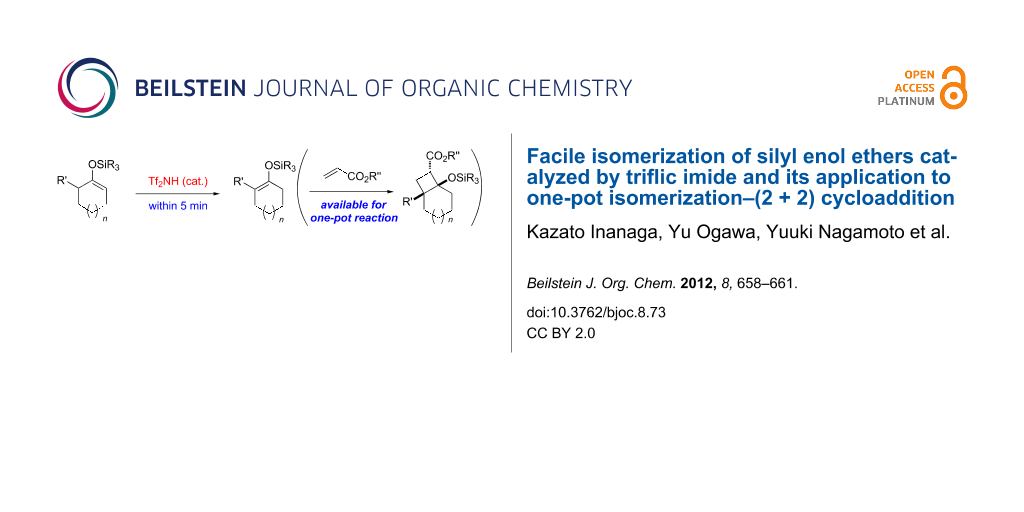
Abstract
A triflic imide (Tf2NH) catalyzed isomerization of kinetically favourable silyl enol ethers into thermodynamically stable ones was developed. We also demonstrated a one-pot catalytic reaction consisting of (2 + 2) cycloaddition and isomerization. In the reaction sequence, Tf2NH catalyzes both of the reactions.

Graphical Abstract
Introduction
Silyl enol ethers, which are isolable equivalents of metal enolates, are useful and important intermediates in synthetic chemistry [1-3]. They react as a good nucleophile for the introduction of a carbon skeleton or a functional group at the α-position of a carbonyl group under appropriate conditions. Although silyl enol ethers are easily prepared from the corresponding ketones, the regiochemical issue would arise in the case of asymmetric ketones. Treatment with a strong base such as lithium diisopropylamide (LDA), followed by silyl chloride, under cryogenic conditions selectively affords kinetically favourable silyl enol ethers. On the other hand, thermodynamically stable ones can be predominantly obtained by the reaction with a silylating agent in the presence of a weak base, such as triethylamine, under equilibration conditions. Although the preparation of silyl enol ethers has been extensively studied, there have only been a limited number of studies on their isomerization [4-7]. Deyine reported that a catalytic amount of triethylammonium chloride promotes the isomerization to give thermodynamically favourable ones in moderate yield [5]. However, harsh conditions (reaction temperature: ca. 100 to 200 °C) were required for the complete equilibration. Yamamoto and co-workers reported that a SnCl4–(BINOL monomethyl ether) complex (5–10 mol %) catalyzes the isomerization of silyl enol ethers at −78 °C [6]. By using this catalyst, they remarkably achieved the kinetic resolution of racemic silyl enol ethers. To make this isomerization synthetically useful and valuable, the development of more-reactive catalysts and a facile procedure would be required. In this communication, we describe isomerization of silyl enol ethers by an organocatalyst under mild conditions and its application to a one-pot catalytic reaction involving isomerization of silyl enol ethers and (2 + 2) cycloaddition.
Results and Discussion
During our research on triflic imide (Tf2NH)-catalyzed reactions [8], we accidentally found that the isomerization of kinetically favourable silyl enol ethers into thermodynamically stable ones occurs smoothly in the presence of Tf2NH. When the TBS enol ether 1a was treated with a catalytic amount of Tf2NH (1.0 mol %) in CH2Cl2 at ambient temperature, isomerization resulted in the thermodynamically stable 2a in 92% yield along with the recovered 1a and ketone 3 (Table 1, entry 1). Equilibrium was reached within 5 min. The reaction using 20 mol % of Tf2NH resulted in an increase of decomposition into 3 (entry 2). When the reaction was performed at −10 °C, the chemical yield of 2 was slightly improved (entry 3). In contrast, no isomerization was observed at −78 °C even after 1 h (entry 4). The catalytic isomerization reaction also proceeded in toluene (entry 5), but no (or almost no) isomerization occurred in CH3CN (entry 6). When 10-camphorsulfonic acid (5 mol %) was used as a catalyst for 1 h, the isomerization was incomplete (entry 7). Enol ethers bearing typical silyl groups were also isomerized (entries 8–12). The decomposition of TMS enol ether 1b into 3b slightly increased at ambient temperature compared to that at −10 °C (entries 8 and 9). In the reaction of TIPS enol ether 1d, the reaction rate decreased and more catalyst (5 mol %) was necessary to achieve equilibrium within 5 min (entry 12).
Table 1: Tf2NH-catalyzed isomerization of silyl enol ethers.a,b
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-73-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | 1 (SiR3) | solvent | temp. (°C) | % yield | ||
|---|---|---|---|---|---|---|
| 2 | 1 (recovd.) | 3 | ||||
| 1 | 1a (TBS) | CH2Cl2 | rt | 92 | 6 | 2 |
| 2c | 1a | CH2Cl2 | rt | 71 | 4 | 25 |
| 3 | 1a | CH2Cl2 | −10 | 93 | 4 | 3 |
| 4d | 1a | CH2Cl2 | −78 | 1 | 97 | 2 |
| 5 | 1a | toluene | −10 | 92 | 5 | 2 |
| 6 | 1a | CH3CN | −10 | 2 | 96 | 2 |
| 7d,e,f | 1a | CH2Cl2 | −10 | 25 | 64 | 11 |
| 8 | 1b (TMS) | CH2Cl2 | rt | 85 | 6 | 9 |
| 9 | 1b | CH2Cl2 | −10 | 91 | 4 | 5 |
| 10 | 1b | CH2Cl2 | −78 | 0 | 95 | 5 |
| 11 | 1c (TES) | CH2Cl2 | −10 | 78 | 5 | 17 |
| 12e | 1d (TIPS) | CH2Cl2 | −10 | 92 | 3 | 5 |
aYields were determined by GC–MS. bRegioisomer 1 (>99% purity) was used as a substrate. c20 mol % of catalyst was used. dReactions were carried out for 1 h. e5 mol % of catalyst was used. f10-Camphorsulfonic acid was used as a catalyst.
Several silyl enol ethers were explored for catalytic isomerization under the optimized conditions (1 mol % of Tf2NH, −10 °C, CH2Cl2). The results are summarized in Table 2. All the kinetically favourable silyl enol ethers 1 were smoothly isomerized to the thermodynamically stable 2 in the presence of Tf2NH.
Table 2: Substrate scope for Tf2NH-catalyzed isomerization.a,b
| entry | substrate | product | % yield of 2 | recovd. 1 (%) |
|---|---|---|---|---|
| 1c |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-73-i5.svg?max-width=637&scale=1.0)
1e |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-73-i6.svg?max-width=637&scale=1.0)
2e |
83 | 7 |
| 2d |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-73-i7.svg?max-width=637&scale=1.0)
1f |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-73-i8.svg?max-width=637&scale=1.0)
2f |
89 | 11 |
| 3e |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-73-i9.svg?max-width=637&scale=1.0)
1g |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-73-i10.svg?max-width=637&scale=1.0)
2g |
95 | 3 |
| 4f,g |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-73-i11.svg?max-width=637&scale=1.0)
1h |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-73-i12.svg?max-width=637&scale=1.0)
2h |
99 | 1 |
aReactions were performed under the same conditions as given in Table 1, entry 3. bYields were determined by GC–MS. cPurity of 1e is 99% (including isomer 2e (1%)). dPurity of 1f is 100% (no isomer 2f). ePurity of 1g is 95% (including isomer 2g (5%)). fPurity of 1h is 93% (including isomer 2h (7%)). g5 mol % of Tf2NH was used.
A plausible mechanism for the catalytic isomerization is shown in Scheme 1. Silyl enol ether 1 is rapidly protonated by a catalytic amount of Tf2NH to give the corresponding siloxonium cation 4, and, then, another molecule of silyl enol ether 1 deprotonates the α-position of 4. Equilibration results in the selective production of the thermodynamically more stable 2. As a side reaction, the counter anion, Tf2N−, could attack the silicon atom of 2 to produce silyl triflic imide (R3SiNTf2) [8-12] and the corresponding ketone 3. Therefore, the use of a large amount of Tf2NH causes decomposition into 3 (Table 1, entry 2).
![[1860-5397-8-73-i1]](/bjoc/content/inline/1860-5397-8-73-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Plausible mechanism for Tf2NH-catalyzed isomerization of silyl enol ethers.
Scheme 1: Plausible mechanism for Tf2NH-catalyzed isomerization of silyl enol ethers.
We have previously reported the Tf2NH catalyzed (2 + 2) cycloaddition of silyl enol ethers with acrylates generating substituted cyclobutanes [10]. We are intrigued that the isomerization of silyl enol ethers and successive (2 + 2) cycloaddition could be promoted by Tf2NH in a one-pot reaction. When 1a was treated with Tf2NH (1 mol %) under the isomerization conditions (−10 °C), followed by the addition of methyl acrylate (5) at −78 °C, 6-methylbicyclo[4.2.0]octane 6 and its diastereomer were obtained in 86% and 6%, respectively (Scheme 2a). No formation of their regioisomers was observed. The obtained compound 6 is identical to the product in the reaction of 2a with 5 [10,13]. It is noteworthy that two different reactions, isomerization and (2 + 2) cycloaddition, are catalyzed by Tf2NH [14-18]. By contrast, when 1a reacted with 5 in the presence of Tf2NH at −78 °C, (2 + 2) cycloaddition directly proceeded to give 2-methylbicyclo[4.2.0]octane 7 in 66% yield along with the formation of two diastereomers (Scheme 2b). Obviously, at this temperature, no isomerization of 1a occurred.
![[1860-5397-8-73-i2]](/bjoc/content/inline/1860-5397-8-73-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Regioselective formation of bicyclo[4.2.0]octanes from the same substrates by the isomerization–(2 + 2) cycloaddition procedure.
Scheme 2: Regioselective formation of bicyclo[4.2.0]octanes from the same substrates by the isomerization–(2 ...
The above finding can be applied to (2 + 2) cycloaddition, even if a mixture of regioisomeric silyl enol ethers is used as a substrate (Scheme 3). Thus, the reaction of ketone 3h with TBSOTf in the presence of NEt3 afforded a regioisomeric mixture of silyl enol ethers 1h and 2h (ca. 7:3). After extraction to remove the amine reagent, the crude regioisomeric mixture was subjected to Tf2NH at −10 °C and subsequently reacted with acrylate 5 to afford (2 + 2) cycloadduct 8 in 70% yield. This result indicates that the Tf2NH catalyzed reaction can save not only the separation to remove the corresponding kinetically favourable regioisomer, but also loss of the undesired regioisomer.
![[1860-5397-8-73-i3]](/bjoc/content/inline/1860-5397-8-73-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of bicyclo[5.2.0]octane from the regioisomeric mixture of silyl enol ethers.
Scheme 3: Formation of bicyclo[5.2.0]octane from the regioisomeric mixture of silyl enol ethers.
Conclusion
In summary, we have developed a new catalytic isomerization reaction of silyl enol ethers. Kinetically favourable silyl enol ethers were smoothly converted into thermodynamically stable ones by treatment with a catalytic amount of Tf2NH under mild conditions. Moreover, we demonstrated that the one-pot reaction involves two different catalytic reactions, an isomerization and a (2 + 2) cycloaddition.
Supporting Information
| Supporting Information File 1: Experimental details and spectral data. | ||
| Format: PDF | Size: 264.8 KB | Download |
References
-
Brownbridge, P. Synthesis 1983, 1–28. doi:10.1055/s-1983-30204
Return to citation in text: [1] -
Brownbridge, P. Synthesis 1983, 85–104. doi:10.1055/s-1983-30234
Return to citation in text: [1] -
Poirier, J.-M. Org. Prep. Proced. Int. 1988, 20, 317–369. doi:10.1080/00304948809355878
Return to citation in text: [1] -
Stork, G.; Hudrlik, P. F. J. Am. Chem. Soc. 1968, 90, 4462–4464. doi:10.1021/ja01018a051
Return to citation in text: [1] -
Deyine, A.; Dujardin, G.; Mammeri, M.; Poirier, J.-M. Synth. Commun. 1998, 28, 1817–1821. doi:10.1080/00397919808007012
Return to citation in text: [1] [2] -
Ishihara, K.; Nakamura, H.; Nakamura, S.; Yamamoto, H. J. Org. Chem. 1998, 63, 6444–6445. doi:10.1021/jo9812936
Return to citation in text: [1] [2] -
Aikawa, H.; Kaneko, T.; Asao, N.; Yamamoto, Y. Beilstein J. Org. Chem. 2011, 7, 648–652. doi:10.3762/bjoc.7.76
Return to citation in text: [1] -
Takasu, K. Synlett 2009, 1905–1914. doi:10.1055/s-0029-1217522
Return to citation in text: [1] [2] -
Ishihara, K.; Hiraiwa, Y.; Yamamoto, H. Synlett 2001, 1851–1854. doi:10.1055/s-2001-18761
Return to citation in text: [1] -
Inanaga, K.; Takasu, K.; Ihara, M. J. Am. Chem. Soc. 2005, 127, 3668–3669. doi:10.1021/ja042661s
Return to citation in text: [1] [2] [3] -
Boxer, M. B.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 48–49. doi:10.1021/ja054725k
Return to citation in text: [1] -
Takasu, K.; Miyakawa, Y.; Ihara, M.; Tokuyama, H. Chem. Pharm. Bull. 2008, 56, 1205–1206. doi:10.1248/cpb.56.1205
Return to citation in text: [1] -
Kurahashi, K.; Takemoto, Y.; Takasu, K. ChemSusChem 2012, 5, 270–273. doi:10.1002/cssc.201100373
Return to citation in text: [1] -
Fogg, D. E.; dos Santos, E. N. Coord. Chem. Rev. 2004, 248, 2365–2379. doi:10.1016/j.ccr.2004.05.012
Return to citation in text: [1] -
Shindoh, N.; Takemoto, Y.; Takasu, K. Chem.–Eur. J. 2009, 15, 12168–12179. doi:10.1002/chem.200901486
Return to citation in text: [1] -
Shindoh, N.; Tokuyama, H.; Takemoto, Y.; Takasu, K. J. Org. Chem. 2008, 73, 7451–7456. doi:10.1021/jo8009243
Return to citation in text: [1] -
Takasu, K.; Tanaka, T.; Azuma, T.; Takemoto, Y. Chem. Commun. 2010, 46, 8246–8248. doi:10.1039/c0cc03336g
Return to citation in text: [1] -
Azuma, T.; Takemoto, Y.; Takasu, K. Chem. Pharm. Bull. 2011, 59, 1190–1193. doi:10.1248/cpb.59.1190
Return to citation in text: [1]
| 1. | Brownbridge, P. Synthesis 1983, 1–28. doi:10.1055/s-1983-30204 |
| 2. | Brownbridge, P. Synthesis 1983, 85–104. doi:10.1055/s-1983-30234 |
| 3. | Poirier, J.-M. Org. Prep. Proced. Int. 1988, 20, 317–369. doi:10.1080/00304948809355878 |
| 6. | Ishihara, K.; Nakamura, H.; Nakamura, S.; Yamamoto, H. J. Org. Chem. 1998, 63, 6444–6445. doi:10.1021/jo9812936 |
| 5. | Deyine, A.; Dujardin, G.; Mammeri, M.; Poirier, J.-M. Synth. Commun. 1998, 28, 1817–1821. doi:10.1080/00397919808007012 |
| 4. | Stork, G.; Hudrlik, P. F. J. Am. Chem. Soc. 1968, 90, 4462–4464. doi:10.1021/ja01018a051 |
| 5. | Deyine, A.; Dujardin, G.; Mammeri, M.; Poirier, J.-M. Synth. Commun. 1998, 28, 1817–1821. doi:10.1080/00397919808007012 |
| 6. | Ishihara, K.; Nakamura, H.; Nakamura, S.; Yamamoto, H. J. Org. Chem. 1998, 63, 6444–6445. doi:10.1021/jo9812936 |
| 7. | Aikawa, H.; Kaneko, T.; Asao, N.; Yamamoto, Y. Beilstein J. Org. Chem. 2011, 7, 648–652. doi:10.3762/bjoc.7.76 |
| 14. | Fogg, D. E.; dos Santos, E. N. Coord. Chem. Rev. 2004, 248, 2365–2379. doi:10.1016/j.ccr.2004.05.012 |
| 15. | Shindoh, N.; Takemoto, Y.; Takasu, K. Chem.–Eur. J. 2009, 15, 12168–12179. doi:10.1002/chem.200901486 |
| 16. | Shindoh, N.; Tokuyama, H.; Takemoto, Y.; Takasu, K. J. Org. Chem. 2008, 73, 7451–7456. doi:10.1021/jo8009243 |
| 17. | Takasu, K.; Tanaka, T.; Azuma, T.; Takemoto, Y. Chem. Commun. 2010, 46, 8246–8248. doi:10.1039/c0cc03336g |
| 18. | Azuma, T.; Takemoto, Y.; Takasu, K. Chem. Pharm. Bull. 2011, 59, 1190–1193. doi:10.1248/cpb.59.1190 |
| 10. | Inanaga, K.; Takasu, K.; Ihara, M. J. Am. Chem. Soc. 2005, 127, 3668–3669. doi:10.1021/ja042661s |
| 13. | Kurahashi, K.; Takemoto, Y.; Takasu, K. ChemSusChem 2012, 5, 270–273. doi:10.1002/cssc.201100373 |
| 10. | Inanaga, K.; Takasu, K.; Ihara, M. J. Am. Chem. Soc. 2005, 127, 3668–3669. doi:10.1021/ja042661s |
| 8. | Takasu, K. Synlett 2009, 1905–1914. doi:10.1055/s-0029-1217522 |
| 9. | Ishihara, K.; Hiraiwa, Y.; Yamamoto, H. Synlett 2001, 1851–1854. doi:10.1055/s-2001-18761 |
| 10. | Inanaga, K.; Takasu, K.; Ihara, M. J. Am. Chem. Soc. 2005, 127, 3668–3669. doi:10.1021/ja042661s |
| 11. | Boxer, M. B.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 48–49. doi:10.1021/ja054725k |
| 12. | Takasu, K.; Miyakawa, Y.; Ihara, M.; Tokuyama, H. Chem. Pharm. Bull. 2008, 56, 1205–1206. doi:10.1248/cpb.56.1205 |
© 2012 Inanaga et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)