46-2]](/bjoc/content/figures/1860-5397-8-46-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Benzylated glycopyranosyl halide donors 7–9.
Figure 2: Benzylated glycopyranosyl halide donors 7–9.
The synthesis of the monobenzylated glucopyranosyl acceptors 1–4 started with benzylidenation [4] of compound 10 and 11, respectively (Scheme 1). As the next step, monobenzylation of 12 and 13 by phase-transfer catalysis [5] afforded derivatives 14–17. Subsequent cleavage of the benzylidene protecting group [4] gave the target compounds 1–4 in yields of up to 95%.
![[1860-5397-8-46-i1]](/bjoc/content/inline/1860-5397-8-46-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of monobenzylated glucopyranosyl acceptors 1–4. Reagents and conditions: (a) Benzaldehyde dimethylacetal, camphorsulfonic acid, ACN, 80 °C, 0.5–19 h; (b) NaOH, Bu4NHSO4, DCM, BnBr, 14–17 h, reflux; (c) MeOH, H2O, HCl, 2–22 h, reflux.
Scheme 1: Synthesis of monobenzylated glucopyranosyl acceptors 1–4. Reagents and conditions: (a) Benzaldehyde...
Preparation of the 3-O-monobenzylated methyl α- and β-D-galactopyranosyl acceptors 5 and 6 was achieved via stannylidene acetals [6] (Scheme 2). Compound 18 was subjected to a one-step synthesis with Bu2SnO and BnBr, affording target compound 5. The synthetic route for the methyl β-glycopyranoside acceptor 6 started with benzylidenation [4] of 19, followed by monobenzylation via intermediate stannylidene acetal formation [6] and subsequent cleavage of the benzylidene group [4].
![[1860-5397-8-46-i2]](/bjoc/content/inline/1860-5397-8-46-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 3-O-monobenzylated gluco- und galactopyranosyl acceptors 5 and 6. Reagents and conditions: (a) Benzaldehyde dimethylacetal, camphorsulfonic acid, ACN, 80 °C, 2.5 h; (b) dibutyltin oxide, toluene, BnBr, 24 h, reflux; (c) MeOH, H2O, HCl, 2–22 h, reflux.
Scheme 2: Synthesis of 3-O-monobenzylated gluco- und galactopyranosyl acceptors 5 and 6. Reagents and conditi...
Formation of the perbenzylated α-fucopyranosyl chloride was achieved according to [2]. Starting with peracetylated derivatives 22 and 23, a five-step synthesis was performed in each case to obtain the galacto- and glucopyranosyl donors 8 and 9 (Scheme 3). Initially, peracetates 22 and 23 were converted into the thioglycopyranosides 24 and 25 with BF3∙Et2O/thiophenole [7]. Subsequent deacetylation [8] and benzylation [9] afforded compounds 28 and 29, whose thioglycosidic bonds were cleaved with NBS in water/acetone [10]. Finally, treatment with oxalyl chloride [11] gave the α-glycopyranosyl chlorides 8 and 9 in high yields.
![[1860-5397-8-46-i3]](/bjoc/content/inline/1860-5397-8-46-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl chloride (8) and 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl chloride (9). Reagents and conditions: (a) 1. DCM, PhSH, BF3·Et2O, 0 °C, 1 h; 2. 4–5 d, rt; (b) MeOH, NaOMe, Amberlite IR 120 (H+), rt, 4–15 h; (c) 1. DMF, NaH, 0 °C, 1–2 h; 2. BnBr, 0 °C → rt, 16–40 h; (d) acetone/H2O (10:1), NBS, rt, −45 °C, 10 min to 5 h; (e) DCM, DMF, oxalyl chloride, 1 h, rt.
Scheme 3: Synthesis of 2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl chloride (8) and 2,3,4,6-tetra-O-benzyl-α-...
The results of the base-promoted glycosylation of monobenzylated methyl α- and β-D-glycopyranosides with fucopyranosyl chloride 7 are summarized in Table 1. Fucosylation of the 2-O-benzylated α-derivative 1 gave predominantly the β-(1→6)-linked disaccharide 33. Conversion of the 3-O-benzylated methyl α-D-glucopyranoside 3 resulted in β-(1→2) 34 and β-(1→6) disaccharides 35 in a ratio of ca. 1:9. By glycosylation of the 3-O-benzylated methyl α-D-galactopyranoside 5 with 7 only the β-(1→6)-linked disaccharide 37 was found. Thus, in all α-methyl glycopyranoside acceptors 1, 3 and 5, successful glycosidic bond formations with high regioselectivity towards position 6 were observed. These findings can be understood by the presence of adjacent hydroxy groups on acceptors 1, 3 and 5, such as 4,6-diol and/or 3,4,6-triol structures, respectively. After deprotonation the negative charge is dispersed by hydrogen bonding of unreacted hydroxy groups and located predominantly at position 6 due to the higher acidity of OH-6 [3]. Additionally, the basicity of the oxyanions stabilized by hydrogen bonding is decreased in comparison to isolated oxyanions, and this favours glycosidic bond formation over elimination.
Table 1: Glycosylation results for acceptors 1–6 with NaH as base and 2,3,4-tri-O-methyl-α-L-fucopyranosyl chloride (Fuc-Cl, 7) as donor.a
| Acceptor | Products | Yield [%]b | Relative yield [%]c | |||
|---|---|---|---|---|---|---|
| β-(1→2) | β-(1→3) | β-(1→4) | β-(1→6) | |||
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-46-i4.svg?max-width=637&scale=1.0)
1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-46-i5.svg?max-width=637&scale=1.0)
|
25 | ■d | – | 40 | 60 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-46-i6.svg?max-width=637&scale=1.0)
2 |
no reaction | – | ■ | – | – | – |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-46-i7.svg?max-width=637&scale=1.0)
3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-46-i8.svg?max-width=637&scale=1.0)
|
48e | 13 | ■ | – | 87 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-46-i9.svg?max-width=637&scale=1.0)
4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-46-i10.svg?max-width=637&scale=1.0)
36 |
30 | – | ■ | – | >98 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-46-i11.svg?max-width=637&scale=1.0)
5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-46-i12.svg?max-width=637&scale=1.0)
37 |
33 | – | ■ | – | >98 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-46-i13.svg?max-width=637&scale=1.0)
6 |
no reaction | – | – | ■ | – | – |
aGeneral reaction conditions: NaH (1.1–1.3 equiv), DMF, rt, 1 h, then donor 7 (1.0 equiv), rt, 4 h to 7 d; btotal yield of possible disaccharide regioisomers; cafter separation by column chromatography; d■: benzylated position, no linkage possible; e [1].
Surprisingly glycosylation of the corresponding β-glucopyranosides 2, 4, and 6 only showed conversion for the 3-O-benzylated glucopyranosyl derivative 4. Acceptors 2 and 6 did not react at all with the fucopyranosyl chloride 7. Obviously, the anomeric configuration in the acceptor structure has a noticeable effect on the glycosylation, resulting in lower oxyanion reactivity for methyl β-glycopyranosides. The considerable effect of the anomeric configuration on the reactivity of remote hydroxy groups may be rationalized by stereoelectronic effects [12]. Moreover, the lower reactivities of the methyl β-glycopyranosides may also be dependent on the decreased solubility of the partially deprotonated methyl β-glycospyranosides in the solvent used.
The results of base-promoted glycosylations with donor mixtures are shown in Table 2. Reaction of the 3-O-benzylated α-D-glucopyranoside 3 with an equimolar mixture of the fuco- and galactopyranosyl donors 7 and 8 gave both β-(1→6)-linked disaccharides 35 and 38 in 20% yield, with a ratio of 85% in favour of the fucosylated product. The corresponding β-configured acceptor 4 yielded only the β-(1→6)-linked galactosylated disaccharide 39 in poor yield (5%). With the same acceptors, equimolar mixtures of the perbenzylated fuco- and glucopyranosyldonors 7 and 9 gave just the β-(1→6)-fucosylated products 35 and 36 in corresponding yields of 15% and 14%. Furthermore, a mixture of galacto- and glucopyranosyl donors 8 and 9 was employed, but this did not show any reaction at all. In all cases, disaccharide yields from donor mixtures were significantly lower; as rational of which, some unknown kind of interaction between the donors may be assumed.
Table 2: Reactivity studies of acceptors 3 and 4 with donors 7–9.a
| Acceptor | Donor (equimolar mixture) | Products | Yield [%]b | Ratio [%] |
|---|---|---|---|---|
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-46-i14.svg?max-width=637&scale=1.0)
3 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-46-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-46-i16.svg?max-width=637&scale=1.0)
|
20 |
Gal/Fuc
15:85c |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-46-i17.svg?max-width=637&scale=1.0)
4 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-46-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-46-i19.svg?max-width=637&scale=1.0)
39 |
5 | Gal only |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-46-i20.svg?max-width=637&scale=1.0)
3 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-46-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-46-i22.svg?max-width=637&scale=1.0)
35 |
15 | Fuc only |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-46-i23.svg?max-width=637&scale=1.0)
4 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-46-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-46-i25.svg?max-width=637&scale=1.0)
36 |
14 | Fuc only |
aGeneral reaction conditions: NaH (1.1–1.3 equiv), DMF, rt, 1h, then donor 7 and 8 or 7 and 9 (1.0 equiv), rt, 4 h to 7 d; btotal yield of possible disaccharide regioisomers; cratio determined by 1H NMR.
Conclusion
These studies gave evidence that the configuration of the anomeric centre of acceptors has a decisive influence on the base-promoted glycosylation approach, but which so far cannot be explained. Several α-monobenzyl acceptors (1, 3 and 5) and their β counterparts (2, 4 and 6) were synthesized and coupled with the perbenzylated fucopyranosyl donor 7. Whereas all α-glycosidic acceptors gave disaccharides in moderate to acceptable yields, only the β-glycosidic acceptor 4 reacted, to give the dissacharide in much lower yield.
Furthermore, a comparison between the reactivities of several donor mixtures was established. For this purpose the perbenzylated donors 7–9 were synthesized and reacted in equimolar mixtures with acceptors 3 and 4. The fucopyranosyl donor 7 showed the highest reactivity, followed by the galactopyranosyl donor 8. The glucopyranosyl donor 9 showed no reaction at all. It turned out that the disaccharide yields from the mixtures were significantly lower, indicating unknown interactions between donors.
In cases with low yields or even no glycosylation products at all, elimination became the dominant reaction and led to the corresponding glycals. Hence, and based on the workup, the recovery of glycosyl halides was neither practicable nor achieved.
Experimental
General: All reagents were purchased from commercial sources and used as received. Sodium hydride (NaH) was used as a 60% suspension in paraffin. TLC was performed on Merck silica gel 60 F254 plates. Compounds were detected by UV and/or by treatment with EtOH/H2SO4 (9:1) and subsequent heating. Column chromatography was performed with Merck/Fluka silica gel 60 (230–400 mesh). Solvents for column chromatography were distilled prior to use. 1H and 13C NMR spectra were recorded with Bruker AMX-400 or Bruker AV-400 spectrometers (400 MHz for 1H, 100 MHz for 13C) and calibrated by using the solvent residual peak. In CDCl3 TMS was used for calibration. Melting points were measured with an Apotec melting point apparatus. Optical rotations were obtained by using a Krüss Optronic P8000 polarimeter (589 nm, 25 °C). HRMS (ESI) were recorded with a Thermo Finnigan MAT 95XL mass spectrometer. MS (MALDI–TOF) were recorded with a Bruker Biflex II (positive reflection mode, matrix: 2,5-dihydroxybenzoic acid). Relative yields of disaccharide mixtures were determined by integration of the signals in the 1H NMR spectra of the anomeric or other well-separated protons. The abbreviation “dd~vt” stands for a double doublet that turns into a virtual triplet because of similar coupling constants. Physical and NMR data for 34 and 35 were published previously [1].
General Procedure: Base-promoted glycosylations: The acceptor molecule (1.0 mmol) was dissolved in dry dimethylformamide (2–4 mL), and sodium hydride (1.1–1.3 mmol for each hydroxy group; as a 60% suspension in paraffin) was added. After being stirred for one hour under argon at room temperature, the donor (1.0 mmol for each hydroxy group; donor-mixtures 0.5 mmol of each donor) dissolved in dry dimethylformamide (2–4 mL) was successively added to the reaction. After stirring for 4–100 h, methanol (2 mL) was added. The crude product was purified by flash column chromatography (petroleum ether/ethyl acetate, 2:1 v/v).
Methyl 3-O-benzyl-β-D-glucopyranoside (4): Prepared by hydrolysis of 17 [5] with hydrochloric acid [2]. Compound 17 (3.49 g, 9.37 mmol), MeOH (80 mL), H2O (8.0 mL), 2 N HCl (1.0 mL), 21.5 h, reflux. Yield: 80% (2.13 g, 7.49 mmol), colourless solid, mp 105 °C. ![[Graphic 23]](/bjoc/content/inline/1860-5397-8-46-i26.svg?max-width=637&scale=1.18182) −7.7 (c 1.0, H2O); 1H NMR (400 MHz, MeOD) δ 7.47–7.43 (m, 2H, Har), 7.36–7.24 (m, 3H, Har), 4.92 (d, JC(3)OCH2Ph-A,B = 11.1 Hz, 1H, C(3)-O-CH2-Ph-A), 4.88 (d, 1H, C(3)-O-CH2-Ph-B), 4.21 (d, J1,2 = 7.3 Hz, 1H, H-1), 3.90 (dd, J6A,6B = 11.8 Hz, 1H, H-6A), 3.69 (dd, 1H, H-6B), 3.48–3.42 (m, 1H, H-3), 3.41–3.38 (m, 1H, H-4), 3.37–3.34 (m, 1H, H-2), 3.32–3.28 (m, J5,6A = 2.3 Hz, J5,6B = 5.8 Hz, 1H, H-5) ppm; 13C NMR (100 MHz, MeOD) δ 140.4, 129.2, 129.0, 128.5 (Car), 105.5 (C-1), 86.3 (C-4), 77.9 (C-5), 76.0 (C(3)-O-CH2-Ph), 75.3 (C-2), 71.5 (C-3), 62.7 (C-6), 57.3 (-O-CH3) ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C14H20O6Na, 307.1158; found, 307.1150.
−7.7 (c 1.0, H2O); 1H NMR (400 MHz, MeOD) δ 7.47–7.43 (m, 2H, Har), 7.36–7.24 (m, 3H, Har), 4.92 (d, JC(3)OCH2Ph-A,B = 11.1 Hz, 1H, C(3)-O-CH2-Ph-A), 4.88 (d, 1H, C(3)-O-CH2-Ph-B), 4.21 (d, J1,2 = 7.3 Hz, 1H, H-1), 3.90 (dd, J6A,6B = 11.8 Hz, 1H, H-6A), 3.69 (dd, 1H, H-6B), 3.48–3.42 (m, 1H, H-3), 3.41–3.38 (m, 1H, H-4), 3.37–3.34 (m, 1H, H-2), 3.32–3.28 (m, J5,6A = 2.3 Hz, J5,6B = 5.8 Hz, 1H, H-5) ppm; 13C NMR (100 MHz, MeOD) δ 140.4, 129.2, 129.0, 128.5 (Car), 105.5 (C-1), 86.3 (C-4), 77.9 (C-5), 76.0 (C(3)-O-CH2-Ph), 75.3 (C-2), 71.5 (C-3), 62.7 (C-6), 57.3 (-O-CH3) ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C14H20O6Na, 307.1158; found, 307.1150.
Methyl 2-O-benzyl-4-O-(2,3,4-tri-O-benzyl-β-L-fucopyranosyl)-α-D-glucopyranoside (32) and Methyl 2-O-benzyl-6-O-(2,3,4-tri-O-benzyl-β-L-fucopyranosyl)-α-D-glucopyranoside (33): Prepared according to the general procedure. Compound 1 (70 mg, 0.25 mmol), NaH (31 mg, 0.77 mmol), 7 (335 mg, 0.739 mmol), DMF (20 mL). Yield: 43 mg (0.061 mmol, 25%), yellow sirup, relative yield 32:33 = 40:60 (1H NMR).
32: 1H NMR (400 MHz, CDCl3) δ 7.31–7.19 (m, 20H, Har), 4.89 (d, 1H, C(4’)-CH2-Ph-A), 4.80 (d, JC(2’)OCH2Ph-A,B = 10.9 Hz, 1H, C(2’)-CH2-Ph-A), 4.77 (d, 1H, C(2’)-CH2-Ph-B), 4.70–4.53 (m, 5H, JC(4’)OCH2Ph-A,B = 11.6 Hz, C(2)-CH2-Ph-A/B, C(3’)-CH2-Ph-A/B, C(4’)-CH2-Ph-B), 4.59 (d, J1’,2’ = 7.8 Hz, 1H, H-1’), 4.50 (d, J1,2 = 3.8 Hz, 1H, H-1), 4.02–3.96 (m, 1H, H-3), 3.96–3.90 (m, 1H, H-6A), 3.74 (dd~vt, 1H, H-2’), 3.62–3.56 (m, 1H, H-6B), 3.53–3.50 (m, 2H, H-4, H-5), 3.48–3.46 (m, 1H, H-4’), 3.46–3.40 (m, J5’,6’ = 6.7 Hz, 2H, H-3’, H-5’), 3.28 (dd, J2,3 = 9.6 Hz, 1H, H-2), 3.27 (s, 3H, -OCH3), 1.10 (d, 3H, H-6’) ppm; 13C NMR (100 MHz, CDCl3) δ 128.4, 128.4, 128.3, 128.2, 128.2, 128.1, 127.9, 127.8, 127.7, 127.7, 127.6 (Car), 104.7 (C-1’), 98.4 (C-1), 82.4 (C-3’), 79.1 (C-2’), 78.9 (C-4), 78.5 (C-2), 76.2 (C-4’), 75.5 (C(2’)-O-CH2-Ph), 74.8 (C(4’)-O-CH2-Ph), 73.6 (C-3), 73.2 (C(3’)-O-CH2-Ph), 73.1 (C(2)-O-CH2-Ph), 70.9 (C-5’), 69.6 (C-5), 61.8 (C-6), 55.2 (-OCH3), 16.7 (C-6’) ppm.
33: 1H NMR (400 MHz, CDCl3) δ 7.33–7.19 (m, 20H, Har), 4.89 (d, 1H, C(4’)-CH2-Ph-A), 4.85 (d, 1H, C(2’)-CH2-Ph-A), 4.75–4.53 (m, JC(2’)OCH2Ph-A,B = 10.9 Hz, JC(4’)OCH2Ph-A,B = 11.6 Hz, 6H, C(2’)-CH2-Ph-B, C(2)-CH2-Ph-A/B, C(3’)-CH2-Ph-A/B, C(4’)-CH2-Ph-B), 4.55 (d, J1,2 = 3.8 Hz, 1H, H-1), 4.28 (d, J1’,2’ = 7.8 Hz, 1H, H-1’), 4.07–3.99 (m, 1H, H-6A/B), 3.88–3.85 (m, 1H, H-3), 3.76–3.70 (m, 1H, H-2’, H-5), 3.62–3.59 (m, 1H, H-4), 3.47–3.45 (m, 1H, H-4’), 3.44–3.39 (m, J5’,6 = 6.3 Hz, 2H, H-3’, H-5’), 3.27–3.24 (m, 1H, H-2), 3.23 (s, 1H, -OCH3), 1.10 (d, 1H, H-6’) ppm; 13C NMR (100 MHz, CDCl3) δ 138.8, 138.4, 137.8, 128.5, 128.5, 128.4, 128.2, 128.0, 127.9, 127.7, 127.6, 127.6, 127.5 (Car), 104.2 (C-1’), 97.9 (C-1), 82.3 (C-3’), 79.4 (C-2’), 79.1 (C-2), 79.1 (C-5), 76.3 (C-4’), 75.0, 74.7, 73.2, 73.0 (-O-CH2-Ph), 72.6 (C-3), 70.6 (C-5’), 70.1 (C-4), 68.8 (C-6), 55.2 (-OCH3), 16.7 (C-6’) ppm; MALDI–TOF–MS (DHB, positive mode; m/z): [M + Na]+ calcd for C41H48O10Na, 723.3; found, 723.5.
Methyl 3-O-benzyl-6-O-(2,3,4-tri-O-benzyl-β-L-fucopyranosyl)-β-D-glucopyranoside (36): (a) Prepared according to the general procedure: Compound 4 (71 mg, 0.25 mmol), NaH (31 mg, 0.71 mmol), 7 (350 mg, 0.774 mmol), DMF (16 mL). Yield: 53 mg (0.074 mmol, 30%). (b) Prepared according to the general procedure by using a donor mixture: Compound 4 (65 mg, 0.23 mmol), NaH (30 mg, 0.75 mmol), 7 (150 mg, 0.330 mmol)/9 (182 mg, 0.325 mmol), DMF (18 mL). Yield: 22 mg (0.031 mmol, 14%), yellow sirup. −106 (c 0.31, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42–7.27 (m, 20H, Har), 4.99 (d, JC(4’)OCH2Ph-A,B = 11.6 Hz, 1H, C(4’)-O-CH2-Ph-A), 4.93 (d, JC(2’)OCH2Ph-A,B = 10.7 Hz, 1H, C(2’)-O-CH2-Ph-A), 4.91 (d, JC(3)OCH2Ph-A,B = 11.3 Hz, 1H, C(3)-O-CH2-Ph-A), 4.86 (d, 1H, C(3)-O-CH2-Ph-B), 4.79 (d, JC(3’)OCH2Ph-A,B = 11.7 Hz, 1H, C(3’)-O-CH2-Ph-A), 4.76 (d, 1H, C(2’)-O-CH2-Ph-B), 4.72 (d, 1H, C(3’)-O-CH2-Ph-B), 4.69 (d, 1H, C(4’)-O-CH2-Ph-B), 4.36 (d, J1’,2’ = 7.5 Hz, 1H, H-1’), 4.19 (d, J1,2 = 7.5 Hz, 1H, H-1), 4.15 (dd, J6A,6B = 11.0 Hz, 1H, H-6A), 3.86–3.79 (m, 3H, H-2’, H-4, H-6B), 3.57–3.38 (m, J5,6A = 4.1 Hz, J5’,6A = 6.6 Hz, 6H, H-2, H-3, H-3’, H-4’, H-5, H-5’), 3.51 (s, 1H, -OCH3), 1.17 (d, 3H, H-6’) ppm; 13C NMR (100 MHz, CDCl3) δ 138.7, 138.6, 138.5, 138.5, 138.4, 128.7, 128.5, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.9, 127.8, 127.6 (Car), 104.0 (C-1’), 103.7 (C-1), 83.5 (C-3) 82.4 (C-3’), 79.0 (C-2’), 76.2 (C-4’), 75.2, 74.7, 74.6, 73.2 (-O-CH2-Ph), 74.6 (C-5), 73.9 (C-2), 70.8 (C-4), 70.6 (C-5’), 69.1 (C-6), 56.9 (-OCH3), 16.7 (-CH3) ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C41H48O10Na, 723.3145; found, 723.3136.
Methyl 3-O-benzyl-6-O-(2,3,4-tri-O-benzyl-β-L-fucopyranosyl)-β-D-galactopyranoside (37): Prepared according to the general procedure. Compound 5 (72 mg, 0.25 mmol), NaH (26 mg, 0.65 mmol), 7 (343 mg, 0.731 mmol), DMF (10 mL). Yield: 58 mg (0.083 mmol, 33%), colourless solid, mp 98 °C. +72 (c 0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42–7.25 (m, 20H, Har), 4.99 (d, 1H, -O-CH2-Ph), 4.90 (d, 1H, -O-CH2-Ph), 4.82–4.75 (m, J1,2 = 4.0 Hz, 3H, H-1, -O-CH2-Ph), 4.73 (d, 1H, -O-CH2-Ph), 4.71 (d, 1H, -O-CH2-Ph), 4.56 (s, 2H, -O-CH2-Ph), 4.40 (d, J1’,2’ = 7.8 Hz, 1H, H-1’), 4.16–4.13 (m, 1H, H-4), 4.01 (dd, J6A,6B = 8.8 Hz, 1H, H-6A), 3.98 (dd, J2,3 = 9.8 Hz, 1H, H-2), 3.93–3.89 (m, J5,6A = 4.3 Hz, J5,6B = 8.6 Hz, 1H, H-5), 3.86 (dd, 1H, H-6B), 3.82 (dd, J2’,3’ = 9.5 Hz, 1H, H-2’), 3.62–3.55 (m, 2H, H-3, H-4’), 3.53 (dd, J3’,4’ = 3.0 Hz, 1H, H-3’), 3.51–3.44 (m, J5’,6’ = 6.5 Hz, 1H, H-5’), 3.39 (s, 3H, OCH3), 1.19 (d, 3H, H-6’) ppm; 13C NMR (100 MHz, CDCl3) δ 128.6, 128.5, 128.4, 128.3, 127.9, 127.8, 127.6, 127.6, 127.6 (Car), 103.9 (C-1’), 99.5 (C-1), 82.5 (C-3’), 79.4 (C-2’), 78.4 (C-3), 76.2 (C-4’), 75.1, 74.6, 73.2, 71.7 (-O-CH2-Ph), 70.5 (C-5’), 68.6 (C-2), 68.1 (C-5), 67.3 (C-6), 66.0 (C-4), 55.4 (OCH3), 16.8 (C-6’) ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C41H48O10Na, 723.3135; found, 723.3140.
Methyl 3-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl)-α-D-glucopyranoside (38): (a) Prepared according to the general procedure: Compound 3 (70 mg, 0.25 mmol), NaH (32 mg, 0.80 mmol), 8 (415 mg, 0.742 mmol), DMF (20 mL). Yield: 20 mg (0.025 mmol, 10%). (b) Prepared according to the general procedure by using a donor mixture: Compound 3 (70 mg, 0.25 mmol), NaH (30 mg, 0.75 mmol), 7 (170 mg, 0.375 mmol)/8 (208 mg, 0.372 mmol), DMF (20 mL). Yield: 34 mg (0.042 mmol, 20%). Relative yield 35:38 = 85:15. Slightly yellow sirup. +53.4 (c 0.70, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.34–7.19 (m, 25H, Har), 4.89–4.84 (m, 3H, -O-CH2-Ph), 4.73–4.61 (m, 5H, H-1, -O-CH2-Ph), 4.52 (d, 1H, -O-CH2-Ph), 4.41–4.31 (m, J1’,2’ = 7.5 Hz, 3H, H-1’, -O-CH2-Ph), 4.09–4.01 (m, 1H, H-6A), 3.83–3.80 (m, 1H, H-4), 3.77 (dd~vt, 1H, H-2’), 3.73–3.66 (m, 2H, H-5’, H-6B), 3.56–3.42 (m, 7H, H-2, H-3, H-3’, H-4’, H-5, H-6’A/B), 3.29 (s, 1H, -OCH3) ppm; 13C NMR (100 MHz, CDCl3) δ 138.7, 138.7, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 127.5 (Car), 104.2 (C-1’), 99.3 (C-1), 82.7 (C-3), 82.3 (C-3’), 79.3 (C-2’), 75.2, 74.9, 74.5, 73.6, 73.0 (-O-CH2-Ph), 73.5 (C-4), 73.5 (C-4’), 72.6 (C-2), 70.8 (C-5), 70.2 (C-5’), 69.2 (C-6), 68.7 (C-6’), 55.3 (-OCH3) ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C48H54O11Na, 829.3564; found, 829.3545.
Methyl 3-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-β-D-galactopyranosyl)-β-D-glucopyranoside (39): Prepared according to the general procedure by using a donor mixture: Compound 4 (65 mg, 0.23 mmol), NaH (29 mg, 0.73 mmol), 7 (151 mg, 0.334 mmol)/8 (175 mg, 0.313 mmol), DMF (16 mL). Yield: 8.6 mg (0.011 mmol, 5%). Yellow sirup. −9.6 (c 0.40, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.31–7.19 (m, 25H, Har), 4.88–4.82 (m, 3H, -O-CH2-Ph), 4.74–4.69 (m, 2H, -O-CH2-Ph), 4.66–4.62 (m, 2H, -O-CH2-Ph), 4.52 (d, 1H, -O-CH2-Ph), 4.42–4.30 (m, 2H, -O-CH2-Ph), 4.37 (d, J1,2 = 3.5 Hz, 1H, H-1), 4.08–4.03 (m, J6A,6B = 10.8 Hz, 1H, H-6A), 4.06 (d, J1’,2’ = 7.6 Hz, 1H, H-1’), 3.82–3.80 (m, 1H, H-4), 3.78–3.74 (m, 1H, H-2), 3.74 (dd, 1H, H-6B), 3.56–3.42 (m, J5,6B = 6.0 Hz, 6H, H-3, H-4’, H-5, H-5’, H-6’A/B), 3.39 (dd, J2’,3’ = 9.8 Hz, 1H, H-2’), 3.36 (s, 1H, -OCH3), 3.31 (dd~vt, 1H, H-3’) ppm; 13C NMR (100 MHz, CDCl3) δ 138.6, 128.6, 128.4, 128.4, 128.3, 128.3, 128.2, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8, 127.6, 127.5 (Car), 104.0 (C-1), 103.6 (C-1’), 83.7 (C-3’), 82.2 (C-3), 79.1 (C-2), 75.2, 74.7, 74.5, 73.5, 72.9 (-O-CH2-Ph), 73.5 (C-4), 74.6 (C-5), 74.2 (C-2’), 73.4 (C-4), 71.4 (C-5’), 69.3 (C-6), 68.7 (C-6’), 57.1 (-OCH3) ppm; MALDI–TOF–MS (DHB, positive mode; m/z): [M + Na]+ calcd for C48H54O11Na, 829.36; found, 830.1.
References
-
Matwiejuk, M.; Thiem, J. Chem. Commun. 2011, 47, 8379–8381. doi:10.1039/c1cc11690h
Return to citation in text: [1] [2] [3] -
Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2011, 5860–5878. doi:10.1002/ejoc.201100861
Return to citation in text: [1] [2] [3] -
Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2012. doi:10.1002/ejoc.201101708
Return to citation in text: [1] [2] -
Demchenko, A. V.; Pornsuriyasak, P.; de Meo, C. J. Chem. Educ. 2006, 83, 782–784. doi:10.1021/ed083p782
Return to citation in text: [1] [2] [3] [4] -
Garegg, J.; Iversen, T.; Oscarson, S. Carbohydr. Res. 1976, 50, C12–C14. doi:10.1016/S0008-6215(00)83867-7
Return to citation in text: [1] [2] -
David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. doi:10.1016/S0040-4020(01)96443-9
Return to citation in text: [1] [2] -
Charbonnier, F.; Penadés, S. Eur. J. Org. Chem. 2004, 3650–3656. doi:10.1002/ejoc.200400141
Return to citation in text: [1] -
Zemplén, G.; Pacsu, E. Ber. Dtsch. Chem. Ges. 1929, 62, 1613–1614. doi:10.1002/cber.19290620640
Return to citation in text: [1] -
Girard, C.; Miramon, M.-L.; de Solminihac, T.; Herscovici, J. Carbohydr. Res. 2002, 337, 1769–1774. doi:10.1016/S0008-6215(02)00206-9
Return to citation in text: [1] -
Nicolaou, K. C.; Seitz, S. P.; Papahatjis, D. P. J. Am. Chem. Soc. 1983, 105, 2430–2434. doi:10.1021/ja00346a053
Return to citation in text: [1] -
Iversen, T.; Bundle, D. R. Carbohydr. Res. 1982, 103, 29–40. doi:10.1016/S0008-6215(82)80005-0
Return to citation in text: [1] -
Pedersen, C. M.; Olsen, J.; Brka, A. B.; Bols, M. Chem.–Eur. J. 2011, 17, 7080–7086. doi:10.1002/chem.201100020
Return to citation in text: [1]
| 5. | Garegg, J.; Iversen, T.; Oscarson, S. Carbohydr. Res. 1976, 50, C12–C14. doi:10.1016/S0008-6215(00)83867-7 |
| 12. | Pedersen, C. M.; Olsen, J.; Brka, A. B.; Bols, M. Chem.–Eur. J. 2011, 17, 7080–7086. doi:10.1002/chem.201100020 |
| 1. | Matwiejuk, M.; Thiem, J. Chem. Commun. 2011, 47, 8379–8381. doi:10.1039/c1cc11690h |
| 1. | Matwiejuk, M.; Thiem, J. Chem. Commun. 2011, 47, 8379–8381. doi:10.1039/c1cc11690h |
| 2. | Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2011, 5860–5878. doi:10.1002/ejoc.201100861 |
| 3. | Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2012. doi:10.1002/ejoc.201101708 |
| 6. | David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. doi:10.1016/S0040-4020(01)96443-9 |
| 4. | Demchenko, A. V.; Pornsuriyasak, P.; de Meo, C. J. Chem. Educ. 2006, 83, 782–784. doi:10.1021/ed083p782 |
| 1. | Matwiejuk, M.; Thiem, J. Chem. Commun. 2011, 47, 8379–8381. doi:10.1039/c1cc11690h |
| 5. | Garegg, J.; Iversen, T.; Oscarson, S. Carbohydr. Res. 1976, 50, C12–C14. doi:10.1016/S0008-6215(00)83867-7 |
| 10. | Nicolaou, K. C.; Seitz, S. P.; Papahatjis, D. P. J. Am. Chem. Soc. 1983, 105, 2430–2434. doi:10.1021/ja00346a053 |
| 4. | Demchenko, A. V.; Pornsuriyasak, P.; de Meo, C. J. Chem. Educ. 2006, 83, 782–784. doi:10.1021/ed083p782 |
| 11. | Iversen, T.; Bundle, D. R. Carbohydr. Res. 1982, 103, 29–40. doi:10.1016/S0008-6215(82)80005-0 |
| 2. | Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2011, 5860–5878. doi:10.1002/ejoc.201100861 |
| 8. | Zemplén, G.; Pacsu, E. Ber. Dtsch. Chem. Ges. 1929, 62, 1613–1614. doi:10.1002/cber.19290620640 |
| 4. | Demchenko, A. V.; Pornsuriyasak, P.; de Meo, C. J. Chem. Educ. 2006, 83, 782–784. doi:10.1021/ed083p782 |
| 9. | Girard, C.; Miramon, M.-L.; de Solminihac, T.; Herscovici, J. Carbohydr. Res. 2002, 337, 1769–1774. doi:10.1016/S0008-6215(02)00206-9 |
| 6. | David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. doi:10.1016/S0040-4020(01)96443-9 |
| 2. | Matwiejuk, M.; Thiem, J. Eur. J. Org. Chem. 2011, 5860–5878. doi:10.1002/ejoc.201100861 |
| 4. | Demchenko, A. V.; Pornsuriyasak, P.; de Meo, C. J. Chem. Educ. 2006, 83, 782–784. doi:10.1021/ed083p782 |
| 7. | Charbonnier, F.; Penadés, S. Eur. J. Org. Chem. 2004, 3650–3656. doi:10.1002/ejoc.200400141 |
© 2012 Boettcher et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)