Abstract
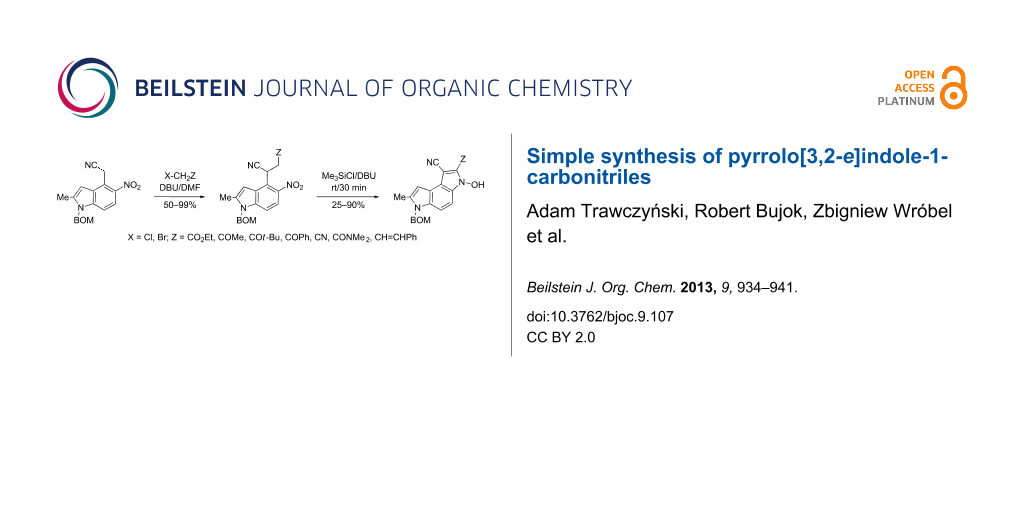
Alkylation of 5-nitroindol-4-ylacetonitriles with ethyl chloroacetate, α-halomethyl ketones, and chloroacetonitrile followed by a treatment of the products with chlorotrimethylsilane in the presence of DBU gives 1-cyanopyrrolo[3,2-e]indoles substituted in position 2 with electron-withdrawing groups.

Graphical Abstract
Introduction
Indole and its analogues bearing condensed arene and heteroarene rings are privileged structures amongst biologically active compounds. The 1,2-dihydropyrrolo[3,2-e]indole fragment is present in anticancer agents, such as CC-1065 [1], duocarmycin [1], and yatakemycin [2]. Some pyrrolo[3,2-e]indole derivatives show antimicrobial activity [3]. One method of synthesis of the 1,2-dihydropyrrolo[2,3-e]indoles is reduction of pyrrolo[3,2-e]indoles with sodium cyanoborohydride [4]. On the other hand there are many methods of synthesis of pyrrolo[3,2-e]indoles such as the copper-catalyzed transformation of tetrahydroquinoline derivatives [4], photochemical cyclization of 1,2-bis(2-pyrrolo)ethylenes [5], the Fischer indole synthesis from indol-5-ylhydrazones [3], or a palladium-catalyzed hydrogenation of 5-nitroindol-4-ylacetonitriles 2 [6]. In the latter synthesis of pyrrolo[3,2-e]indole 3 the starting nitrile 2 was obtained by the vicarious nucleophilic substitution (VNS) [7-11] of hydrogen in 1-alkyl-5-nitroindole 1 with 4-chlorophenoxyacetonitrile [12] (Scheme 1).
![[1860-5397-9-107-i1]](/bjoc/content/inline/1860-5397-9-107-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyrrolo[3,2-e]indoles via VNS in 5-nitroindoles [6,12].
Scheme 1: Synthesis of pyrrolo[3,2-e]indoles via VNS in 5-nitroindoles [6,12].
In our previous papers [13-16] we have shown that o-nitroarylacetonitriles alkylated and alkenylated at the α-position to the cyano group can be converted into indoles under basic conditions in the presence of a silylating agent.
Results and Discussion
Here we report a simple two-step procedure for the transformation of 5-nitroindol-4-ylacetonitriles into pyrrolo[3,2-e]indole-1-carbonitriles 6 bearing an additional electron-withdrawing substituent at position 2. In our approach the starting material was 1-benzyloxymethyl-4-cyanomethyl-2-methyl-5-nitroindole (4) obtained via the VNS of hydrogen in 1-benzyloxymethyl-2-methyl-5-nitroindole with 4-chlorophenoxyacetonitrile according to our earlier elaborated method [12]. Alkylation of the nitrile 4 with ethyl bromoacetate in the presence of K2CO3 led to the expected cyanoester 5a in 68% yield, but the product contained some contaminants difficult to separate by crystallization or column chromatography. Searching for more convenient reaction conditions, we have found that this reaction proceeds satisfactorily in almost quantitative yield when diazabicycloundecene (DBU) was used as the base. Analogous alkylation with α-halomethyl ketones, chloroacetonitrile, chloroacetamide and cinnamyl bromide provided the expected alkylation products 5b–g in good yields (Scheme 2 and Table 1).
![[1860-5397-9-107-i2]](/bjoc/content/inline/1860-5397-9-107-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of pyrrolo[3,2-e]indoles 6.
Scheme 2: Synthesis of pyrrolo[3,2-e]indoles 6.
Table 1: Alkylation products 5 and synthesized 1-cyano-3-hydroxy-pyrrolo[3,2-e]indoles 6.
| Entry | X–CH2–Z | Indole 5 | Yield (%) | Pyrrolo[3,2-e]indole 6 | Yield (%) |
|---|---|---|---|---|---|
| 1 | Br–CH2CO2Et |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-107-i5.svg?max-width=637&scale=1.0)
5a |
99 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-107-i6.svg?max-width=637&scale=1.0)
6a |
90 |
| 2 | Cl–CH2COMe |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-107-i7.svg?max-width=637&scale=1.0)
5b |
88 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-107-i8.svg?max-width=637&scale=1.0)
6d |
61 |
| 3 | Cl–CH2COCMe3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-107-i9.svg?max-width=637&scale=1.0)
5c |
82 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-107-i10.svg?max-width=637&scale=1.0)
6c |
55 |
| 4 | Br–CH2COPh |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-107-i11.svg?max-width=637&scale=1.0)
5d |
98 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-107-i12.svg?max-width=637&scale=1.0)
6d |
30 |
| 5 | Cl–CH2CN |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-107-i13.svg?max-width=637&scale=1.0)
5e |
86 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-107-i14.svg?max-width=637&scale=1.0)
6e |
30 |
| 6 | Cl–CH2CONMe2 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-107-i15.svg?max-width=637&scale=1.0)
5f |
95 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-107-i16.svg?max-width=637&scale=1.0)
6f |
44 |
| 7 | Br–CH2CH=CH2Ph |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-107-i17.svg?max-width=637&scale=1.0)
5g |
50 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-107-i18.svg?max-width=637&scale=1.0)
6g |
25 |
To find optimal conditions for cyclization of the model compound 5a we screened various combinations of base and a reagent promoting the cyclization. With chlorotrimethylsilane–triethylamine the reaction proceeded slowly, and the starting material was completely consumed after 24 h, but the product 6a was isolated in moderate 30% yield. However, when we replaced triethylamine with a stronger base, such as DBU, the reaction was completed in 30 min, and the product was isolated in 90% yield. Similarly, with N,O-bis(trimethylsilyl)acetamide (BSA) the reaction was completed in 30 min giving 6a in 67% yield. With tributylchlorostannane combined with DBU the reaction proceeded slowly to form after 24 h product 6a in 72% yield. Methanesulfonyl and pivaloyl chlorides, in combination with DBU proved ineffective in this reaction giving a very low rate of conversion after 24 h. Thus, transformations of other nitriles 5b–g into pyrrolo[3,2-e]indoles 6 were performed in the DBU–chlorotrimethylsilane system, and the results are presented in Table 1. It is worth mentioning that the ketone 5d upon reduction with SnCl2 cyclized to pyrrolo[3,2-f]quinoline-9-carbonitrile 7 [17].
The removal of the benzyloxymethyl group from 1-(benzyloxymethyl)pyrrolo[3,2-e]indoles by catalytic hydrogenation has been described by Macor [6]. The hydroxy group from the N-hydroxypyrrole fragment can be removed by a procedure elaborated by us [18] employing α-bromoacetophenone in the presence of triethylamine as exemplified for pyrroloindoles 6a and 6d that were transformed under these conditions into the corresponding derivatives 8a and 8d (Scheme 2). The crude 3-hydroxy-pyrrolo[3,2-e]indole 6d without isolation and purification was subjected to dehydroxylation giving compound 8d in 47% yield.
A plausible route to the formation of 3-hydroxy-1-cyanopyrrolo[3,2-e]indoles is exemplified by the synthesis of 1,2-dicyano derivative 6e from the dinitrile 5e (Scheme 3). In the first step the o-nitrobenzylic carbanion is silylated with trimethylchlorosilane to form trimethylsilyl nitronate A. Then a consecutive deprotonation forms another carbanion B at the β-position to the ring. The attack of this carbanion on the trimethylsilylnitronate results in the substitution of trimethoxysiloxyl and formation of C in that the five-membered ring finally isomerizes to the N-hydroxypyrrole fragment of 6e.
![[1860-5397-9-107-i3]](/bjoc/content/inline/1860-5397-9-107-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible route for transformation of indoles 5 into pyrrolo[3,2-e]indoles 6.
Scheme 3: Plausible route for transformation of indoles 5 into pyrrolo[3,2-e]indoles 6.
To remove the benzyloxymethyl group from the compound 8a we adopted the procedure proposed by Macor [6]. Heating 8a with ammonium formate and 10% palladium on carbon as a catalyst in isopropanol in a sealed tube (95 °C) led to a mixture of the expected product 9a and the product 10a in that the cyano group was reduced to a methyl substituent (Scheme 4). There is a literature precedence [19] for similar transformations of cyanoarenes into corresponding methyl derivatives upon transfer hydrogenation with ammonium formate in the presence of palladium on a carbon catalyst.
![[1860-5397-9-107-i4]](/bjoc/content/inline/1860-5397-9-107-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Removal of the benzyloxymethyl group from the compound 8a.
Scheme 4: Removal of the benzyloxymethyl group from the compound 8a.
Conclusion
In conclusion, the approach presented herein can be useful for the synthesis of variously substituted pyrrolo[3,2-e]indoles. The method does not require reductive conditions for the formation of the pyrrole ring and, thus, can be applicable for derivatives bearing sensitive substituents.
Experimental
Melting points (mp) are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 500 or Varian vnmr s500 (both 500 MHz for 1H and 125 MHz for 13C spectra) instruments at 298 K. Chemical shifts δ are expressed in parts per million referenced to TMS; coupling constants J in hertz. IR spectra were recorded in KBr on a Perkin Elmer PE Spectrum 2000 spectrometer. Electron impact mass spectra (EI, 70 eV) were obtained on AMD-604 and AutoSpec Premier spectrometer. Electrospray mass spectra (ESI) were obtained on 4000 Q-TRAP and SYNAPT G2-S HDMS. Silica gel (Merck 60, 230–400 mesh) was used for column chromatography (CC). All reagents and solvents were of reagent grade or purified according to standard methods before use. 1-Benzyloxymethyl-4-(cyanomethyl)-2-methyl-5-nitroindole (4) was obtained by VNS of hydrogen in 1-benzyloxymethyl-2-methyl-5-nitroindole with 4-chlorophenoxyacetonitrile following our previously elaborated method [12].
Alkylation of indolylacetonitrile 4 with ethyl bromoacetate. Synthesis of 3-(1-benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-3-cyanopropionic acid ethyl ester (5a) – Typical procedure
A solution of (1-benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)acetonitrile (4, 0.335 g, 1 mmol) and ethyl bromoacetate (0.25 g, 1.5 mmol) were stirred in DMF (5 mL) and DBU (0.30 g, 2 mmol) at 60 °C until the starting material 4 disappeared (usually 24 h, TLC control). Then the reaction mixture was poured into diluted HCl and the product was extracted with EtOAc (3 × 30 mL) and dried with Na2SO4. After evaporation of the solvent the residue was purified by column chromatography (SiO2, hexane–EtOAc, 2:1). Yellow crystals; mp 76–78 °C; 1H NMR (500 MHz, CDCl3) δ 1.28 (t, J = 7.3 Hz, 3H), 2.52 (s, 3H), 3.06 (dd, J = 16.9, 6.1 Hz, 1H), 3.28 (dd, J = 16.9, 9.0 Hz, 1H), 4.21 (q, J = 7.3 Hz, 2H), 4.47 (s, 2H), 5.42 (dd, J = 9.0, 6.1 Hz, 1H), 5.55 (s, 2H), 6.80 (br s, 1H), 7.23–7.27 (m, 2H), 7.31–7.37 (m, 4H), 7.84 (d, J = 9.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 12.82, 14.10, 27.65, 37.67, 61.66, 70.12, 71.96, 102.16, 109.93, 118.79, 119.02, 120.50, 126.92, 127.66, 128.29, 128.63, 136.43, 139.48, 141.32, 142.22, 168.98; IR (KBr, cm−1) ν: 2979, 2243, 1728, 1603, 1561, 1510, 1461, 1450, 1409, 1373, 1339, 1272, 1244, 1205, 1188, 1130, 1096, 959, 813, 800, 769, 759, 741; EIMS (70 eV) m/z (% relative intensity): 421 (14) [M] +∙, 404 (5), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C23H23N3O5, 421.1638; found, 421.1630.
2-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-4-oxopentanenitrile (5b). Yellow crystals; mp 102–104 °C; 1H NMR (500 MHz, CDCl3) δ 2.21 (s, 3H), 2.52 (d, J = 1.0 Hz, 3H), 3.12 (dd, J = 18.3, 5.2 Hz, 1H), 3.50 (dd, J = 18.3, 8.7 Hz, 1H), 4.47 (s, 2H), 5.37 (dd, J = 8.7, 5.2 Hz, 1H), 5.54 (s, 2H), 6.77 (br s, 1H), 7.24–7.27 (m, 2H), 7.31–7.37 (m, 4H), 7.83 (d, J = 8.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 12.80, 26.20, 29.42, 46.23, 70.06, 71.92, 102.12, 109.73, 118.82, 119.21, 121.08, 127.03, 127.64, 128.25, 128.62, 136.39, 139.40, 141.21, 142.19, 202.77; IR (KBr, cm−1) ν: 2915, 2250, 2240, 1714, 1607, 1559, 1517, 1504, 1451, 1400, 1330, 1257, 1238, 1170, 1081, 1060, 1006, 948, 817, 733; EIMS (70 eV) m/z (% relative intensity): 391 (3) [M]+∙, 357 (11), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C22H21N3O4, 391.1532; found, 391.1540.
2-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-5,5-dimethyl-4-oxohexanenitrile (5c). Yellow crystals; mp 92–94 °C; 1H NMR (500 MHz, CDCl3) δ 1.15 (s, 9H), 2.53 (d, J = 0.8 Hz, 3H), 3.14 (dd, J = 17.9, 4.9 Hz, 1H), 3.57 (dd, J = 17.9, 9.1 Hz, 1H), 4.47 (s, 2H), 5.32 (dd, J = 9.1, 4.9 Hz, 1H), 5.55 (s, 2H), 6.78 (br s, 1H), 7.24–7.37 (m, 6H), 7.83 (d, J = 9.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 12.84, 26.11, 26.91, 40.21, 44.06, 70.05, 71.92, 102.10, 109.66, 118.83, 119.42, 121.47, 127.10, 127.65, 128.26, 128.62, 136.40, 139.37, 141.18, 142.21, 210.44; IR (KBr, cm−1) ν: 2969, 2244, 1707, 1606, 1652, 1519, 1477, 1340, 1071, 1029, 818, 757, 740; EIMS (70 eV) m/z (% relative intensity): 433 (3) [M]+∙, 399 (6), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C25H27N3O4, 433.2002; found, 433.2004.
2-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-4-oxo-4-phenylbutyronitrile (5d). Yellow crystals; mp 123–125 °C; 1H NMR (500 MHz, CDCl3) δ 2.52 (d, J = 1.0 Hz, 3H), 3.68 (dd, J = 18.0, 5.1 Hz, 1H), 4.05 (dd, J = 18.0, 8.7 Hz, 1H), 4.47 (s, 2H), 5.55 (s, 2H), 5.58 (dd, J = 8.7, 5.1 Hz, 1H), 6.83 (m, 1H), 7.25 (s, 1H), 7.26 (s, 1H), 7.30–7.37 (m, 4H), 7.44–7.48 (m, 2H), 7.56–7.60 (m, 1H), 7.85 (d, J = 9.0 Hz, 1H), 7.93–7.96 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 12.86, 26.64, 42.01, 70.09, 71.96, 102.24, 109.78, 118.88, 119.46, 121.36, 127.15, 127.67, 128.17, 128.27, 128.64, 128.78, 133.85, 135.57, 136.44, 139.45, 141.27, 142.37, 194.46; IR (KBr, cm−1) ν: 2921, 2246, 1690, 1559, 1518, 1447, 1343, 1213, 1086, 1071, 803, 751, 689; EIMS (70 eV) m/z (% relative intensity): 453 (2) [M]+∙, 419 (9), 105 (35), 92 (8), 91 (100), 77 (13); HRMS–EI (70 eV, m/z): [M]+ calcd for C27H23N3O4, 453.1689; found, 453.1671.
2-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-succinonitrile (5e). Brown oil; 1H NMR (500 MHz, CDCl3) δ 2.54 (d, J = 1.0 Hz, 3H), 3.25, 3.31, 5.34 (ABX, J = 17.0, 8.0, 6.8 Hz, 3H), 4.48 (s, 2H), 5.56 (s, 2H), 6.87 (br s, 1H), 7.24–7.27 (m, 2H), 7.31–7.37 (m, 3H), 7.41 (d, J = 8.9 Hz, 1H), 7.93 (d, J = 8.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 12.88, 22.04, 28.75, 70.20, 71.98, 102.00, 110.78, 115.41, 117.06, 118.19, 119.09, 127.02, 127.65, 128.31, 128.64, 136.25, 139.80, 141.77, 142.14; IR (KBr, cm−1) ν: 2949, 2249, 2225, 1607, 1562, 1519, 1446, 1399, 1337, 1071, 821, 738, 700; EIMS (70 eV) m/z (% relative intensity): 374 (11) [M]+∙, 340 (8), 92 (14), 91 (100), 65 (8); HRMS–EI (70 eV, m/z): [M]+ calcd for C21H18N4O3, 374.1379; found, 374.1385.
3-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-3-cyano-N,N-dimethylpropionamide (5f). Orange oil; 1H NMR (500 MHz, CDCl3) δ 2.52 (s, 3H), 2.97 (br s, 1H), 2.96 (s, 3H), 2.98 (s, 3H), 3.40 (dd, J = 16.4, 8.8, Hz, 1H), 4.47 (s, 2H), 5.44 (dd, J = 8.8, 5.4 Hz, 1H), 5.54 (s, 2H), 6.81 (m, 1H), 7.25–7.30 (m, 2H), 7.31–7.37 (m, 4H), 7.83 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 12.83, 28.10, 35.69, 36.93, 36.99, 70.07, 71.94, 102.26, 109.66, 118.88, 119.58, 121.67, 127.32, 127.66, 128.26, 128.64, 136.45, 139.39, 141.10, 142.39, 167.99; IR (KBr, cm−1) ν: 2932, 2244, 1651, 1561, 1519, 1400, 1340, 1267, 1241, 1150, 1070, 822, 737, 700; EIMS (70 eV) m/z (% relative intensity): 420 (2) [M]+∙, 375 (6), 374 (16), 195 (8), 108 (8), 107 (6), 92 (12), 91 (49); HRMS–EI (70 eV, m/z): [M]+ calcd for C23H24N4O4, 420.1798; found, 420.1796.
2-(1-Benzyloxymethyl-2-methyl-5-nitro-1H-indol-4-yl)-5-phenylpent-4-enenitrile (5g). Yellow crystals; mp 130–132 °C; 1H NMR (500 MHz, CDCl3) δ 2.52 (s, 3H), 2.98 (ddd, J = 13.8, 9.1, 7.5 Hz, 1H), 3.13 (ddd, J = 13.8, 7.5, 6.0 Hz, 1H), 5.09 (dd, J = 9.1, 6.0 Hz, 1H), 6.31 (ddd, 15.4, 7.5, 7.5 Hz, 1H), 6.58 (d, J = 15.4 Hz, 1H), 6.90 (s, 1H), 7.21–7.40 (m, 11H), 7.85 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 12.82, 32.17, 37.05, 70.06, 71.94, 102.88, 109.67, 118.86, 119.73, 121.77, 124.03, 126.43, 126.92, 127.70, 127.73, 128.29, 128.55, 129.45, 134.43, 136.44, 136.64, 139.54, 140.92, 142.10; IR (KBr, cm−1) ν: 3026, 2239, 2206, 1605, 1556, 1512, 1437, 1400, 1335, 1080, 1070, 967, 817, 764, 742, 734, 693; EIMS (70 eV) m/z (% relative intensity): 451 (9) [M]+, 434 (4), 405 (4), 334 (15), 117 (22), 115 (13), 105 (6), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C28H25N3O3, 451.1896; found, 451.1890.
Cyclization of 5-nitroindol-4-ylacetonitriles to pyrrolo[3,2-e]indoles – Typical procedure
To a solution of indole derivative 5 (1 mmol) and DBU (0.75 g, 5 mmol) in DMF (5 mL) Me3SiCl (0.54 g, 5 mmol) was added at room temperature. The reaction mixture was stirred for 20–30 min (TLC control), quenched with diluted HCl, extracted with EtOAc (3 × 30 mL) and dried with Na2SO4. After evaporation of the solvent the residue was purified by column chromatography (SiO2, hexane–EtOAc, 2:1).
6-Benzyloxymethyl-1-cyano-3-hydroxy-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-2-carboxylic acid ethyl ester (6a). Yellow crystals; mp 119–121 °C; 1H NMR (500 MHz, DMSO-d6) δ 1.37 (t, J = 7.1 Hz, 3H), 2.58 (s, 3H), 4.41 (q, J = 7.1 Hz, 2H), 4.48 (s, 2H), 5.77 (s, 2H), 6.84 (m, 1H), 7.23–7.36 (m, 5H), 7.73 (d, J = 9.1 Hz, 1H), 11.22 (br s, 1H); 13C NMR (125 MHz, CD3COCD3) δ 12.64, 14.44, 62.30, 70.32, 73.18, 85.95, 100.46, 104.26, 112.46, 116.13, 116.59, 119.80, 126.68, 128.37, 128.41, 128.45, 129.12, 131.11, 134.06, 138.27, 138.73,159.36; IR (KBr, cm−1) ν: 2987, 2212, 1753, 1682, 1618, 1597, 1499, 1453, 1430, 1368, 1333, 1321, 1265, 1136, 1062, 1028, 775; EIMS (70 eV) m/z (% relative intensity): 403 (38) [M]+∙, 387 (13), 311 (11), 297 (14), 283 (13), 281 (6), 192 (5), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C23H21N3O4, 403.1532; found, 403.1519.
2-Acetyl-6-benzyloxymethyl-3-hydroxy-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-1-carbonitrile (6b). Yellow crystals; mp 194–196 °C; 1H NMR (500 MHz, DMSO-d6) δ 2.53 (d, J = 0.7 Hz, 3H), 2.74 (s, 3H), 4.85 (s, 2H), 5.74 (s, 2H), 6.75 (br s, 1H), 7.24–7.29 (m, 3H), 7.30–7.34 (m, 2H), 7.35 (d, J = 9.1 Hz, 1H), 7.76 (d, J = 9.1 Hz, 1H), 12.49 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.23, 29.97, 69.15, 72.20, 82.41, 99.30, 103.81, 112.04, 115.60, 116.54, 118.09, 127.44, 127.59, 128.27, 129.90, 132.66, 134.36, 137.32, 137.53, 187.55; IR (KBr, cm−1) ν: 2917, 2216, 1645, 1594, 1545, 1510, 1483, 1444, 1426, 1366, 1314, 1236, 1121, 1088, 1020, 990; EIMS (70 eV) m/z (% relative intensity): 373 (9) [M]+∙, 358 (9), 357 (39), 327 (15), 326 (5), 252 (6), 251 (23), 250 (8), 237 (6), 236 (7), 208 (8), 194 (7), 108 (5), 106 (5), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C22H19N3O3, 373.1426; found, 373.1421.
6-Benzyloxymethyl-2-(2,2-dimethylpropionyl)-3-hydroxy-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-1-carbonitrile (6c). Brown solid; mp 155–157 °C; 1H NMR (500 MHz, DMSO-d6) δ 1.32 (s, 9H), 2.52 (d, J = 0.8 Hz, 3H), 4.47 (s, 2H), 5.73 (s, 2H), 6.68 (br s, 1H), 7.24–7.35 (m, 6H), 7.67 (d, J = 9.0 Hz, 1H), 12.50 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.27, 26.00, 44.68, 69.08, 72.21, 78.41, 98.67, 103.64, 109.66, 114.83, 116.34, 118.20, 127.43, 127.59, 128.00, 128.28, 132.87, 137.25, 137.30, 137.58, 202.17; IR (KBr, cm−1) ν: 2969, 2244, 1707, 1562, 1519, 1477, 1398, 1340, 1071, 1029; EIMS (70 eV) m/z (% relative intensity): 415 (9) [M]+∙, 400 (15), 399 (59), 369 (6), 342 (7), 313 (5), 312 (17), 294 (5), 293 (19), 292 (8), 285 (7), 284 (8), 236 (8), 208 (5), 194 (6), 193 (5), 108 (6), 92 (8), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C25H25N3O3, 415.1896; found, 415.1878.
2-Benzoyl-6-benzyloxymethyl-7-methyl-3-hydroxy-3,6-dihydropyrrolo[3,2-e]indole-1-carbonitrile (6d). Red crystals; mp 130–132 °C; 1H NMR (500 MHz, CDCl3) δ 2.51 (s, 3H), 4.44 (s, 2H), 5.60 (s, 2H), 6.93 (s, 1H), 7.24–7.28 (m, 2H), 7.29–7.39 (m, 4H), 7.53 (d, J = 9.1 Hz, 1H), 7.58–7.63 (m, 2H), 7.69–7.75 (m, 1H), 7.97–8.02 (m, 2H), 12.56 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 12.67, 69.59, 72.04, 85.65, 101.26, 103.34, 113.27, 115.64, 117.69, 119.00, 126.39, 127.74, 128.08, 128.56, 128.69, 128.87, 129.95, 132.94, 134.15, 135.87, 136.85, 137.15, 189.1; IR (KBr, cm−1) ν: 2921, 2215, 1639, 1599, 1569, 1496, 1479, 1424, 1367, 1329, 1314, 1260, 1085, 1072, 778, 732, 693; ESIMS (MeOH) m/z: 436 [M + H]+, 458 [M + Na]+; HRMS–EI (70 eV, m/z): [M + Na]+ calcd for C27H21N3O3Na, 458.1475; found, 458.1495.
6-Benzyloxymethyl-3-hydroxy-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-1,2-dicarbonitrile (6e). Brownish crystals; mp 150–152 °C; 1H NMR (500 MHz, DMSO-d6) δ 2.53 (s, 3H), 4.47 (s, 2H), 5.75 (s, 2H), 6.70 (br s, 1H), 7.22–7.34 (m, 5H), 7.36 (d, J = 9.1 Hz, 1H), 7.83 (d, J = 9.1 Hz, 1H), 13.29 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.26, 69.22, 72.30, 99.16, 103.44, 108.90, 110.30, 112.65, 114.14, 115.21, 117.70, 127.47, 127.64, 128.31, 128.33, 129.46, 132.86, 137.52, 138.01; IR (KBr, cm−1) ν: 2922, 2224, 1544, 1484, 1393, 1351, 1319, 1209, 1145, 1064, 1024, 774, 753, 699; ESIMS (MeOH/CH2Cl2) m/z: 357 [M + H]+, 379 [M + Na]+, 735 [2M + Na]+.
6-Benzyloxymethyl-1-cyano-3-hydroxy-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-2-carboxylic acid dimethylamide (6f). Brown semisolid; 1H NMR (500 MHz, DMSO-d6) δ 2.49 (s, 3H), 3.02 (s, 3H), 3.30 (br s, 3H), 4.44 (s, 2H), 5.68 (s, 2H), 6.61 (br s, 1H), 7.22–7.33 (m, 6H), 7.52 (d, J = 9.0 Hz, 1H), 13.24 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.21, 34.38, 37.24, 68.92, 72.06, 98.73, 104.84, 107.15, 115.29, 117.24, 117.99, 127.35, 127.49, 127.93, 128.22, 132.58, 135.89, 137.64 (3 peaks not visible); IR (KBr, cm−1) ν: 2930, 2208, 1615, 1525, 1445, 1390, 1363, 1319, 1258, 1209, 1121, 1066, 778, 738, 698; EIMS (70 eV) m/z (% relative intensity): 402 (10) [M]+∙, 387 (7), 386 (27), 312 (6), 311 (17), 280 (5), 265 (6), 221 (5), 220 (6), 213 (6), 193 (6), 192 (5), 165 (14), 135 (7), 108 (39), 107 (10), 105 (7), 92 (10), 91 (100); HRMS–EI (70 eV, m/z): [M]+ calcd for C23H22N4O3, 402.1692; found, 402.1684.
6-Benzyloxymethyl-3-hydroxy-7-methyl-2-((E)-styryl)-3,6-dihydropyrrolo[3,2-e]indole-1-carbonitrile (6g). Dark green solid; mp 130–132 °C; 1H NMR (500 MHz, DMSO-d6) δ 2.52 (d, J = 0.8 Hz, 3H), 4.48 (s, 2H), 5.71 (s, 2H), 6.69 (br s, 1H), 7.21–7.40 (m, 8H), 7.44–7.48 (m, 2H), 7.56 (d, J = 9.0 Hz, 1H), 7.69 (m, 2H), 7.77 (d, J = 16.5 Hz, 1H), 12.10 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.26, 69.07, 72.18, 98.74, 103.21, 108.35, 113.73, 115.56, 117.83, 117.85, 126.83, 127.42, 127.56, 127.66, 128.27, 128.60, 128.91, 129.05, 132.84, 133.04, 135.95, 136.87, 136.99, 137.63; IR (KBr, cm−1) ν: 2923, 2200, 1739, 1547, 1467, 1389, 1361, 1339, 1317, 1203, 1142, 1066, 955, 746; EIMS (70 eV) m/z (% relative intensity): 433 (2) [M]+∙, 431 (16), 325 (10), 297 (11), 165 (28), 108 (92), 107 (68), 106 (35), 105 (46), 92 (8), 91 (82); HRMS–ESI (m/z): [M + Na]+ calcd for C28H23N3O2Na, 456.1688; found, 456.1685.
6-Benzyloxymethyl-1-cyano-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-2-carboxylic acid ethyl ester (8a). Pale creamy crystals; mp 215–217 °C; 1H NMR (500 MHz, DMSO-d6) δ 1.40 (t, J = 7.1 Hz, 3H), 2.52 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 4.47 (s, 2H), 5.71 (s, 2H), 6.74 (br s, 1H), 7.24–7.35 (m, 6H), 12.99 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.23, 14.07, 61.35, 69.09, 72.18, 87.59, 99.08, 106.70, 111.12, 115.93, 118.34, 119.90, 127.42, 127.57, 128.27, 129.34, 131.58, 132.28, 136.77, 137.57, 159.10; IR (KBr, cm−1) ν: 3264, 2219, 1689, 1527, 1455, 1434, 1362, 1315, 1261, 1064, 1055, 1025, 743; EIMS (70 eV) m/z (% relative intensity): 387 (50) [M]+∙, 312 (8), 311 (37), 281 (14), 280 (5), 266 (5), 235 (6), 220 (12), 206 (5), 192 (6), 165 (5), 92 (8), 91 (100); HRMS–EI (m/z): [M]+ calcd for C23H21N3O3, 387.1583; found, 387.1587.
2-Benzoyl-6-benzyloxymethyl-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-1-carbonitrile (8d). Yellow crystals; mp 180–182 ºC; 1H NMR (500 MHz, DMSO-d6) δ 2.53 (s, 3H), 4.47 (s, 2H), 5.74 (s, 2H), 6.75 (s, 1H), 7.23–7.38 (m, 6H), 7.60–7.67 (m, 2H), 7.69–7.78 (m, 2H), 7.89–7.94 (m, 2H), 12.93 (br s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 12.24, 69.07, 72.17, 88.59, 99.28, 106.82, 111.83, 116.11, 118.42, 120.51, 127.44, 127.59, 128.28, 128.67, 129.38, 132.20, 132.38, 133.11, 136.36, 136.86, 136.97, 137.57, 185.27; IR (KBr, cm−1) ν: 3265, 2217, 1710, 1624, 1540, 1510, 1408, 1329, 1254, 1062, 942, 739, 696; ESIMS (MeOH) m/z: 420 [M + H]+, 442 [M + Na]+, 861 [2M + Na]+; HRMS–ESI (m/z): [M + Na]+ calcd for C27H21N3O2Na, 442.1526; found, 442.1544.
Debenzyloxymethylation of compound 8a
Compound 8 (100 mg, 0.26 mmol), ammonium formate (0.16 g, 10 mmol) and 10% palladium on charcoal (90 mg) were suspended in isopropanol (5 mL), flushed with argon for 5 min and then heated in a sealed tube at 95 °C overnight. Then the reaction mixture was passed through Celite, washed with dichloromethane–methanol, 1:1 (15 mL). After evaporation the residue was purified by column chromatography on silica gel with hexane–ethyl acetate (gradient 4:1 to 1:1). The following compounds were obtained:
1-Cyano-7-methyl-3,6-dihydropyrrolo[3,2-e]indole-2-carboxylic acid ethyl ester (9a). Yield 22%; mp > 280 °C; 1H NMR (500 MHz, DMSO-d6) δ 1.39 (t, J = 7.1 Hz, 3H), 2.45 (s, 3H), 4.42 (q, J = 7.1 Hz, 2H), 6.56 (s, 1H), 7.19 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.8 Hz, 1H), 11.33 (s, 1H), 12.85 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 13.27, 14.07, 61.19, 87.56, 97.30, 105.78, 112.34, 116.06, 118.75, 120.01, 128.49, 131.03, 131.09, 135.00, 159.14; IR (film, cm−1) ν: 3265, 2256, 1690, 1549, 1526, 1439, 1363, 1338, 1254, 1115, 1073,1016; EIMS (70 eV) m/z (% relative intensity): 267 (75) [M]+∙, 222 (26), 221 (100), 194 (25), 193 (55), 167 (17).
1,7-Dimethyl-3,6-dihydropyrrolo[3,2-e]indole-2-carboxylic acid ethyl ester (10a). Yield 34%; mp 225–227 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 1.35 (t, J = 7.0 Hz, 3H), 2.42 (s, 3H), 2.72 (s, 3H), 4.32 (q, J = 7.0 Hz, 2H), 6.46 (s, 1H), 7.04 (d, J = 8.8 Hz, 1H), 7.23 (d, J = 8.8 Hz, 1H), 10.97 (s, 1H), 11.21 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 11.57, 13.35, 14.40, 59.47, 98.24, 105.38, 110.99, 118.48, 119.62, 120.46, 120.54, 129.65, 131.77, 133.23, 162.01; IR (KBr, cm−1) ν: 3318, 1675, 1548, 1534, 1437, 1362, 1334, 1291, 1243, 1209, 1190, 1117, 1092, 1017, 772.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR, IR and mass spectra for compounds 4, 5a–g, 6a–g, 8a, 8d, 9a and 10a. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Ghosh, N.; Sheldrake, H. M.; Searcey, M.; Pors, K. Curr. Top. Med. Chem. 2009, 9, 1494–1524. doi:10.2174/156802609789909812
Return to citation in text: [1] [2] -
Tichenor, M. S.; Boger, D. L. Nat. Prod. Rep. 2008, 25, 220–226. doi:10.1039/b705665f
Return to citation in text: [1] -
Samsoniya, Sh. A.; Kadzhrishvili, D. O.; Chikvaidze, I. Sh. Pharm. Chem. J. 2011, 45, 22–25. doi:10.1007/s11094-011-0553-7
Return to citation in text: [1] [2] -
Okano, K.; Mitsuhashi, N.; Tokuyama, H. Chem. Commun. 2010, 46, 2641–2643. doi:10.1039/b926965g
Return to citation in text: [1] [2] -
Rawal, V. H.; Jones, R. J.; Cava, M. P. J. Org. Chem. 1987, 52, 19–28. doi:10.1021/jo00377a004
Return to citation in text: [1] -
Macor, J. E.; Forman, J. T.; Post, R. J.; Ryan, K. Tetrahedron Lett. 1997, 38, 1673–1676. doi:10.1016/S0040-4039(97)00198-6
Return to citation in text: [1] [2] [3] [4] -
Mąkosza, M. Synthesis 2011, 2341–2356. doi:10.1055/s-0030-1260668
Return to citation in text: [1] -
Mąkosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen - an Efficient Tool in Synthesis of Heterocyclic Compounds. In Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, O., Ed.; Soc. Chimica Italiana: Rome, 2011; Vol. 14, pp 19–48.
Return to citation in text: [1] -
Mąkosza, M. Chem. Soc. Rev. 2010, 39, 2855–2868. doi:10.1039/b822559c
Return to citation in text: [1] -
Mąkosza, M.; Wojciechowski, K. Chem. Rev. 2004, 104, 2631–2666. doi:10.1021/cr020086+
Return to citation in text: [1] -
Mąkosza, M.; Wojciechowski, K. Heterocycles 2001, 54, 445–474. doi:10.3987/REV-00-SR(I)2
Return to citation in text: [1] -
Wojciechowski, K.; Mąkosza, M. Synthesis 1989, 106–109. doi:10.1055/s-1989-34082
Return to citation in text: [1] [2] [3] [4] -
Wróbel, Z.; Mąkosza, M. Synlett 1993, 597–598. doi:10.1055/s-1993-22544
Return to citation in text: [1] -
Wróbel, Z.; Mąkosza, M. Tetrahedron 1993, 49, 5315–5326. doi:10.1016/S0040-4020(01)82380-2
Return to citation in text: [1] -
Wróbel, Z.; Mąkosza, M. Tetrahedron 1997, 53, 5501–5514. doi:10.1016/S0040-4020(97)00208-1
Return to citation in text: [1] -
Wróbel, Z.; Wojciechowski, K.; Kwast, A.; Gajda, N. Synlett 2010, 2435–2438. doi:10.1055/s-0030-1258555
Return to citation in text: [1] -
Bujok, R.; Trawczyński, A.; Wróbel, Z.; Wojciechowski, K. Synlett 2012, 2682–2686. doi:10.1055/s-0032-1317380
Return to citation in text: [1] -
Bujok, R.; Wróbel, Z.; Wojciechowski, K. Synlett 2012, 1315–1320. doi:10.1055/s-0031-1291044
Return to citation in text: [1] -
Brown, G. R.; Foubister, A. J. Synthesis 1982, 1036–1037. doi:10.1055/s-1982-30054
Return to citation in text: [1]
| 19. | Brown, G. R.; Foubister, A. J. Synthesis 1982, 1036–1037. doi:10.1055/s-1982-30054 |
| 18. | Bujok, R.; Wróbel, Z.; Wojciechowski, K. Synlett 2012, 1315–1320. doi:10.1055/s-0031-1291044 |
| 6. | Macor, J. E.; Forman, J. T.; Post, R. J.; Ryan, K. Tetrahedron Lett. 1997, 38, 1673–1676. doi:10.1016/S0040-4039(97)00198-6 |
| 1. | Ghosh, N.; Sheldrake, H. M.; Searcey, M.; Pors, K. Curr. Top. Med. Chem. 2009, 9, 1494–1524. doi:10.2174/156802609789909812 |
| 4. | Okano, K.; Mitsuhashi, N.; Tokuyama, H. Chem. Commun. 2010, 46, 2641–2643. doi:10.1039/b926965g |
| 17. | Bujok, R.; Trawczyński, A.; Wróbel, Z.; Wojciechowski, K. Synlett 2012, 2682–2686. doi:10.1055/s-0032-1317380 |
| 3. | Samsoniya, Sh. A.; Kadzhrishvili, D. O.; Chikvaidze, I. Sh. Pharm. Chem. J. 2011, 45, 22–25. doi:10.1007/s11094-011-0553-7 |
| 6. | Macor, J. E.; Forman, J. T.; Post, R. J.; Ryan, K. Tetrahedron Lett. 1997, 38, 1673–1676. doi:10.1016/S0040-4039(97)00198-6 |
| 2. | Tichenor, M. S.; Boger, D. L. Nat. Prod. Rep. 2008, 25, 220–226. doi:10.1039/b705665f |
| 13. | Wróbel, Z.; Mąkosza, M. Synlett 1993, 597–598. doi:10.1055/s-1993-22544 |
| 14. | Wróbel, Z.; Mąkosza, M. Tetrahedron 1993, 49, 5315–5326. doi:10.1016/S0040-4020(01)82380-2 |
| 15. | Wróbel, Z.; Mąkosza, M. Tetrahedron 1997, 53, 5501–5514. doi:10.1016/S0040-4020(97)00208-1 |
| 16. | Wróbel, Z.; Wojciechowski, K.; Kwast, A.; Gajda, N. Synlett 2010, 2435–2438. doi:10.1055/s-0030-1258555 |
| 1. | Ghosh, N.; Sheldrake, H. M.; Searcey, M.; Pors, K. Curr. Top. Med. Chem. 2009, 9, 1494–1524. doi:10.2174/156802609789909812 |
| 12. | Wojciechowski, K.; Mąkosza, M. Synthesis 1989, 106–109. doi:10.1055/s-1989-34082 |
| 6. | Macor, J. E.; Forman, J. T.; Post, R. J.; Ryan, K. Tetrahedron Lett. 1997, 38, 1673–1676. doi:10.1016/S0040-4039(97)00198-6 |
| 12. | Wojciechowski, K.; Mąkosza, M. Synthesis 1989, 106–109. doi:10.1055/s-1989-34082 |
| 3. | Samsoniya, Sh. A.; Kadzhrishvili, D. O.; Chikvaidze, I. Sh. Pharm. Chem. J. 2011, 45, 22–25. doi:10.1007/s11094-011-0553-7 |
| 6. | Macor, J. E.; Forman, J. T.; Post, R. J.; Ryan, K. Tetrahedron Lett. 1997, 38, 1673–1676. doi:10.1016/S0040-4039(97)00198-6 |
| 12. | Wojciechowski, K.; Mąkosza, M. Synthesis 1989, 106–109. doi:10.1055/s-1989-34082 |
| 5. | Rawal, V. H.; Jones, R. J.; Cava, M. P. J. Org. Chem. 1987, 52, 19–28. doi:10.1021/jo00377a004 |
| 12. | Wojciechowski, K.; Mąkosza, M. Synthesis 1989, 106–109. doi:10.1055/s-1989-34082 |
| 4. | Okano, K.; Mitsuhashi, N.; Tokuyama, H. Chem. Commun. 2010, 46, 2641–2643. doi:10.1039/b926965g |
| 7. | Mąkosza, M. Synthesis 2011, 2341–2356. doi:10.1055/s-0030-1260668 |
| 8. | Mąkosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen - an Efficient Tool in Synthesis of Heterocyclic Compounds. In Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, O., Ed.; Soc. Chimica Italiana: Rome, 2011; Vol. 14, pp 19–48. |
| 9. | Mąkosza, M. Chem. Soc. Rev. 2010, 39, 2855–2868. doi:10.1039/b822559c |
| 10. | Mąkosza, M.; Wojciechowski, K. Chem. Rev. 2004, 104, 2631–2666. doi:10.1021/cr020086+ |
| 11. | Mąkosza, M.; Wojciechowski, K. Heterocycles 2001, 54, 445–474. doi:10.3987/REV-00-SR(I)2 |
© 2013 Trawczyński et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)