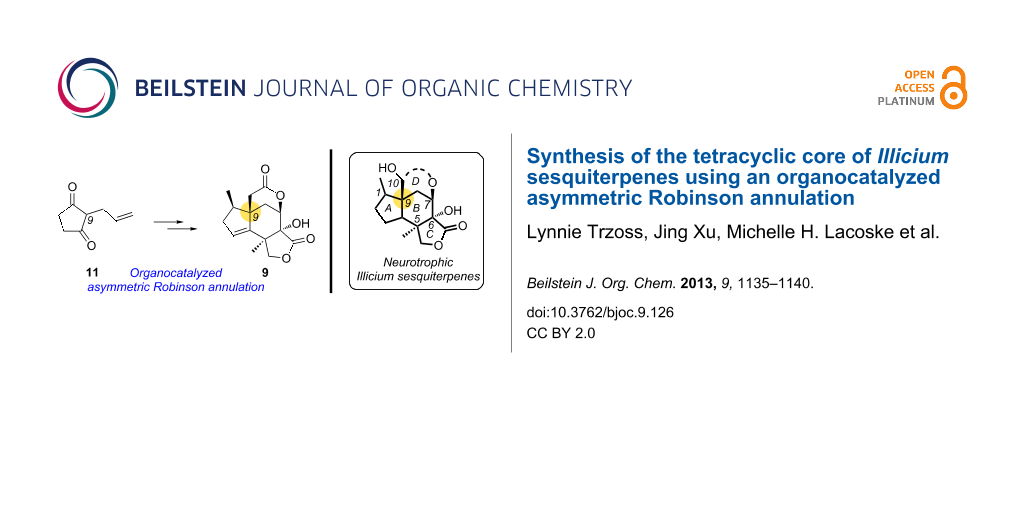
Abstract
An enantioselective synthesis of the core framework of neurotrophic Illicium majucin-type sesquiterpenes is described here. This strategy is based on an organocatalyzed asymmetric Robinson annulation and provides an efficient approach for a diversity-oriented synthesis of Illicium natural products that holds remarkable therapeutic potential for neurodegenerative diseases.

Graphical Abstract
Introduction
Neurotrophins are a family of endogenous proteins that are vital for neuron function, survival, and regeneration [1-3]. As such, they have prompted intense studies toward the treatment of various neurodegenerative diseases including Alzheimer’s disease [4] and Parkinson’s disease [5]. Despite their unambiguous importance, approaches to neurotrophin-based drug development have encountered problems associated with their limited oral availability, insufficient delivery to the central neural system and considerable manufacturing cost [6,7]. These limitations have stimulated the search for small molecules that can enhance or mimic neurotrophin activity as potential drug leads [8-12].
Majucin-type Illicium sesquiterpenes (Figure 1) [13], such as majucin (1) [14,15], jiadifenolide (2) [16], jiadifenin (3) [17], jiadifenoxolane A (4) [16] and (2R)-hydroxynorneomajucin (5) [18], share a caged tetracyclic scaffold (6). These compounds (2–5) have shown a great potential in enhancing neurite outgrowth in primary cultured rat cortical neurons at low nanomolar to low micromolar concentrations. Thus, to develop an efficient synthetic approach toward the complex core skeleton of these natural products is of paramount importance. Consequently, this family of neurotrophic sesquiterpenes has been the focus of extensive synthetic studies in which asymmetric and efficient construction of the tetracyclic core presents the principal challenge [19-23].
![[1860-5397-9-126-1]](/bjoc/content/figures/1860-5397-9-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative majucin-type Illicium sesquiterpenes.
Figure 1: Representative majucin-type Illicium sesquiterpenes.
We have recently reported a unified synthetic strategy of 2, 3 and designed analogues using scaffold 7 as the key intermediate (Figure 2) [24-26]. A potential drawback of this strategy is the late-stage modification of the A ring motif of 7 that requires additional steps for the synthesis of the target molecules. In an effort to overcome this issue, we describe here a second-generation strategy of framework 9 in which the C-1 center has been methylated early in the synthesis. As such, it represents an efficient route toward a diversity-oriented synthesis of several Illicium sesquiterpenes. The enantioselective entry to these molecules is based on an organocatalyzed asymmetric Robinson annulation that allows access to the enantiomerically enriched bicyclic motif 8 from achiral diketone 11 (Figure 2).
![[1860-5397-9-126-2]](/bjoc/content/figures/1860-5397-9-126-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Comparison of core skeleton synthetic strategies.
Figure 2: Comparison of core skeleton synthetic strategies.
Results and Discussion
During the past 20 years, organocatalysis has emerged as an important field in asymmetric stereoselective synthesis due to its advantages, which include high enantioselectivity, environmental friendliness and ease of handling [27-50]. Organocatalyzed asymmetric Robinson annulation has long been proven to be one of the most powerful strategies to construct bicyclic systems with a chiral quaternary center [51-58]. Among them, the Hajos–Wiechert and Wieland–Miescher ketones represent two of the most famous examples [59-65]. With this background information in mind, we devised an enantioselective synthesis of 8 starting from commercially available dione 12, and the synthesis of 8 was previously published [25,26]. Tsuji–Trost allylation [66-68] of 12 produced compound 11, which was readily converted to 13 by an acid-catalyzed Michael addition with methyl vinyl ketone (MVK) (two steps, 63% overall yield) [69-71]. The organocatalyzed cyclization of 13 was achieved by optimizing the previously reported Tu/Zhang conditions [71] using D-prolinamide as the organocatalyst (Scheme 1). Performing this reaction at 80 °C gave rise to bicyclic motif 8 in about 70% ee (70 % yield after 12 h), while decreasing the temperature to 25 °C increased the enantioselectivity to over 99% (70% yield after 60 days). To compromise between high enantioselectivity and short reaction time, we decided to pursue this conversion at 40 °C where we obtained an enantiomeric excess of 90% (70% yield after 14 days).
![[1860-5397-9-126-i1]](/bjoc/content/inline/1860-5397-9-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Organocatalyzed asymmetric Robinson annulation.
Scheme 1: Organocatalyzed asymmetric Robinson annulation.
The enantiomerically enriched Hajos–Wiechert-like diketone 8 (ee > 90%) was then subjected to a selective protection of the C-6 enone motif to yield dithioketal 14 (86% yield) [72-74]. Wittig olefination of the C-1 ketone with methoxymethylenetriphenylphosphine [75] yielded the corresponding enol methyl ether, which was hydrolyzed to the aldehyde under acidic conditions and reduced with NaBH4 to form alcohol 15 with desired diastereoselectivity at the C-1 center (dr = 9:1) in 81% yield (over three steps) [76]. The stereochemistry of 15 was unambiguously confirmed by single-crystal X-ray analysis of the related tosylate derivative 16 [77]. Deoxygenation of the C-15 primary alcohol was performed by: (a) mesylation of the alcohol with MsCl; and (b) reductive deoxygenation with LiEt3BH (super hydride). The thioketal protecting group was then removed under oxidative conditions with [bis(trifluoroacetoxy)iodo]benzene (PIFA) to yield ketone 10 in good yield (66% over three steps, Scheme 2) [78]. This approach allowed us to produce a sufficient amount of enone 10 (>10 grams) for further functionalization.
![[1860-5397-9-126-i2]](/bjoc/content/inline/1860-5397-9-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Early stage A-ring functionalization.
Scheme 2: Early stage A-ring functionalization.
Conversion of 10 to 9 was accomplished based on our previously reported strategy (Scheme 3) [25]. Treatment of 10 with magnesium methyl carbonate (MMC) [79-81] yielded the C-5 carboxylic acid that, without further purification, was esterified under Meerwein’s conditions to afford β-ketoester 17. Treatment of 17 with TMSOTf/Et3N followed by enolate alkylation [82] under TBAF/MeI conditions afforded the desired C-5 quaternary center of 18 as a single isomer (35% over four steps). Global reduction of 18 with lithium aluminium hydride produced the corresponding C-6/C-14 diol motif. Selective TBS protection of the C-14 primary alcohol followed by an IBX oxidation of the C-6 secondary alcohol yielded ketone 19 in 80% combined yield over three steps. Triflation of the C-6 ketone with McMurry’s reagent (PhNTf2) [83-86] followed by a Pd(0)-catalyzed carbomethoxylation [87-90] produced the desired C-ring lactone 20 in 61% yield. Epoxidation of the C-6/C-7 enone with NaOH/H2O2 followed by oxidative cleavage of the C-11 terminal alkene under OsO4/NaIO4 conditions [91,92] afforded the corresponding C-11 aldehyde. Exposure of this intermediate to Jones oxidation triggered a highly efficient oxidation–epoxide opening [93-98] reaction cascade [99,100] to construct the critical D-ring of 9 (46% yield, over 3 steps). Notably, this scalable approach rendered us several hundred milligrams of compound 9, paving the way for a diversity-oriented synthesis. For example, a Mn(III) promoted C-2 allylic oxidation [24,101,102] would provide a C-2 oxygenated functionality. Similarly, C-10 α-substitution would provide a large diversity of neurotrophic analogues based on our recent findings [26].
![[1860-5397-9-126-i3]](/bjoc/content/inline/1860-5397-9-126-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
We describe here an efficient and enantioselective approach to tetracyclic lactone 9 representing a key motif toward the synthesis of various neurotrophic [103-110] Illicium sesquiterpenes. Key to the strategy was a highly enantioselective Robinson annulation reaction that proceeded under organocatalytic conditions to form the Hajos–Wiechert-like enone 8. The overall strategy highlights the importance of organocatalytic approaches in the modern synthesis of bioactive natural products [111-116].
Supporting Information
| Supporting Information File 1: Experimental procedures for the syntheses of all new compounds. | ||
| Format: PDF | Size: 1.6 MB | Download |
Acknowledgements
We gratefully acknowledge the National Institutes of Health (NIH) for financial support of this work through Grant Number CA 133002. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr. Anthony Mrse (UCSD NMR Facility), Dr. Yongxuan Su (UCSD MS Facility) and Dr. Arnold L. Rheingold and Dr. Curtis E. Moore (UCSD X-Ray Facility).
References
-
Sofroniew, M. V.; Howe, C. L.; Mobley, W. C. Annu. Rev. Neurosci. 2001, 24, 1217–1281. doi:10.1146/annurev.neuro.24.1.1217
Return to citation in text: [1] -
Chao, M. V. Nat. Rev. Neurosci. 2003, 4, 299–309. doi:10.1038/nrn1078
Return to citation in text: [1] -
Huang, E. J.; Reichardt, L. F. Annu. Rev. Neurosci. 2001, 24, 677–736. doi:10.1146/annurev.neuro.24.1.677
Return to citation in text: [1] -
Querfurth, H. W.; LaFerla, F. M. N. Engl. J. Med. 2010, 362, 329–344. doi:10.1056/NEJMra0909142
Return to citation in text: [1] -
Shulman, J. M.; De Jager, P. L.; Feany, M. B. Annu. Rev. Pathol.: Mech. Dis. 2011, 6, 193–222. doi:10.1146/annurev-pathol-011110-130242
Return to citation in text: [1] -
Skaper, S. D. CNS Neurol. Disord.: Drug Targets 2008, 7, 46–62. doi:10.2174/187152708783885174
Return to citation in text: [1] -
Skaper, S. D. Curr. Pharm. Des. 2011, 17, 2704–2718. doi:10.2174/138161211797415995
Return to citation in text: [1] -
Me, Y.; Longo, F. M. Prog. Brain Res. 2000, 128, 333–347. doi:10.1016/S0079-6123(00)28030-8
Return to citation in text: [1] -
Massa, S. M.; Xie, Y. M.; Longo, F. M. J. Mol. Neurosci. 2003, 20, 323–326. doi:10.1385/JMN:20:3:323
Return to citation in text: [1] -
Longo, F. M.; Yang, T.; Knowles, J. K.; Xie, Y. M.; Moore, L. A.; Massa, S. M. Curr. Alzheimer Res. 2007, 4, 503–506. doi:10.2174/156720507783018316
Return to citation in text: [1] -
Joyner, P. M.; Cichewicz, R. H. Nat. Prod. Rep. 2011, 28, 26–47. doi:10.1039/c0np00017e
Return to citation in text: [1] -
Williams, P.; Sorribas, A.; Howes, M.-J. R. Nat. Prod. Rep. 2011, 28, 48–77. doi:10.1039/c0np00027b
Return to citation in text: [1] -
Urabe, D.; Inoue, M. Tetrahedron 2009, 65, 6271–6289. doi:10.1016/j.tet.2009.06.010
Return to citation in text: [1] -
Yang, C.-S.; Kouno, I.; Kawano, N.; Sato, S. Tetrahedron Lett. 1988, 29, 1165–1168. doi:10.1016/S0040-4039(00)86678-2
Return to citation in text: [1] -
Kouno, I.; Baba, N.; Hashimoto, M.; Kawano, N.; Takahashi, M.; Kaneto, H.; Yang, C.-S.; Sato, S. Chem. Pharm. Bull. 1989, 37, 2448–2451. doi:10.1248/cpb.37.2448
Return to citation in text: [1] -
Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett. 2009, 11, 5190–5193. doi:10.1021/ol9021029
Return to citation in text: [1] [2] -
Yokoyama, R.; Huang, J. M.; Yang, C. S.; Fukuyama, Y. J. Nat. Prod. 2002, 65, 527–531. doi:10.1021/np010571k
Return to citation in text: [1] -
Kubo, M.; Kobayashi, K.; Huang, J.-M.; Harada, K.; Fukuyama, Y. Tetrahedron Lett. 2012, 53, 1231–1235. doi:10.1016/j.tetlet.2011.12.107
Return to citation in text: [1] -
Cho, Y. S.; Carcache, D. A.; Tian, Y.; Li, Y. M.; Danishefsky, S. J. J. Am. Chem. Soc. 2004, 126, 14358–14359. doi:10.1021/ja045939p
Return to citation in text: [1] -
Carcache, D. A.; Cho, Y. S.; Hua, Z.; Tian, Y.; Li, Y.-M.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 1016–1022. doi:10.1021/ja056980a
Return to citation in text: [1] -
Harada, K.; Imai, A.; Uto, K.; Carter, R. G.; Kubo, M.; Hioki, H.; Fukuyama, Y. Org. Lett. 2011, 13, 988–991. doi:10.1021/ol103024z
Return to citation in text: [1] -
Mehta, G.; Shinde, H. M.; Kumaran, R. S. Tetrahedron Lett. 2012, 53, 4320–4323. doi:10.1016/j.tetlet.2012.06.001
Return to citation in text: [1] -
Yang, Y.; Fu, X.; Chen, J.; Zhai, H. Angew. Chem., Int. Ed. 2012, 51, 9825–9828. doi:10.1002/anie.201203176
Return to citation in text: [1] -
Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Org. Lett. 2011, 13, 4554–4557. doi:10.1021/ol201742j
Return to citation in text: [1] [2] -
Xu, J.; Trzoss, L.; Chang, W. K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2011, 50, 3672–3676. doi:10.1002/anie.201100313
Return to citation in text: [1] [2] [3] -
Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem.–Eur. J. 2013, 20, 6398–6408. doi:10.1002/chem.201300198
Return to citation in text: [1] [2] [3] -
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650
Return to citation in text: [1] -
List, B. Acc. Chem. Res. 2004, 37, 548–557. doi:10.1021/ar0300571
Return to citation in text: [1] -
Lelais, G.; MacMillan, D. W. C. Aldrichimica Acta 2006, 39, 79–87.
Return to citation in text: [1] -
Taylor, M. S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; Nolan, I. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380
Return to citation in text: [1] -
Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. Drug Discovery Today 2007, 12, 8–27. doi:10.1016/j.drudis.2006.11.004
Return to citation in text: [1] -
Bertelsen, S.; Jorgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g
Return to citation in text: [1] -
List, B.; Lerner, R. A.; Barbas, C. F., III. J. Am. Chem. Soc. 2000, 122, 2395–2396. doi:10.1021/ja994280y
Return to citation in text: [1] -
Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 9874–9875. doi:10.1021/ja005517p
Return to citation in text: [1] -
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s
Return to citation in text: [1] -
Beeson, T. D.; Mastracchio, A.; Hong, J. B.; Ashton, K.; MacMillan, D. W. C. Science 2007, 316, 582–585.
Return to citation in text: [1] -
Zhu, S. L.; Wang, Y.; Ma, D. W. Adv. Synth. Catal. 2009, 351, 2563–2566. doi:10.1002/adsc.200900449
Return to citation in text: [1] -
Marques-Lopez, E.; Herrera, R. P.; Christmann, M. Nat. Prod. Rep. 2010, 27, 1138–1167. doi:10.1039/b924964h
Return to citation in text: [1] -
Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167–178. doi:10.1038/nchem.539
Return to citation in text: [1] -
Zhu, S. L.; Yu, S. Y.; Wang, Y.; Ma, D. W. Angew. Chem., Int. Ed. 2010, 49, 4656–4660. doi:10.1002/anie.201001644
Return to citation in text: [1] -
Knowles, R. R.; Carpenter, J.; Blakey, S. B.; Kayano, A.; Mangion, I. K.; Sinz, C. J.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 308–311. doi:10.1039/c0sc00577k
Return to citation in text: [1] -
Sun, X.; Ma, D. Chem.–Asian J. 2011, 6, 2157–2164. doi:10.1002/asia.201100219
Return to citation in text: [1] -
Pham, P. V.; Ashton, K.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 1470–1473. doi:10.1039/c1sc00176k
Return to citation in text: [1] -
Wang, Y.; Zhu, S.; Ma, D. Org. Lett. 2011, 13, 1602–1605. doi:10.1021/ol200004s
Return to citation in text: [1] -
Jones, S. B.; Simmons, B.; Mastracchio, A.; MacMillan, D. W. C. Nature 2011, 475, 183–188. doi:10.1038/nature10232
Return to citation in text: [1] -
Zi, W.; Xie, W.; Ma, D. J. Am. Chem. Soc. 2012, 134, 9126–9129. doi:10.1021/ja303602f
Return to citation in text: [1] -
Huo, L.; Ma, A.; Zhang, Y.; Ma, D. Adv. Synth. Catal. 2012, 354, 991–994. doi:10.1002/adsc.201100903
Return to citation in text: [1] -
Simonovich, S. P.; Van Humbeck, J. F.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 58–61. doi:10.1039/c1sc00556a
Return to citation in text: [1] -
Zhang, Y.; Xing, H.; Xie, W.; Wan, X.; Lai, Y.; Ma, D. Adv. Synth. Catal. 2013, 355, 68–72. doi:10.1002/adsc.201200782
Return to citation in text: [1] -
Ling, T.; Xiang, A. X.; Theodorakis, E. A. Angew. Chem., Int. Ed. 1999, 38, 3089–3091. doi:10.1002/(SICI)1521-3773(19991018)38:20<3089::AID-ANIE3089>3.0.CO;2-W
Return to citation in text: [1] -
Ling, T.; Poupon, E.; Rueden, E. J.; Kim, S. H.; Theodorakis, E. A. J. Am. Chem. Soc. 2002, 124, 12261–12267. doi:10.1021/ja027517q
Return to citation in text: [1] -
Brady, T. P.; Kim, S. H.; Wen, K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 739–742. doi:10.1002/anie.200352868
Return to citation in text: [1] -
Ling, T.; Poupon, E.; Rueden, E. J.; Theodorakis, E. A. Org. Lett. 2002, 4, 819–822. doi:10.1021/ol025501z
Return to citation in text: [1] -
Ghosh, S.; Rivas, F.; Fischer, D.; González, M. A.; Theodorakis, E. A. Org. Lett. 2004, 6, 941–944. doi:10.1021/ol036492c
Return to citation in text: [1] -
Brady, T. P.; Kim, S. H.; Wen, K.; Kim, C.; Theodorakis, E. A. Chem.–Eur. J. 2005, 11, 7175–7190. doi:10.1002/chem.200500513
Return to citation in text: [1] -
Nguyen, T. X.; Dakanali, M.; Trzoss, L.; Theodorakis, E. A. Org. Lett. 2011, 13, 3308–3311. doi:10.1021/ol200966z
Return to citation in text: [1] -
Peng, F.; Dai, M.-J.; Angeles, A. R.; Danishefsky, S. J. Chem. Sci. 2012, 3, 3076–3080. doi:10.1039/c2sc20868g
See for a recent update of Robinson annulation.
Return to citation in text: [1] -
Wieland, P.; Miescher, K. Helv. Chim. Acta 1950, 33, 2215–2228. doi:10.1002/hlca.19500330730
Return to citation in text: [1] -
Eder, U.; Sauer, G.; Weichert, R. Angew. Chem., Int. Ed. Engl. 1971, 10, 496–497. doi:10.1002/anie.197104961
Return to citation in text: [1] -
Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1973, 38, 3239–3243. doi:10.1021/jo00959a002
Return to citation in text: [1] -
Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615–1621. doi:10.1021/jo00925a003
Return to citation in text: [1] -
Bradshaw, B.; Bonjoch, J. Synlett 2012, 337–356. doi:10.1055/s-0031-1290107
Return to citation in text: [1] -
Zhou, P.; Zhang, L.; Luo, S.; Cheng, J.-P. J. Org. Chem. 2012, 77, 2526–2530. doi:10.1021/jo202433v
Return to citation in text: [1] -
Winterfeldt, E. Angew. Chem., Int. Ed. 2013, 52, 4723. doi:10.1002/anie.201301415
Return to citation in text: [1] -
Tsuji, J.; Takahashi, H.; Morikawa, M. Tetrahedron Lett. 1965, 6, 4387–4388. doi:10.1016/S0040-4039(00)71674-1
Return to citation in text: [1] -
Trost, B. M.; Fullerto, T. J. Am. Chem. Soc. 1973, 95, 292–294. doi:10.1021/ja00782a080
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804
Return to citation in text: [1] -
Ruprah, P. K.; Cros, J.-P.; Pease, J. E.; Whittingham, W. G.; Williams, J. M. J. Eur. J. Org. Chem. 2002, 3145–3152. doi:10.1002/1099-0690(200209)2002:18<3145::AID-EJOC3145>3.0.CO;2-3
Return to citation in text: [1] -
Lacoste, E.; Vaique, E.; Berlande, M.; Pianet, I.; Vincent, J.-M.; Landais, Y. Eur. J. Org. Chem. 2007, 167–177. doi:10.1002/ejoc.200600664
Return to citation in text: [1] -
Zhang, X.-M.; Wang, M.; Tu, Y.-Q.; Fan, C.-A.; Jiang, Y.-J.; Zhang, S.-Y.; Zhang, F.-M. Synlett 2008, 2831–2835. doi:10.1055/s-0028-1083542
Return to citation in text: [1] [2] -
Coates, R. M.; Shaw, J. E. Chem. Commun. 1968, 515–516. doi:10.1039/c19680000515
Return to citation in text: [1] -
Williams, J. R.; Sarkisia, G. M. Synthesis 1974, 32–33. doi:10.1055/s-1974-23227
Return to citation in text: [1] -
Bosch, M. P.; Camps, F.; Coll, J.; Guerrero, A.; Tatsuoka, T.; Meinwald, J. J. Org. Chem. 1986, 51, 773–784. doi:10.1021/jo00356a002
Return to citation in text: [1] -
Pu, X.; Ma, D. Angew. Chem., Int. Ed. 2004, 43, 4222–4225. doi:10.1002/anie.200460128
Return to citation in text: [1] -
Paquette, L. A.; Wang, T.-Z.; Philippo, C. M. G.; Wang, S. J. Am. Chem. Soc. 1994, 116, 3367–3374. doi:10.1021/ja00087a023
Return to citation in text: [1] -
CCDC 931875 contains the supplementary crystallographic data for compound 16. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/products/csd/request/.
Return to citation in text: [1] -
Angeles, A. R.; Dorn, D. C.; Kou, C. A.; Moore, M. A. S.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2007, 46, 1451–1454. doi:10.1002/anie.200604308
Return to citation in text: [1] -
Finkbeiner, H. L.; Stiles, M. J. Am. Chem. Soc. 1963, 85, 616–622. doi:10.1021/ja00888a031
Return to citation in text: [1] -
Micheli, R. A.; Hajos, Z. G.; Cohen, N.; Parrish, D. R.; Portland, L. A.; Sciamanna, W.; Scott, M. A.; Wehrli, P. A. J. Org. Chem. 1975, 40, 675–681. doi:10.1021/jo00894a003
Return to citation in text: [1] -
Frie, J. L.; Jeffrey, C. S.; Sorensen, E. J. Org. Lett. 2009, 11, 5394–5397. doi:10.1021/ol902168g
Return to citation in text: [1] -
Lee, H. M.; Nieto-Oberhuber, C.; Shair, M. D. J. Am. Chem. Soc. 2008, 130, 16864–16865. doi:10.1021/ja8071918
Return to citation in text: [1] -
Mcmurry, J. E.; Scott, W. J. Tetrahedron Lett. 1983, 24, 979–982. doi:10.1016/S0040-4039(00)81581-6
Return to citation in text: [1] -
Scott, W. J.; Mcmurry, J. E. Acc. Chem. Res. 1988, 21, 47–54. doi:10.1021/ar00146a001
Return to citation in text: [1] -
Nicolaou, K. C.; Peng, X.-S.; Sun, Y.-P.; Polet, D.; Zou, B.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2009, 131, 10587–10597. doi:10.1021/ja902939t
Return to citation in text: [1] -
Ding, H.; Chen, D. Y. K. Angew. Chem., Int. Ed. 2011, 50, 676–679. doi:10.1002/anie.201006367
Return to citation in text: [1] -
Cowell, A.; Stille, J. K. J. Am. Chem. Soc. 1980, 102, 4193–4198. doi:10.1021/ja00532a034
Return to citation in text: [1] -
Cacchi, S.; Morera, E.; Ortar, G. Tetrahedron Lett. 1985, 26, 1109–1112. doi:10.1016/S0040-4039(00)98525-3
Return to citation in text: [1] -
Magro, A. A. N.; Robb, L. M.; Pogorzelec, P. J.; Slawin, A. M. Z.; Eastham, G. R.; Cole-Hamilton, D. J. Chem. Sci. 2010, 1, 723–730. doi:10.1039/c0sc00276c
Return to citation in text: [1] -
Nicolaou, K. C.; Ding, H.; Richard, J.-A.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 3815–3818. doi:10.1021/ja9093988
Return to citation in text: [1] -
Zuo, Z.; Xie, W.; Ma, D. J. Am. Chem. Soc. 2010, 132, 13226–13228. doi:10.1021/ja106739g
Return to citation in text: [1] -
Richard, J.-A.; Chen, D. Y.-K. Eur. J. Org. Chem. 2012, 484–487. doi:10.1002/ejoc.201101629
Return to citation in text: [1] -
Nicolaou, K. C.; Majumder, U.; Roche, S. P.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2007, 46, 4715–4718. doi:10.1002/anie.200701947
Return to citation in text: [1] -
Nicolaou, K. C.; Dalby, S. M.; Li, S.; Suzuki, T.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 7616–7620. doi:10.1002/anie.200904588
Return to citation in text: [1] -
Nicolaou, K. C.; Wu, T. R.; Kang, Q.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 3440–3443. doi:10.1002/anie.200900438
Return to citation in text: [1] -
Nicolaou, K. C.; Kang, Q.; Wu, T. R.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 7540–7548. doi:10.1021/ja102623j
Return to citation in text: [1] -
Peixoto, P. A.; Richard, J.-A.; Severin, R.; Chen, D. Y.-K. Org. Lett. 2011, 13, 5724–5727. doi:10.1021/ol202053m
Return to citation in text: [1] -
Peixoto, P. A.; Severin, R.; Tseng, C.-C.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2011, 50, 3013–3016. doi:10.1002/anie.201008000
Return to citation in text: [1] -
Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872
Return to citation in text: [1] -
Wasilke, J.-C.; Obrey, S. J.; Baker, R.-T.; Bazan, G. C. Chem. Rev. 2005, 105, 1001–1020. doi:10.1021/cr020018n
Return to citation in text: [1] -
Nicolaou, K. C.; Toh, Q.-Y.; Chen, D. Y.-K. J. Am. Chem. Soc. 2008, 130, 11292–11293. doi:10.1021/ja804588r
Return to citation in text: [1] -
Leung, G. Y. C.; Li, H.; Toh, Q.-Y.; Ng, A. M.-Y.; Sum, R. J.; Bandow, J. E.; Chen, D. Y.-K. Eur. J. Org. Chem. 2011, 183–196. doi:10.1002/ejoc.201001281
Return to citation in text: [1] -
Yuan, C.; Chang, C.-T.; Axelrod, A.; Siegel, D. J. Am. Chem. Soc. 2010, 132, 5924–5925. doi:10.1021/ja101956x
Return to citation in text: [1] -
Fischer, D. F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926–5927. doi:10.1021/ja101893b
Return to citation in text: [1] -
Jana, C. K.; Hoecker, J.; Woods, T. M.; Jessen, H. J.; Neuburger, M.; Gademann, K. Angew. Chem., Int. Ed. 2011, 50, 8407–8411. doi:10.1002/anie.201101869
Return to citation in text: [1] -
Scott, L. E.; Telpoukhovskaia, M.; Rodriguez-Rodriguez, C.; Merkel, M.; Bowen, M. L.; Page, B. D. G.; Green, D. E.; Storr, T.; Thomas, F.; Allen, D. D.; Lockman, P. R.; Patrick, B. O.; Adam, M. J.; Orvig, C. Chem. Sci. 2011, 2, 642–648. doi:10.1039/c0sc00544d
Return to citation in text: [1] -
Tun, M. K. M.; Wüstmann, D.-J.; Herzon, S. B. Chem. Sci. 2011, 2, 2251–2253. doi:10.1039/c1sc00455g
Return to citation in text: [1] -
Cheng, X.; Harzdorf, N.; Khaing, Z.; Kang, D.; Camelio, A. M.; Shaw, T.; Schmidt, C. E.; Siegel, D. Org. Biomol. Chem. 2012, 10, 383–393. doi:10.1039/c1ob06363d
Return to citation in text: [1] -
Elamparuthi, E.; Fellay, C.; Neuburger, M.; Gademann, K. Angew. Chem., Int. Ed. 2012, 51, 4071–4073. doi:10.1002/anie.201200515
Return to citation in text: [1] -
Newton, J. N.; Fischer, D. F.; Sarpong, R. Angew. Chem., Int. Ed. 2013, 52, 1726–1730. doi:10.1002/anie.201208571
Return to citation in text: [1] -
Drouet, K. E.; Theodorakis, E. A. J. Am. Chem. Soc. 1999, 121, 456–457. doi:10.1021/ja983429n
Return to citation in text: [1] -
Tisdale, E. J.; Slobodov, I.; Theodorakis, E. A. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12030–12035. doi:10.1073/pnas.0401932101
Return to citation in text: [1] -
Vong, B. G.; Kim, S. H.; Abraham, S.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 3947–3951. doi:10.1002/anie.200460203
Return to citation in text: [1] -
Guizzunti, G.; Brady, T. P.; Malhotra, V.; Theodorakis, E. A. J. Am. Chem. Soc. 2006, 128, 4190–4191. doi:10.1021/ja058259a
Return to citation in text: [1] -
Xu, J.; Caro-Diaz, E. J. E.; Trzoss, L.; Theodorakis, E. A. J. Am. Chem. Soc. 2012, 134, 5072–5075. doi:10.1021/ja300807e
Return to citation in text: [1] -
Xu, J.; Caro-Diaz, E. J. E.; Lacoske, M. H.; Hung, C.-I.; Jamora, C.; Theodorakis, E. A. Chem. Sci. 2012, 3, 3378–3386. doi:10.1039/c2sc21308g
Return to citation in text: [1]
| 82. | Lee, H. M.; Nieto-Oberhuber, C.; Shair, M. D. J. Am. Chem. Soc. 2008, 130, 16864–16865. doi:10.1021/ja8071918 |
| 83. | Mcmurry, J. E.; Scott, W. J. Tetrahedron Lett. 1983, 24, 979–982. doi:10.1016/S0040-4039(00)81581-6 |
| 84. | Scott, W. J.; Mcmurry, J. E. Acc. Chem. Res. 1988, 21, 47–54. doi:10.1021/ar00146a001 |
| 85. | Nicolaou, K. C.; Peng, X.-S.; Sun, Y.-P.; Polet, D.; Zou, B.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2009, 131, 10587–10597. doi:10.1021/ja902939t |
| 86. | Ding, H.; Chen, D. Y. K. Angew. Chem., Int. Ed. 2011, 50, 676–679. doi:10.1002/anie.201006367 |
| 87. | Cowell, A.; Stille, J. K. J. Am. Chem. Soc. 1980, 102, 4193–4198. doi:10.1021/ja00532a034 |
| 88. | Cacchi, S.; Morera, E.; Ortar, G. Tetrahedron Lett. 1985, 26, 1109–1112. doi:10.1016/S0040-4039(00)98525-3 |
| 89. | Magro, A. A. N.; Robb, L. M.; Pogorzelec, P. J.; Slawin, A. M. Z.; Eastham, G. R.; Cole-Hamilton, D. J. Chem. Sci. 2010, 1, 723–730. doi:10.1039/c0sc00276c |
| 90. | Nicolaou, K. C.; Ding, H.; Richard, J.-A.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 3815–3818. doi:10.1021/ja9093988 |
| 1. | Sofroniew, M. V.; Howe, C. L.; Mobley, W. C. Annu. Rev. Neurosci. 2001, 24, 1217–1281. doi:10.1146/annurev.neuro.24.1.1217 |
| 2. | Chao, M. V. Nat. Rev. Neurosci. 2003, 4, 299–309. doi:10.1038/nrn1078 |
| 3. | Huang, E. J.; Reichardt, L. F. Annu. Rev. Neurosci. 2001, 24, 677–736. doi:10.1146/annurev.neuro.24.1.677 |
| 8. | Me, Y.; Longo, F. M. Prog. Brain Res. 2000, 128, 333–347. doi:10.1016/S0079-6123(00)28030-8 |
| 9. | Massa, S. M.; Xie, Y. M.; Longo, F. M. J. Mol. Neurosci. 2003, 20, 323–326. doi:10.1385/JMN:20:3:323 |
| 10. | Longo, F. M.; Yang, T.; Knowles, J. K.; Xie, Y. M.; Moore, L. A.; Massa, S. M. Curr. Alzheimer Res. 2007, 4, 503–506. doi:10.2174/156720507783018316 |
| 11. | Joyner, P. M.; Cichewicz, R. H. Nat. Prod. Rep. 2011, 28, 26–47. doi:10.1039/c0np00017e |
| 12. | Williams, P.; Sorribas, A.; Howes, M.-J. R. Nat. Prod. Rep. 2011, 28, 48–77. doi:10.1039/c0np00027b |
| 51. | Ling, T.; Xiang, A. X.; Theodorakis, E. A. Angew. Chem., Int. Ed. 1999, 38, 3089–3091. doi:10.1002/(SICI)1521-3773(19991018)38:20<3089::AID-ANIE3089>3.0.CO;2-W |
| 52. | Ling, T.; Poupon, E.; Rueden, E. J.; Kim, S. H.; Theodorakis, E. A. J. Am. Chem. Soc. 2002, 124, 12261–12267. doi:10.1021/ja027517q |
| 53. | Brady, T. P.; Kim, S. H.; Wen, K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 739–742. doi:10.1002/anie.200352868 |
| 54. | Ling, T.; Poupon, E.; Rueden, E. J.; Theodorakis, E. A. Org. Lett. 2002, 4, 819–822. doi:10.1021/ol025501z |
| 55. | Ghosh, S.; Rivas, F.; Fischer, D.; González, M. A.; Theodorakis, E. A. Org. Lett. 2004, 6, 941–944. doi:10.1021/ol036492c |
| 56. | Brady, T. P.; Kim, S. H.; Wen, K.; Kim, C.; Theodorakis, E. A. Chem.–Eur. J. 2005, 11, 7175–7190. doi:10.1002/chem.200500513 |
| 57. | Nguyen, T. X.; Dakanali, M.; Trzoss, L.; Theodorakis, E. A. Org. Lett. 2011, 13, 3308–3311. doi:10.1021/ol200966z |
| 58. |
Peng, F.; Dai, M.-J.; Angeles, A. R.; Danishefsky, S. J. Chem. Sci. 2012, 3, 3076–3080. doi:10.1039/c2sc20868g
See for a recent update of Robinson annulation. |
| 111. | Drouet, K. E.; Theodorakis, E. A. J. Am. Chem. Soc. 1999, 121, 456–457. doi:10.1021/ja983429n |
| 112. | Tisdale, E. J.; Slobodov, I.; Theodorakis, E. A. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12030–12035. doi:10.1073/pnas.0401932101 |
| 113. | Vong, B. G.; Kim, S. H.; Abraham, S.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 3947–3951. doi:10.1002/anie.200460203 |
| 114. | Guizzunti, G.; Brady, T. P.; Malhotra, V.; Theodorakis, E. A. J. Am. Chem. Soc. 2006, 128, 4190–4191. doi:10.1021/ja058259a |
| 115. | Xu, J.; Caro-Diaz, E. J. E.; Trzoss, L.; Theodorakis, E. A. J. Am. Chem. Soc. 2012, 134, 5072–5075. doi:10.1021/ja300807e |
| 116. | Xu, J.; Caro-Diaz, E. J. E.; Lacoske, M. H.; Hung, C.-I.; Jamora, C.; Theodorakis, E. A. Chem. Sci. 2012, 3, 3378–3386. doi:10.1039/c2sc21308g |
| 6. | Skaper, S. D. CNS Neurol. Disord.: Drug Targets 2008, 7, 46–62. doi:10.2174/187152708783885174 |
| 7. | Skaper, S. D. Curr. Pharm. Des. 2011, 17, 2704–2718. doi:10.2174/138161211797415995 |
| 59. | Wieland, P.; Miescher, K. Helv. Chim. Acta 1950, 33, 2215–2228. doi:10.1002/hlca.19500330730 |
| 60. | Eder, U.; Sauer, G.; Weichert, R. Angew. Chem., Int. Ed. Engl. 1971, 10, 496–497. doi:10.1002/anie.197104961 |
| 61. | Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1973, 38, 3239–3243. doi:10.1021/jo00959a002 |
| 62. | Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615–1621. doi:10.1021/jo00925a003 |
| 63. | Bradshaw, B.; Bonjoch, J. Synlett 2012, 337–356. doi:10.1055/s-0031-1290107 |
| 64. | Zhou, P.; Zhang, L.; Luo, S.; Cheng, J.-P. J. Org. Chem. 2012, 77, 2526–2530. doi:10.1021/jo202433v |
| 65. | Winterfeldt, E. Angew. Chem., Int. Ed. 2013, 52, 4723. doi:10.1002/anie.201301415 |
| 5. | Shulman, J. M.; De Jager, P. L.; Feany, M. B. Annu. Rev. Pathol.: Mech. Dis. 2011, 6, 193–222. doi:10.1146/annurev-pathol-011110-130242 |
| 24. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Org. Lett. 2011, 13, 4554–4557. doi:10.1021/ol201742j |
| 25. | Xu, J.; Trzoss, L.; Chang, W. K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2011, 50, 3672–3676. doi:10.1002/anie.201100313 |
| 26. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem.–Eur. J. 2013, 20, 6398–6408. doi:10.1002/chem.201300198 |
| 26. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem.–Eur. J. 2013, 20, 6398–6408. doi:10.1002/chem.201300198 |
| 4. | Querfurth, H. W.; LaFerla, F. M. N. Engl. J. Med. 2010, 362, 329–344. doi:10.1056/NEJMra0909142 |
| 27. | Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650 |
| 28. | List, B. Acc. Chem. Res. 2004, 37, 548–557. doi:10.1021/ar0300571 |
| 29. | Lelais, G.; MacMillan, D. W. C. Aldrichimica Acta 2006, 39, 79–87. |
| 30. | Taylor, M. S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132 |
| 31. | Marion, N.; Díez-González, S.; Nolan, I. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380 |
| 32. | Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. Drug Discovery Today 2007, 12, 8–27. doi:10.1016/j.drudis.2006.11.004 |
| 33. | Bertelsen, S.; Jorgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g |
| 34. | List, B.; Lerner, R. A.; Barbas, C. F., III. J. Am. Chem. Soc. 2000, 122, 2395–2396. doi:10.1021/ja994280y |
| 35. | Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 9874–9875. doi:10.1021/ja005517p |
| 36. | Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s |
| 37. | Beeson, T. D.; Mastracchio, A.; Hong, J. B.; Ashton, K.; MacMillan, D. W. C. Science 2007, 316, 582–585. |
| 38. | Zhu, S. L.; Wang, Y.; Ma, D. W. Adv. Synth. Catal. 2009, 351, 2563–2566. doi:10.1002/adsc.200900449 |
| 39. | Marques-Lopez, E.; Herrera, R. P.; Christmann, M. Nat. Prod. Rep. 2010, 27, 1138–1167. doi:10.1039/b924964h |
| 40. | Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167–178. doi:10.1038/nchem.539 |
| 41. | Zhu, S. L.; Yu, S. Y.; Wang, Y.; Ma, D. W. Angew. Chem., Int. Ed. 2010, 49, 4656–4660. doi:10.1002/anie.201001644 |
| 42. | Knowles, R. R.; Carpenter, J.; Blakey, S. B.; Kayano, A.; Mangion, I. K.; Sinz, C. J.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 308–311. doi:10.1039/c0sc00577k |
| 43. | Sun, X.; Ma, D. Chem.–Asian J. 2011, 6, 2157–2164. doi:10.1002/asia.201100219 |
| 44. | Pham, P. V.; Ashton, K.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 1470–1473. doi:10.1039/c1sc00176k |
| 45. | Wang, Y.; Zhu, S.; Ma, D. Org. Lett. 2011, 13, 1602–1605. doi:10.1021/ol200004s |
| 46. | Jones, S. B.; Simmons, B.; Mastracchio, A.; MacMillan, D. W. C. Nature 2011, 475, 183–188. doi:10.1038/nature10232 |
| 47. | Zi, W.; Xie, W.; Ma, D. J. Am. Chem. Soc. 2012, 134, 9126–9129. doi:10.1021/ja303602f |
| 48. | Huo, L.; Ma, A.; Zhang, Y.; Ma, D. Adv. Synth. Catal. 2012, 354, 991–994. doi:10.1002/adsc.201100903 |
| 49. | Simonovich, S. P.; Van Humbeck, J. F.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 58–61. doi:10.1039/c1sc00556a |
| 50. | Zhang, Y.; Xing, H.; Xie, W.; Wan, X.; Lai, Y.; Ma, D. Adv. Synth. Catal. 2013, 355, 68–72. doi:10.1002/adsc.201200782 |
| 103. | Yuan, C.; Chang, C.-T.; Axelrod, A.; Siegel, D. J. Am. Chem. Soc. 2010, 132, 5924–5925. doi:10.1021/ja101956x |
| 104. | Fischer, D. F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926–5927. doi:10.1021/ja101893b |
| 105. | Jana, C. K.; Hoecker, J.; Woods, T. M.; Jessen, H. J.; Neuburger, M.; Gademann, K. Angew. Chem., Int. Ed. 2011, 50, 8407–8411. doi:10.1002/anie.201101869 |
| 106. | Scott, L. E.; Telpoukhovskaia, M.; Rodriguez-Rodriguez, C.; Merkel, M.; Bowen, M. L.; Page, B. D. G.; Green, D. E.; Storr, T.; Thomas, F.; Allen, D. D.; Lockman, P. R.; Patrick, B. O.; Adam, M. J.; Orvig, C. Chem. Sci. 2011, 2, 642–648. doi:10.1039/c0sc00544d |
| 107. | Tun, M. K. M.; Wüstmann, D.-J.; Herzon, S. B. Chem. Sci. 2011, 2, 2251–2253. doi:10.1039/c1sc00455g |
| 108. | Cheng, X.; Harzdorf, N.; Khaing, Z.; Kang, D.; Camelio, A. M.; Shaw, T.; Schmidt, C. E.; Siegel, D. Org. Biomol. Chem. 2012, 10, 383–393. doi:10.1039/c1ob06363d |
| 109. | Elamparuthi, E.; Fellay, C.; Neuburger, M.; Gademann, K. Angew. Chem., Int. Ed. 2012, 51, 4071–4073. doi:10.1002/anie.201200515 |
| 110. | Newton, J. N.; Fischer, D. F.; Sarpong, R. Angew. Chem., Int. Ed. 2013, 52, 1726–1730. doi:10.1002/anie.201208571 |
| 17. | Yokoyama, R.; Huang, J. M.; Yang, C. S.; Fukuyama, Y. J. Nat. Prod. 2002, 65, 527–531. doi:10.1021/np010571k |
| 18. | Kubo, M.; Kobayashi, K.; Huang, J.-M.; Harada, K.; Fukuyama, Y. Tetrahedron Lett. 2012, 53, 1231–1235. doi:10.1016/j.tetlet.2011.12.107 |
| 99. | Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872 |
| 100. | Wasilke, J.-C.; Obrey, S. J.; Baker, R.-T.; Bazan, G. C. Chem. Rev. 2005, 105, 1001–1020. doi:10.1021/cr020018n |
| 16. | Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett. 2009, 11, 5190–5193. doi:10.1021/ol9021029 |
| 19. | Cho, Y. S.; Carcache, D. A.; Tian, Y.; Li, Y. M.; Danishefsky, S. J. J. Am. Chem. Soc. 2004, 126, 14358–14359. doi:10.1021/ja045939p |
| 20. | Carcache, D. A.; Cho, Y. S.; Hua, Z.; Tian, Y.; Li, Y.-M.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 1016–1022. doi:10.1021/ja056980a |
| 21. | Harada, K.; Imai, A.; Uto, K.; Carter, R. G.; Kubo, M.; Hioki, H.; Fukuyama, Y. Org. Lett. 2011, 13, 988–991. doi:10.1021/ol103024z |
| 22. | Mehta, G.; Shinde, H. M.; Kumaran, R. S. Tetrahedron Lett. 2012, 53, 4320–4323. doi:10.1016/j.tetlet.2012.06.001 |
| 23. | Yang, Y.; Fu, X.; Chen, J.; Zhai, H. Angew. Chem., Int. Ed. 2012, 51, 9825–9828. doi:10.1002/anie.201203176 |
| 24. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Org. Lett. 2011, 13, 4554–4557. doi:10.1021/ol201742j |
| 101. | Nicolaou, K. C.; Toh, Q.-Y.; Chen, D. Y.-K. J. Am. Chem. Soc. 2008, 130, 11292–11293. doi:10.1021/ja804588r |
| 102. | Leung, G. Y. C.; Li, H.; Toh, Q.-Y.; Ng, A. M.-Y.; Sum, R. J.; Bandow, J. E.; Chen, D. Y.-K. Eur. J. Org. Chem. 2011, 183–196. doi:10.1002/ejoc.201001281 |
| 14. | Yang, C.-S.; Kouno, I.; Kawano, N.; Sato, S. Tetrahedron Lett. 1988, 29, 1165–1168. doi:10.1016/S0040-4039(00)86678-2 |
| 15. | Kouno, I.; Baba, N.; Hashimoto, M.; Kawano, N.; Takahashi, M.; Kaneto, H.; Yang, C.-S.; Sato, S. Chem. Pharm. Bull. 1989, 37, 2448–2451. doi:10.1248/cpb.37.2448 |
| 91. | Zuo, Z.; Xie, W.; Ma, D. J. Am. Chem. Soc. 2010, 132, 13226–13228. doi:10.1021/ja106739g |
| 92. | Richard, J.-A.; Chen, D. Y.-K. Eur. J. Org. Chem. 2012, 484–487. doi:10.1002/ejoc.201101629 |
| 13. | Urabe, D.; Inoue, M. Tetrahedron 2009, 65, 6271–6289. doi:10.1016/j.tet.2009.06.010 |
| 16. | Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett. 2009, 11, 5190–5193. doi:10.1021/ol9021029 |
| 93. | Nicolaou, K. C.; Majumder, U.; Roche, S. P.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2007, 46, 4715–4718. doi:10.1002/anie.200701947 |
| 94. | Nicolaou, K. C.; Dalby, S. M.; Li, S.; Suzuki, T.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 7616–7620. doi:10.1002/anie.200904588 |
| 95. | Nicolaou, K. C.; Wu, T. R.; Kang, Q.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 3440–3443. doi:10.1002/anie.200900438 |
| 96. | Nicolaou, K. C.; Kang, Q.; Wu, T. R.; Lim, C. S.; Chen, D. Y.-K. J. Am. Chem. Soc. 2010, 132, 7540–7548. doi:10.1021/ja102623j |
| 97. | Peixoto, P. A.; Richard, J.-A.; Severin, R.; Chen, D. Y.-K. Org. Lett. 2011, 13, 5724–5727. doi:10.1021/ol202053m |
| 98. | Peixoto, P. A.; Severin, R.; Tseng, C.-C.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2011, 50, 3013–3016. doi:10.1002/anie.201008000 |
| 69. | Ruprah, P. K.; Cros, J.-P.; Pease, J. E.; Whittingham, W. G.; Williams, J. M. J. Eur. J. Org. Chem. 2002, 3145–3152. doi:10.1002/1099-0690(200209)2002:18<3145::AID-EJOC3145>3.0.CO;2-3 |
| 70. | Lacoste, E.; Vaique, E.; Berlande, M.; Pianet, I.; Vincent, J.-M.; Landais, Y. Eur. J. Org. Chem. 2007, 167–177. doi:10.1002/ejoc.200600664 |
| 71. | Zhang, X.-M.; Wang, M.; Tu, Y.-Q.; Fan, C.-A.; Jiang, Y.-J.; Zhang, S.-Y.; Zhang, F.-M. Synlett 2008, 2831–2835. doi:10.1055/s-0028-1083542 |
| 25. | Xu, J.; Trzoss, L.; Chang, W. K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2011, 50, 3672–3676. doi:10.1002/anie.201100313 |
| 26. | Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem.–Eur. J. 2013, 20, 6398–6408. doi:10.1002/chem.201300198 |
| 66. | Tsuji, J.; Takahashi, H.; Morikawa, M. Tetrahedron Lett. 1965, 6, 4387–4388. doi:10.1016/S0040-4039(00)71674-1 |
| 67. | Trost, B. M.; Fullerto, T. J. Am. Chem. Soc. 1973, 95, 292–294. doi:10.1021/ja00782a080 |
| 68. | Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804 |
| 25. | Xu, J.; Trzoss, L.; Chang, W. K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2011, 50, 3672–3676. doi:10.1002/anie.201100313 |
| 79. | Finkbeiner, H. L.; Stiles, M. J. Am. Chem. Soc. 1963, 85, 616–622. doi:10.1021/ja00888a031 |
| 80. | Micheli, R. A.; Hajos, Z. G.; Cohen, N.; Parrish, D. R.; Portland, L. A.; Sciamanna, W.; Scott, M. A.; Wehrli, P. A. J. Org. Chem. 1975, 40, 675–681. doi:10.1021/jo00894a003 |
| 81. | Frie, J. L.; Jeffrey, C. S.; Sorensen, E. J. Org. Lett. 2009, 11, 5394–5397. doi:10.1021/ol902168g |
| 77. | CCDC 931875 contains the supplementary crystallographic data for compound 16. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/products/csd/request/. |
| 78. | Angeles, A. R.; Dorn, D. C.; Kou, C. A.; Moore, M. A. S.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2007, 46, 1451–1454. doi:10.1002/anie.200604308 |
| 75. | Pu, X.; Ma, D. Angew. Chem., Int. Ed. 2004, 43, 4222–4225. doi:10.1002/anie.200460128 |
| 76. | Paquette, L. A.; Wang, T.-Z.; Philippo, C. M. G.; Wang, S. J. Am. Chem. Soc. 1994, 116, 3367–3374. doi:10.1021/ja00087a023 |
| 71. | Zhang, X.-M.; Wang, M.; Tu, Y.-Q.; Fan, C.-A.; Jiang, Y.-J.; Zhang, S.-Y.; Zhang, F.-M. Synlett 2008, 2831–2835. doi:10.1055/s-0028-1083542 |
| 72. | Coates, R. M.; Shaw, J. E. Chem. Commun. 1968, 515–516. doi:10.1039/c19680000515 |
| 73. | Williams, J. R.; Sarkisia, G. M. Synthesis 1974, 32–33. doi:10.1055/s-1974-23227 |
| 74. | Bosch, M. P.; Camps, F.; Coll, J.; Guerrero, A.; Tatsuoka, T.; Meinwald, J. J. Org. Chem. 1986, 51, 773–784. doi:10.1021/jo00356a002 |
© 2013 Trzoss et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)