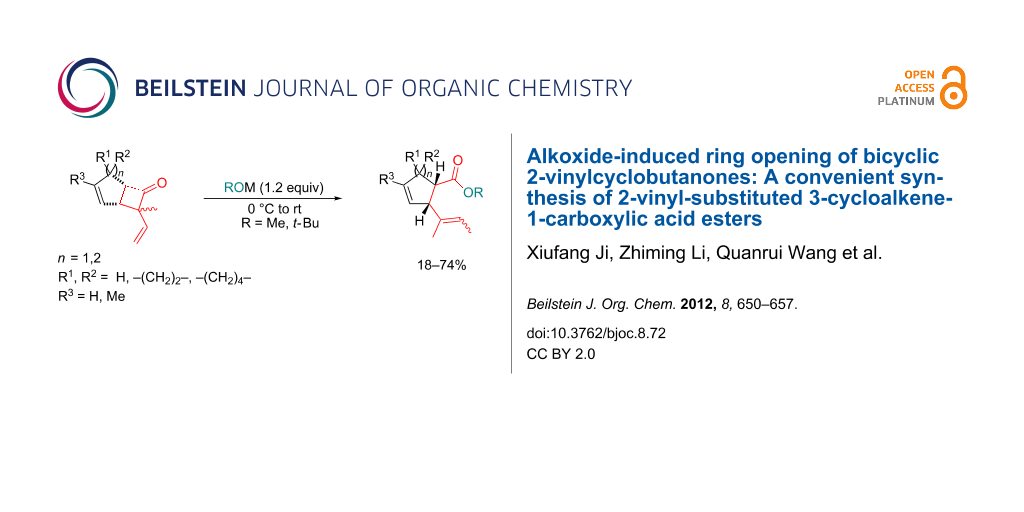
Abstract
The fused 2-vinyl or 2-phenyl substituted cyclobutanones 4 undergo stereoselective ring openings by the action of alkoxide ions (t-BuO− or MeO−) to produce novel vicinally disubstituted cycloalkene derivatives 5 and 6 in moderate to high yields. The ring cleavage usually occurs with complete regioselectivity. The accessibility of γ,δ-unsaturated ester or acid derivatives makes this transformation a good supplementary method for the well-established Johnson–Claisen rearrangement.

Graphical Abstract
Introduction
A great variety of methods are available for the synthesis of cyclobutane derivatives. Frequently employed ones include the thermal [2 + 2] cycloaddition of ketenes to alkenes and the polar addition of cyclopropyl ylides to carbonyls [1,2]. These methods generally allow regioselective as well as stereoselective syntheses of extensively substituted four-membered ring carbocycles. In particular, due to their facile accessibility [3-8] and expeditious ring transformations, the use of cyclobutanones as extremely versatile and useful starting materials or intermediates in the construction of carbon skeletons has flourished over the past three decades [9,10]. In addition, the high electrophilicity of the carbonyl carbon atom, and the puckering of the cyclobutane caused by the substitution at C-2 and C-4, also offer both interesting preparative and mechanistic aspects [11]. The ease of ring opening of cyclobutanones is influenced by the strain of the four-membered rings, the substitution pattern on the ring, and the properties of the reagents as well as the reaction conditions. In particular, the ring opening of certain fused-ring cyclobutanones, which are easily prepared by the well-established [2 + 2] cycloadditions of ketenes with various cycloalkenes, has been extensively investigated [3-8,12-14]. To achieve the ring opening, many reaction conditions have been investigated, such as acidic conditions, basic conditions, nucleophilic attack, thermolysis, and oxidizing as well as reducing conditions [4,11]. Cohen and Matz described the [1,2]- and [1,3]-acyl migration of 2-vinylcyclobutanones, leading to cyclopentenones or cyclohexenones, respectively [15]. Danheiser and co-workers reported an oxyanion-accelerated [1,3]-rearrangement of in situ reduced 2-vinylcyclobutanones, by applying mixtures of LiBu3BH-MeLi and HMPT to give bicyclic cyclohexenols [16], which were also subsequently described by Cohen who elaborated the method into a synthesis of (−)-β-selinene [17]. The core structures of ophiobolins A and other natural products can be synthesized by an alkenylation/oxy-Cope/methylation sequence of vinylcyclobutones in one pot [18-23]. Ring-opening reactions of cyclobutanones are expected to be a valuable methodology for vicinal difunctionalization of double bonds. A prerequisite for this reaction to take place is the presence at C-2 of substituents capable of stabilizing the developing vicinal carbanion [6-8]. In this context, α,α-dihalosubstituents, thioketal, sulfoxide, ester or aryl substituents have been reported to facilitate the ring cleavage [6-8].
We recently reported a novel metathetic cycloreversion of 7-vinyl and aryl bicyclo[3.2.0]hept-2-en-6-ones giving rise to linear polyene ketones. The reaction was triggered by an in situ alkylation with alkyl or aryllithium reagents in the presence of catalytic amounts of Li-chelating agents (e.g., Scheme 1) [24]. The configuration of the newly formed double bond in position 6 was diastereoselectively determined by the configuration of the bicyclic precursor.
![[1860-5397-8-72-i1]](/bjoc/content/inline/1860-5397-8-72-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Metathetic ring opening of 7-methyl-7-vinylbicyclo[3.2.0]hept-2-en-6-one to a linear polyene ketone.
Scheme 1: Metathetic ring opening of 7-methyl-7-vinylbicyclo[3.2.0]hept-2-en-6-one to a linear polyene ketone....
Our ongoing interest in constructing odorants from easily accessible starting materials prompted us to search further for novel transformations of cyclobutanones. We were pleased to find that selective ring cleavage of bicyclic cyclobutanones 4 bearing a vinyl or phenyl group in the alpha position can be achieved by the action of sodium methoxide or potassium tert-butoxide. This approach constitutes an efficient path to vicinally disubstituted ester products 5 or 6.
Results and Discussion
We began our investigation with the preparation of cyclobutanones as shown in Scheme 2. The reaction involves generation of the intermediate vinylketenes or phenylketenes by the triethylamine-promoted 1,4- or 1,2-dehydrochlorination of acyl chlorides 2 [1]. The in situ generated vinylketenes or phenylketenes 3 were trapped by reaction with excessive amount of alkenes 1 in methylene chloride. The reaction of ketenes (R2C=C=O) with alkenes under thermal conditions to give cyclobutanone products is generally described as an [π2s + π2a] process from orbital symmetry considerations. However, there is strong evidence that formation of the Staudinger-type cycloadducts by using cyclic (s-cis) 1,3-dienes proceeds by a two-step process, involving an initial hetero Diels–Alder cycloaddition of the diene and the ketene carbonyl group, followed by a [3,3]-sigmatropic (Claisen) rearrangement [25,26].
![[1860-5397-8-72-i2]](/bjoc/content/inline/1860-5397-8-72-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of vinyl or phenyl substituted cyclobutanones 4a–i.
Scheme 2: Synthesis of vinyl or phenyl substituted cyclobutanones 4a–i.
The most effective conditions were achieved by the employment of a slightly excessive amount of olefins 1. The cycloadditions proceeded with complete regio- and stereoselectivity with respect to the olefin 1, but less so with respect to the ketene 3 (Table 1), such that endo-4 and exo-4 isomers were formed in variable amounts, depending on the nature of the substrates. In general, the conversions of cyclopenta-1,3-diene 1a and the spiroheptadiene (1c) gave high yields of product 4 at 0 °C. However, the yields were moderate to low with cyclic mono olefin 1f or styrene (1g), even under reflux conditions. Mixtures of endo- and exo-isomers of compounds 4 were used in the subsequent ring-opening step without prior separation.
Table 1: Formal [2 + 2] cycloaddition of vinylketenes 2 with olefins.a
| entry | alkene 1 | acid chloride 2 | timeb (h) | product 4 | endo:exoc | yieldd (%) |
|---|---|---|---|---|---|---|
| 1 | 1a | 2a | 3 | 4a | 30:70 | 76 |
| 2 | 1b | 2a | 3 | 4b | 50:50 | 80 |
| 3 | 1c | 2a | 4 | 4c | 25:75 | 40 |
| 4 | 1d | 2a | 8 | 4d | 50:50 | 34 |
| 5 | 1e | 2a | 8 | 4e | 35:65 | 40 |
| 6e | 1f | 2a | 10 | 4f | 25:75 | 60 |
| 7e | 1g | 2a | 12 | 4g | 33:67f | 16 |
| 8 | 1a | 2b | 3 | 4h | 95:5 | 85 |
| 9 | 1e | 2b | 7 | 4i | >95:5 | 21 |
aReagents and conditions (cf. Scheme 2): alkene 1 (1.5 equiv), Et3N (1.2 equiv), acid chloride 2 (1.0 equiv), 0 °C. bTime required for the olefin to achieve maximal conversion. cthe exo/endo ratio was determined by 1H NMR spectroscopy. dIsolated yields after purification by column chromatography on silica gel. eUsing chloroform as solvent under reflux. fcis/trans ratio. Structures are shown in Scheme 2.
The configurations were determined from their 1H NMR spectra and mechanistic considerations [27]. For example, the C-7 methyl signal in the endo-bicyclo[3.2.0]heptenone systems was found in 1H NMR at a lower field (δ = 1.40 for endo-4a), while that for the exo-counterpart appeared at a higher field (δ = 1.10 for exo-4a). The configuration of compound 4g was discernible from 1H NMR spectra and confirmed by 2D NMR spectroscopy. Thus, the NOESY spectrum of trans-4g indicated a correlation between the signal at 6.05 ppm from the α-vinyl proton of the vinyl group and the triplet at 3.74 ppm from the C-3 proton (Figure 1).
![[1860-5397-8-72-1]](/bjoc/content/figures/1860-5397-8-72-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Determination of the structure of 3-phenyl-2-vinyl substituted cyclobutanone 4g.
Figure 1: Determination of the structure of 3-phenyl-2-vinyl substituted cyclobutanone 4g.
Taking the high degree of ring strain in substrates 4 into account, an alkoxide-promoted ring opening to vicinally functionalized products 5 or 6 was expected. To validate this proposal, the 7-methyl-7-vinyl substituted bicyclo[3.2.0]heptenone 4a was employed as a model substrate with sodium methoxide or potassium tert-butoxide as the base. Various reaction conditions, including the amount of metal alkoxide, solvents and reaction temperature were examined. Representative results are listed in Table 2.
Table 2: Cleavage reaction of 4a with metal alkoxide.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-72-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | alkoxide (equiv) | solvent | time (h) | product | E:Zb | yieldc (%) |
|---|---|---|---|---|---|---|
| 1 | NaOMe (1.2) | MeOH | 2 | 5a | 60:40 | 40 |
| 2 | NaOMe (0.010) | MeOH | 16 | 5a | 55:45 | <10 |
| 3 | t-BuOK (1.2) | THF | 2 | 6a | 9:1 | 46 |
| 4 | t-BuOK (0.010) | THF | 16 | 6a | 75:25 | <10 |
| 5d | t-BuOK (1.2) | THF | 0.5 | 6a | — | decomp. |
| 6 | t-BuOK (1.2) | t-BuOH | 24 | 6a | >99:1 | 30 |
| 7 | t-BuOK (0.010) | t-BuOH | 24 | 6a | 95:5 | <10 |
| 8 | t-BuOK (1.2)e | THF | 2 | 6a | 90:10 | 47 |
aAddition of 4a to the alkoxide was performed at 0 °C, then continued at rt. bE/Z ratio was determined by GC–MS prior to chromatography. cIsolated yield after column chromatography on silica gel. dUnder reflux. eUsing 0.05 equiv dibenzo-18-C-6 as a K-chelating agent.
It was found that both MeONa and t-BuOK can promote the ring opening to furnish the alkyl 2-substituted cyclopent-3-enecarboxylate 5a or 6a. Using 1.2 equiv of MeONa with methanol as solvent gave 5a in moderate isolated yield, after 2 h at room temperature, in form of an E/Z isomeric mixture about the 2-vinyl double bond, in a ratio of 6:4 (Table 2, entry 1). Reducing the amount of the base significantly decreased the yield. Using 0.01 equiv of MeONa afforded no more than 10% yield, even though the reaction time was prolonged to 16 h (Table 2, entry 2). The more bulky tert-BuOK was also a suitable base to trigger the ring opening, giving acceptable yields of product 6a. Not surprisingly, the use of tert-BuOK as base provided 6a with a higher diastereoselectivity of 9:1 in favour of the E-isomer. It seems that 1.2 equiv of the base had to be applied, and THF as solvent was superior to tert-butanol in terms of yield and reaction rate (Table 2, entry 3 versus 4, entry 3 versus 6). A detrimental effect of enhancing the temperature was observed. Thus, an attempt to accelerate the reaction led to the decomposition of the cyclobutanone 4a under reflux conditions, without the detection of any desired product 6a (Table 2, entry 5). In addition, the use of a K-chelating agent, i.e., dibenzo-18-crown-6, did not show any improvement of the yield of compound 6a (Table 2, entry 8).
In light of the above promising results, the substrate scope of this base-promoted ring-opening reaction was assessed by using a variety of cyclobutanones 4b–i (Scheme 3, Table 3). Diverse substitution patterns at both the ketene and the olefin moiety were compatible with this ring-opened method. Thus, cyclobutanones carrying both vinyl (Table 3, entries 1–14) and phenyl (Table 3, entries 15–18) substituents underwent the cleavage reaction. The monocyclic butanone 4g was also tolerated to give moderate yields (Table 3, entries 13 and 14). Another noticeable observation is that the use of t-BuOK furnished generally better yields than MeONa, except for compound 4d, which was obtained in a slightly higher yield in the presence of MeONa as the base (Table 3, entries 7 and 8).
![[1860-5397-8-72-i3]](/bjoc/content/inline/1860-5397-8-72-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Ring opening of cyclobutanones 4 to afford products 5 or 6.
Scheme 3: Ring opening of cyclobutanones 4 to afford products 5 or 6.
Table 3: Reaction of 4 with MeONa or t-BuOK.a
| entry | ketone 4 | ROM | time (h) | product | product label | E:Zb | yieldc (%) |
|---|---|---|---|---|---|---|---|
|
1
2 |
4a
4a |
MeONa
t-BuOK |
2
2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-72-i7.svg?max-width=637&scale=1.0)
|
5a
6a |
60:40
9:1 |
40
46 |
|
3
4 |
4b
4b |
MeONa
t-BuOK |
4
6 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-72-i8.svg?max-width=637&scale=1.0)
|
5b
6b |
50:50
95:5 |
41
50 |
|
5
6 |
4c
4c |
MeONa
t-BuOK |
4
16 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-72-i9.svg?max-width=637&scale=1.0)
|
5c
6c |
67:33
80:20 |
18
43 |
|
7
8 |
4d
4d |
MeONa
t-BuOK |
24
24 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-72-i10.svg?max-width=637&scale=1.0)
|
5d
6d |
60:40
>99:1 |
37
29 |
| 9 | 4e | MeONa | 14 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-72-i11.svg?max-width=637&scale=1.0)
|
5e | 55:45 | 41 |
| 10 | 4e | t-BuOK | 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-72-i12.svg?max-width=637&scale=1.0)
|
6e | >99:1 | 72 |
| 11 | 4f | MeONa | 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-72-i13.svg?max-width=637&scale=1.0)
|
5f | 60:40 | 51 |
| 12 | 4f | t-BuOK | 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-72-i14.svg?max-width=637&scale=1.0)
|
6f | - | 60 |
| 13 | 4g | MeONa | 2 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-72-i15.svg?max-width=637&scale=1.0)
|
5g | 60:40 | 45 |
| 14 | 4g | t-BuOK | 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-72-i16.svg?max-width=637&scale=1.0)
|
6g | >99:1 | 65 |
|
15
16 |
4h
4h |
MeONa
t-BuOK |
12
2 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-72-i17.svg?max-width=637&scale=1.0)
|
5h
6h |
-
- |
63
74 |
|
17
18 |
4i
4i |
MeONa
t-BuOK |
6
3 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-72-i18.svg?max-width=637&scale=1.0)
|
5i
6i |
-
- |
57
70 |
a1.2 equiv of base was employed. For MeONa methanol was the solvent, and for t-BuOK THF was the solvent. The progression was monitored by GC. bThe E/Z ratio was determined by GC-MS before chromatography. cIsolated yield after silica-gel column chromatography.
Surprisingly, the reaction of 4f with t-BuOK led to ester 6f (Table 3, entry 12) carrying an allylic instead of a vinylic side chain. In the case of the 2-phenyl substituted cyclobutanone 4g the reaction afforded a 65% yield of γ,δ-unsaturated acid product (E)-6g (Table 3, entry 14). Finally, an attempt using the sterically hindered base LDA only led to the formation of aldol adduct 7 (Scheme 4).
![[1860-5397-8-72-i4]](/bjoc/content/inline/1860-5397-8-72-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Although the mechanism is not fully understood, a tentative mechanistic rationale is depicted in Scheme 5. Due to the high ring strain of the cyclobutanones, the ring is very sensitive to the influence of nucleophiles. As exemplified by 4a, the reaction begins with the nucleophilic exo-facial attack on the carbonyl group by an alkoxide ion to trigger the cleavage of the four-membered ring. The presence of a vinyl group facilitates the ring fission since the resultant carbanion can be stabilized by resonance. Protonation leads to the formation of the isolated products 5a or 6a. When the hindered base t-BuOK is employed, the reaction occurs with a high preference for E-product formation. This may be a result of thermodynamic control.
![[1860-5397-8-72-i5]](/bjoc/content/inline/1860-5397-8-72-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism for ring opening of 4a.
Scheme 5: Plausible mechanism for ring opening of 4a.
Conclusion
In conclusion, our intention to synthesize potential olfactorily useful compounds led us to disclose the facial ring opening of a series of cyclobutanones to produce 1,2-disubstituted cyclopentene derivatives with high selectivity. The economic cycloaddition/ring-opening sequence is significant in that it allows a useful functional group to be easily introduced under mild conditions. Furthermore, the products have a general γ,δ-unsaturated carbonyl skeleton, and hence the protocol should be a good surrogate for the well-established Johnson orthoester Claisen rearrangement [28,29].
References
-
Lee-Ruff, E.; Mladenova, G. Chem. Rev. 2003, 103, 1449–1484. doi:10.1021/cr010013a
Return to citation in text: [1] [2] -
Lee-Ruff, E. Synthesis of Cyclobutanes. In The Chemistry of Cyclobutanes; Rappoport, Z.; Liebman, J. F., Eds.; John Wiley & Sons, Ltd.: West Sussex, UK, 2005; pp 281–356. doi:10.1002/0470864028.ch8
Return to citation in text: [1] -
Brady, W. T. Synthesis 1971, 415–422. doi:10.1055/s-1971-21750
Return to citation in text: [1] [2] -
Ali, S. M.; Lee, T. V.; Roberts, S. M. Synthesis 1977, 155–166. doi:10.1055/s-1977-24302
Return to citation in text: [1] [2] [3] -
Moore, H. W.; Gheorghiu, M. D. Chem. Soc. Rev. 1981, 10, 289–328. doi:10.1039/CS9811000289
Return to citation in text: [1] [2] -
Trost, B. M. Sulfuranes in organic reactions and synthesis. In New Concepts I; Davison, A.; Dewar, M. J. S., Eds.; Topics in Current Chemistry, Vol. 41; Springer: Berlin, Heidelberg, 1973; pp 1–29. doi:10.1007/3-540-06333-1_17
Return to citation in text: [1] [2] [3] [4] -
Conia, J. M.; Robson, M. J. Angew. Chem. 1975, 87, 505–516. doi:10.1002/ange.19750871404
Angew. Chem., Int. Ed. Engl. 1975, 14, 473–485. doi:10.1002/anie.197504731
Return to citation in text: [1] [2] [3] [4] -
Trost, B. M. Acc. Chem. Res. 1974, 7, 85–92. doi:10.1021/ar50075a004
Return to citation in text: [1] [2] [3] [4] -
Wong, H. N. C.; Lau, K.-L.; Tam, K.-F. The application of cyclobutane derivatives in organic synthesis. In Small Ring Compounds in Organic Synthesis I; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 133; Springer: Berlin, Heidelberg, 1986; pp 83–157. doi:10.1007/3-540-16307-7_2
Return to citation in text: [1] -
Moore, H. W.; Decker, O. H. W. Chem. Rev. 1986, 86, 821–830. doi:10.1021/cr00075a006
Return to citation in text: [1] -
Belluš, D.; Ernst, B. Angew. Chem. 1988, 100, 820–850. doi:10.1002/ange.19881000607
Angew. Chem., Int. Ed. Engl. 1988, 27, 797–827. doi:10.1002/anie.198807971
Return to citation in text: [1] [2] -
Baldwin, J. E.; Leber, P. A. Org. Biomol. Chem. 2008, 6, 36–47. doi:10.1039/B711494J
Return to citation in text: [1] -
Leber, P. A.; Baldwin, J. E. Acc. Chem. Res. 2002, 35, 279–287. doi:10.1021/ar010100p
Return to citation in text: [1] -
Bronson, J. J.; Danheiser, R. L. In Comprehensive Organic Synthesis, 1st ed.; Trost, B. M.; Flemming, I.; Paquette, L. A., Eds.; Pergamon Press: Oxford, 1991; pp 1016 ff.
Return to citation in text: [1] -
Matz, J. R.; Cohen, T. Tetrahedron Lett. 1981, 22, 2459–2462. doi:10.1016/S0040-4039(01)92932-6
Return to citation in text: [1] -
Danheiser, R. L.; Martinez-Davila, C.; Sard, H. Tetrahedron 1981, 37, 3943–3950. doi:10.1016/S0040-4020(01)93268-5
Return to citation in text: [1] -
Cohen, T.; Bhupathy, M.; Matz, J. R. J. Am. Chem. Soc. 1983, 105, 520–525. doi:10.1021/ja00341a035
Return to citation in text: [1] -
Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1984, 67, 1506–1514. doi:10.1002/hlca.19840670614
Return to citation in text: [1] -
Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1982, 65, 451–461. doi:10.1002/hlca.19820650203
Return to citation in text: [1] -
Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1982, 65, 1563–1575. doi:10.1002/hlca.19820650530
Return to citation in text: [1] -
Danheiser, R. L.; Gee, S. K.; Sard, H. J. Am. Chem. Soc. 1982, 104, 7670–7672. doi:10.1021/ja00390a054
Return to citation in text: [1] -
Paquette, L. A.; Colapret, J. A.; Andrews, D. R. J. Org. Chem. 1985, 50, 201–205. doi:10.1021/jo00202a012
Return to citation in text: [1] -
Paquette, L. A.; Andrews, D. R.; Springer, J. P. J. Org. Chem. 1983, 48, 1147–1149. doi:10.1021/jo00155a059
Return to citation in text: [1] -
Ji, X.; Wang, Q.; Goeke, A. Chem. Commun. 2010, 46, 8845–8847. doi:10.1039/c0cc02694h
Return to citation in text: [1] -
Machiguchi, T.; Hasegawa, T.; Ishiwata, A.; Terashima, S.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 1999, 121, 4771–4786. doi:10.1021/ja990072u
Return to citation in text: [1] -
Ussing, B. R.; Hang, C.; Singleton, D. A. J. Am. Chem. Soc. 2006, 128, 7594–7607. doi:10.1021/ja0606024
Return to citation in text: [1] -
Rey, M.; Roberts, S.; Dieffenbacher, A.; Dreiding, A. S. Helv. Chim. Acta 1970, 53, 417–432. doi:10.1002/hlca.19700530223
Return to citation in text: [1] -
Johnson, W. S.; Werthemann, L.; Bartlett, W. R.; Brocksom, T. J.; Li, T.-T.; Faulkner, D. J.; Petersen, M. R. J. Am. Chem. Soc. 1970, 92, 741–743. doi:10.1021/ja00706a074
Return to citation in text: [1] -
Daub, G. W.; Edwards, J. P.; Okada, C. R.; Allen, J. W.; Maxey, C. T.; Wells, M. S.; Goldstein, A. S.; Dibley, M. J.; Wang, C. J.; Ostercamp, D. P.; Chung, S.; Cunningham, P. S.; Berliner, M. A. J. Org. Chem. 1997, 62, 1976–1985. doi:10.1021/jo9614250
Return to citation in text: [1]
| 28. | Johnson, W. S.; Werthemann, L.; Bartlett, W. R.; Brocksom, T. J.; Li, T.-T.; Faulkner, D. J.; Petersen, M. R. J. Am. Chem. Soc. 1970, 92, 741–743. doi:10.1021/ja00706a074 |
| 29. | Daub, G. W.; Edwards, J. P.; Okada, C. R.; Allen, J. W.; Maxey, C. T.; Wells, M. S.; Goldstein, A. S.; Dibley, M. J.; Wang, C. J.; Ostercamp, D. P.; Chung, S.; Cunningham, P. S.; Berliner, M. A. J. Org. Chem. 1997, 62, 1976–1985. doi:10.1021/jo9614250 |
| 1. | Lee-Ruff, E.; Mladenova, G. Chem. Rev. 2003, 103, 1449–1484. doi:10.1021/cr010013a |
| 2. | Lee-Ruff, E. Synthesis of Cyclobutanes. In The Chemistry of Cyclobutanes; Rappoport, Z.; Liebman, J. F., Eds.; John Wiley & Sons, Ltd.: West Sussex, UK, 2005; pp 281–356. doi:10.1002/0470864028.ch8 |
| 3. | Brady, W. T. Synthesis 1971, 415–422. doi:10.1055/s-1971-21750 |
| 4. | Ali, S. M.; Lee, T. V.; Roberts, S. M. Synthesis 1977, 155–166. doi:10.1055/s-1977-24302 |
| 5. | Moore, H. W.; Gheorghiu, M. D. Chem. Soc. Rev. 1981, 10, 289–328. doi:10.1039/CS9811000289 |
| 6. | Trost, B. M. Sulfuranes in organic reactions and synthesis. In New Concepts I; Davison, A.; Dewar, M. J. S., Eds.; Topics in Current Chemistry, Vol. 41; Springer: Berlin, Heidelberg, 1973; pp 1–29. doi:10.1007/3-540-06333-1_17 |
| 7. |
Conia, J. M.; Robson, M. J. Angew. Chem. 1975, 87, 505–516. doi:10.1002/ange.19750871404
Angew. Chem., Int. Ed. Engl. 1975, 14, 473–485. doi:10.1002/anie.197504731 |
| 8. | Trost, B. M. Acc. Chem. Res. 1974, 7, 85–92. doi:10.1021/ar50075a004 |
| 12. | Baldwin, J. E.; Leber, P. A. Org. Biomol. Chem. 2008, 6, 36–47. doi:10.1039/B711494J |
| 13. | Leber, P. A.; Baldwin, J. E. Acc. Chem. Res. 2002, 35, 279–287. doi:10.1021/ar010100p |
| 14. | Bronson, J. J.; Danheiser, R. L. In Comprehensive Organic Synthesis, 1st ed.; Trost, B. M.; Flemming, I.; Paquette, L. A., Eds.; Pergamon Press: Oxford, 1991; pp 1016 ff. |
| 25. | Machiguchi, T.; Hasegawa, T.; Ishiwata, A.; Terashima, S.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 1999, 121, 4771–4786. doi:10.1021/ja990072u |
| 26. | Ussing, B. R.; Hang, C.; Singleton, D. A. J. Am. Chem. Soc. 2006, 128, 7594–7607. doi:10.1021/ja0606024 |
| 11. |
Belluš, D.; Ernst, B. Angew. Chem. 1988, 100, 820–850. doi:10.1002/ange.19881000607
Angew. Chem., Int. Ed. Engl. 1988, 27, 797–827. doi:10.1002/anie.198807971 |
| 27. | Rey, M.; Roberts, S.; Dieffenbacher, A.; Dreiding, A. S. Helv. Chim. Acta 1970, 53, 417–432. doi:10.1002/hlca.19700530223 |
| 9. | Wong, H. N. C.; Lau, K.-L.; Tam, K.-F. The application of cyclobutane derivatives in organic synthesis. In Small Ring Compounds in Organic Synthesis I; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 133; Springer: Berlin, Heidelberg, 1986; pp 83–157. doi:10.1007/3-540-16307-7_2 |
| 10. | Moore, H. W.; Decker, O. H. W. Chem. Rev. 1986, 86, 821–830. doi:10.1021/cr00075a006 |
| 24. | Ji, X.; Wang, Q.; Goeke, A. Chem. Commun. 2010, 46, 8845–8847. doi:10.1039/c0cc02694h |
| 3. | Brady, W. T. Synthesis 1971, 415–422. doi:10.1055/s-1971-21750 |
| 4. | Ali, S. M.; Lee, T. V.; Roberts, S. M. Synthesis 1977, 155–166. doi:10.1055/s-1977-24302 |
| 5. | Moore, H. W.; Gheorghiu, M. D. Chem. Soc. Rev. 1981, 10, 289–328. doi:10.1039/CS9811000289 |
| 6. | Trost, B. M. Sulfuranes in organic reactions and synthesis. In New Concepts I; Davison, A.; Dewar, M. J. S., Eds.; Topics in Current Chemistry, Vol. 41; Springer: Berlin, Heidelberg, 1973; pp 1–29. doi:10.1007/3-540-06333-1_17 |
| 7. |
Conia, J. M.; Robson, M. J. Angew. Chem. 1975, 87, 505–516. doi:10.1002/ange.19750871404
Angew. Chem., Int. Ed. Engl. 1975, 14, 473–485. doi:10.1002/anie.197504731 |
| 8. | Trost, B. M. Acc. Chem. Res. 1974, 7, 85–92. doi:10.1021/ar50075a004 |
| 1. | Lee-Ruff, E.; Mladenova, G. Chem. Rev. 2003, 103, 1449–1484. doi:10.1021/cr010013a |
| 17. | Cohen, T.; Bhupathy, M.; Matz, J. R. J. Am. Chem. Soc. 1983, 105, 520–525. doi:10.1021/ja00341a035 |
| 6. | Trost, B. M. Sulfuranes in organic reactions and synthesis. In New Concepts I; Davison, A.; Dewar, M. J. S., Eds.; Topics in Current Chemistry, Vol. 41; Springer: Berlin, Heidelberg, 1973; pp 1–29. doi:10.1007/3-540-06333-1_17 |
| 7. |
Conia, J. M.; Robson, M. J. Angew. Chem. 1975, 87, 505–516. doi:10.1002/ange.19750871404
Angew. Chem., Int. Ed. Engl. 1975, 14, 473–485. doi:10.1002/anie.197504731 |
| 8. | Trost, B. M. Acc. Chem. Res. 1974, 7, 85–92. doi:10.1021/ar50075a004 |
| 16. | Danheiser, R. L.; Martinez-Davila, C.; Sard, H. Tetrahedron 1981, 37, 3943–3950. doi:10.1016/S0040-4020(01)93268-5 |
| 6. | Trost, B. M. Sulfuranes in organic reactions and synthesis. In New Concepts I; Davison, A.; Dewar, M. J. S., Eds.; Topics in Current Chemistry, Vol. 41; Springer: Berlin, Heidelberg, 1973; pp 1–29. doi:10.1007/3-540-06333-1_17 |
| 7. |
Conia, J. M.; Robson, M. J. Angew. Chem. 1975, 87, 505–516. doi:10.1002/ange.19750871404
Angew. Chem., Int. Ed. Engl. 1975, 14, 473–485. doi:10.1002/anie.197504731 |
| 8. | Trost, B. M. Acc. Chem. Res. 1974, 7, 85–92. doi:10.1021/ar50075a004 |
| 15. | Matz, J. R.; Cohen, T. Tetrahedron Lett. 1981, 22, 2459–2462. doi:10.1016/S0040-4039(01)92932-6 |
| 4. | Ali, S. M.; Lee, T. V.; Roberts, S. M. Synthesis 1977, 155–166. doi:10.1055/s-1977-24302 |
| 11. |
Belluš, D.; Ernst, B. Angew. Chem. 1988, 100, 820–850. doi:10.1002/ange.19881000607
Angew. Chem., Int. Ed. Engl. 1988, 27, 797–827. doi:10.1002/anie.198807971 |
| 18. | Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1984, 67, 1506–1514. doi:10.1002/hlca.19840670614 |
| 19. | Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1982, 65, 451–461. doi:10.1002/hlca.19820650203 |
| 20. | Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1982, 65, 1563–1575. doi:10.1002/hlca.19820650530 |
| 21. | Danheiser, R. L.; Gee, S. K.; Sard, H. J. Am. Chem. Soc. 1982, 104, 7670–7672. doi:10.1021/ja00390a054 |
| 22. | Paquette, L. A.; Colapret, J. A.; Andrews, D. R. J. Org. Chem. 1985, 50, 201–205. doi:10.1021/jo00202a012 |
| 23. | Paquette, L. A.; Andrews, D. R.; Springer, J. P. J. Org. Chem. 1983, 48, 1147–1149. doi:10.1021/jo00155a059 |
© 2012 Ji et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)