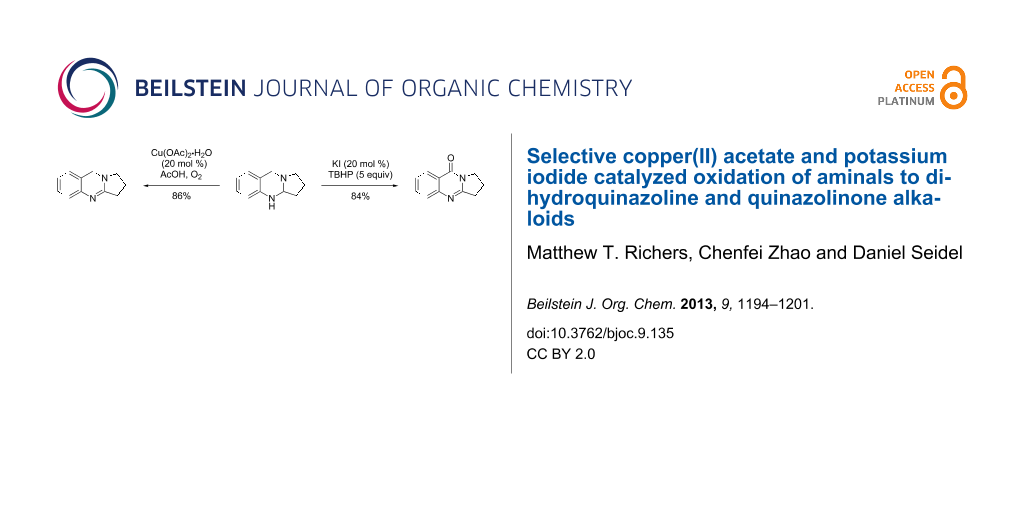
Abstract
Copper(II) acetate/acetic acid/O2 and potassium iodide/tert-butylhydroperoxide systems are shown to affect the selective oxidation of ring-fused aminals to dihydroquinazolines and quinazolinones, respectively. These methods enable the facile preparation of a number of quinazoline alkaloid natural products and their analogues.

Graphical Abstract
Introduction
Quinazoline alkaloids are a class of naturally occurring compounds with a range of medicinal properties and have been indicated for use as bronchodilators, vasodilators, anti-inflammatory agents and acetylcholinesterase inhibitors [1-5]. Many of the plants these products have been isolated from, such as Adhatoda vasica, Peganum harmala and Evodia rutaecarpa, have been used in folk medicine for centuries [6-9]. Since the original isolation of vasicine (1, Figure 1) in 1888 [10], the biological properties of this class of alkaloids have been extensively studied.
![[1860-5397-9-135-1]](/bjoc/content/figures/1860-5397-9-135-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of naturally occurring quinazoline alkaloids.
Figure 1: Examples of naturally occurring quinazoline alkaloids.
A number of synthetic strategies have been employed to gain access to quinazoline alkaloids [5,11-26]. Perhaps the most common method involves the condensation of an ortho-aminobenzoic ester with a lactam promoted by phosphoryl chloride, known as the Niementowski reaction [3,27-30] (Figure 2). The availability, or lack thereof, of the corresponding lactam can determine the length and efficiency of the route. Access to the sometimes more biologically active dihydroquinazolines, such as deoxyvasicine (2), from quinazolinones requires a subsequent reduction of the amide. In 2008, our group reported the syntheses of deoxyvasicinone (4) and rutaecarpine (6) by the potassium permanganate promoted oxidation of aminals, which in turn were obtained from the condensation of ortho-aminobenzaldehydes and simple secondary amines [31,32]. A number of these aminal precursors were prepared in generally good to excellent yields with the scope encompassing various cyclic amines and substituents on the aminobenzaldehyde aryl ring. Since then, we have demonstrated that the reaction can be run on a multigram scale [33] and have shown that dihydroquinazolines vasicine (1) and deoxyvasicine (2) can be synthesized from their corresponding aminals by using an iodine-promoted oxidation [34]. While resulting in good yields, these oxidations have the drawback of requiring large amounts of a strong oxidant for the permanganate oxidation and the necessity of stoichiometric n-butyllithium for the iodine reaction.
![[1860-5397-9-135-2]](/bjoc/content/figures/1860-5397-9-135-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Different approaches to the synthesis of quinazoline alkaloid structures.
Figure 2: Different approaches to the synthesis of quinazoline alkaloid structures.
The conversion of the aminals formed from the condensation of aminobenzaldehydes and secondary amines to the corresponding dihydroquinazoline and quinazolinone structures under mild and catalytic conditions would be preferable to using harsh oxidants and strong bases. Han et al. have recently shown the ability of copper salts, in conjunction with oxygen, to catalyze oxidations of 2-substituted tetrahydroquinazoline aminals to quinazolines [35] (Figure 2). In addition, Reddy and co-workers have developed a catalytic system in which 2,3-substituted tetrahydroquinazoline aminals are converted to quinazolinones using tert-butylhydroperoxide (TBHP) and catalytic potassium iodide [36,37]. While these examples deal with the oxidation of bicyclic aminals, we were interested in developing methods to create dihydroquinazoline and quinazolinone alkaloids from ring-fused aminals. Here we present catalytic methods for the synthesis of both these compound classes from aminals using Cu(OAc)2/O2/AcOH and KI/TBHP systems, respectively.
Results and Discussion
Copper-catalyzed oxidations of aminals to dihydroquinazolines
Copper-catalyzed oxidation reactions have received a great deal of interest in recent years [38-44]. Han’s copper-catalyzed method for the synthesis of aminals to quinazolines results in high yields [35], but the process is not applicable to mono-oxidation as dihydroquinazolines are not isolated as products in these reactions. We set out to develop a method for the synthesis of dihydroquinazolines that would prevent further oxidation at the benzylic position. A factor complicating this effort was that dihydroquinazolines like deoxyvasicine (2) are known to auto-oxidize to their quinazolinone counterparts by exposure to air [3,45-47]. We initiated our efforts by exposing aminal 7 to stoichiometric amounts of CuCl2 in acetonitrile under a nitrogen atmosphere, which led to the formation of 2 in 81% yield (Table 1, entry 1). To improve the efficiency of the process, catalytic conditions were subsequently evaluated. When aminal 7 was heated under reflux in an oxygen atmosphere and in the presence of 20 mol % of CuCl2, 2 was only observed in trace amounts; deoxyvasicinone (4) and peroxide 8 were also formed as products. Switching the catalyst to Cu(OAc)2 led to a 15% yield of the desired product 2, but the process was still unselective.
Table 1: Optimization of conditions for deoxyvasicine (2) formation.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-135-i2.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Solvent (0.2 M) | Catalyst (mol %) | Acid (equiv) | Temp. (°C) | Time (h) | Yield of 2 (%) | Yield of 4 (%) | Yield of 8 (%) |
|---|---|---|---|---|---|---|---|---|
| 1b | MeCN | CuCl2∙2H2O (100) | – | rt | 6 | 81 | – | – |
| 2 | MeCN | CuCl2∙2H2O (20) | – | 81 | 2 | trace | 14 | 10 |
| 3 | MeCN | Cu(OAc)2∙H2O (20) | – | 81 | 3 | 15 | 17 | trace |
| 4 | MeCN | Cu(OAc)2∙H2O (20) | AcOH (1.1) | 81 | 3 | 53 | – | – |
| 5 | MeOH | Cu(OAc)2∙H2O (20) | AcOH (1.1) | 65 | 4 | 81 | – | – |
| 6 | MeOH | Cu(OAc)2∙H2O (20) | – | 65 | 4 | 33 | 6 | 24 |
| 7 | AcOH | Cu(OAc)2∙H2O (20) | – | 80 | 24 | 18c | – | – |
| 8 | DMF | Cu(OAc)2∙H2O (20) | AcOH (1.1) | 80 | 4 | 17 | 20 | – |
| 9 | MeOH | Cu(2-EH)2 (20) | 2-EHA (1.1) | 65 | 12 | 71 | – | – |
| 10 | MeOH | CuBr (20) | AcOH (1.1) | 65 | 8 | 72 | – | – |
| 11 | EtOHd | Cu(OAc)2∙H2O (20) | AcOH (1.1) | 78 | 1.5 | 73 | – | trace |
| 12 | MeOH | Cu(OAc)2∙H2O (10) | AcOH (1.1) | 65 | 18 | 67 | – | trace |
| 13 | MeOH | Cu(OAc)2∙H2O (20) | AcOH (1.1) | 40 | 24 | 61 | trace | trace |
| 14 | MeOH | Cu(acac)2 (10) | AcOH (1.1) | 65 | 24 | 68c | trace | trace |
aReactions were performed on a 0.25 mmol scale. Cu(2-EH)2 = copper(II) 2-ethylhexanoate. 2-EHA = 2-ethylhexanoic acid. bNitrogen atmosphere. cThe reaction was incomplete. d95% Solution.
It appears that the first oxidation occurs exclusively at the aminal site to form deoxyvasicine (2). The presence of the amidine moiety apparently activates the molecule for oxidation at the benzylic position; we have observed that samples of aminal 7 can remain stable in the freezer for years, whereas 2 begins to convert to 4 within a day when exposed to atmospheric oxygen. Considering this, we reasoned that addition of a weak acid to protonate the relatively basic amidine moiety of 2 might deactivate the benzylic position toward oxidation while not interfering with the initial aminal oxidation. Indeed, using 1.1 equivalents of acetic acid as an additive with catalytic Cu(OAc)2 in acetonitrile led to the formation of 2 in 53% yield without formation of 4 and 8 (Table 1, entry 4). A simple change of the solvent from acetonitrile to methanol drastically improved the yield of 2 to 81% (Table 1, entry 5). A number of different copper salts, solvents and acids were then evaluated, but none of the changes led to a further improvement in yield. It appears that under certain conditions catalyst deactivation via copper oxide formation decreased the catalyst turnover and consequently product yields.
Using the optimized reaction conditions, a range of different aminals were selectively oxidized to the corresponding dihydroquinazolines (Table 2). In general, these products were obtained in moderate to good yields. Product 10, containing a piperidine ring, required a higher reaction temperature and resulted in a lower yield than the corresponding pyrrolidine and azepane products (2 and 12, respectively). While differences in conformation may in part account for the observed differences in reactivity (X-ray crystal structures of aminals containing pyrrolidine and piperidine revealed that the pyrrolidine-containing aminal adopts a bent structure, whereas the piperidine aminal appears relatively strain-free [34]), this finding likely relates to the reduced propensity of six-membered rings to engage in reactions that form exocyclic double bonds. The isolation of azepinoquinazoline 12 in 73% yield was gratifying but somewhat unexpected since Decker reported that samples of the compound completely oxidized to quinazolinone 23 when exposed to air for 24 h [3]. This demonstrates the need for acetic acid to protonate the amidine, preventing further oxidation. While product 16 was obtained in good yields from tetrahydroisoquinoline-aminal 15, rutaecarpane-derived product 18 was formed in only 47% yield, apparently due to unidentified side-reactions. The reaction leading to the synthesis of the dibromo- analogue of deoxyvasicine 20, even under elevated temperature and extended reaction time, still did not reach completion after 3 days. The attenuated reactivity of aminal 19 is most likely the result of the decreased electron density on the anilinic nitrogen.
Table 2: Scope of the copper-catalyzed conversion of aminals to dihydroquinazolines.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-135-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Starting material | Product | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-135-i4.svg?max-width=637&scale=1.0)
7 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-135-i5.svg?max-width=637&scale=1.0)
2 |
7 | 86 |
| 2b,c |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-135-i6.svg?max-width=637&scale=1.0)
9 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-135-i7.svg?max-width=637&scale=1.0)
10 |
8 | 57 |
| 3c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-135-i8.svg?max-width=637&scale=1.0)
11 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-135-i9.svg?max-width=637&scale=1.0)
12 |
7 | 73 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-135-i10.svg?max-width=637&scale=1.0)
13 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-135-i11.svg?max-width=637&scale=1.0)
14 |
24 | 72 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-135-i12.svg?max-width=637&scale=1.0)
15 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-135-i13.svg?max-width=637&scale=1.0)
16 |
8 | 82 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-135-i14.svg?max-width=637&scale=1.0)
17 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-135-i15.svg?max-width=637&scale=1.0)
18 |
4 | 47 |
| 7b,d |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-135-i16.svg?max-width=637&scale=1.0)
19 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-135-i17.svg?max-width=637&scale=1.0)
20 |
72 | 18 |
aReactions run on a 1 mmol scale. bEtOH used as solvent. c0.5 mmol scale. dReaction incomplete.
KI-catalyzed oxidations of aminals to quinazolinones
Different conditions for the direct catalytic oxidation of aminals to quinazolinones were also explored. The use of Cu(OAc)2 and methanol, while appropriate for furnishing deoxyvasicine (2) from aminal 7, did not result in satisfactory yields of deoxyvasicinone (4, Table 1). Attempts to use other copper(I) or copper(II) salts and solvents under oxygen without the addition of acid to promote the full oxidation of aminal 21 to deoxyvasicinone (4) were met with disappointment, with yields of 4 for these conditions reaching a maximum of around 40% (Table 3). In most cases, peroxide 8 was observed as a major side product. The Cu/TEMPO/DABCO catalyst system employed by Han et al. [35] for the oxidation of aminals to quinazolines provided an increased yield of 50% (Table 3, entry 9). The best yields were obtained by using the conditions developed by Reddy and co-workers [36], namely the combined use of catalytic amounts of potassium iodide (20 mol %) and excess TBHP (5 equiv), followed by the addition of piperidine. In this instance, deoxyvasicinone was isolated in 80% yield (Table 3, entry 12). In the course of this reaction, the TBHP adduct 22 is formed as an intermediate that is subsequently converted to the quinazolinone upon addition of piperidine. A slight modification of Reddy’s conditions, in which piperidine was added directly to the solution after 36 hours instead of the removal of solvent from the intermediate peroxide beforehand, resulted in identical yields.
Table 3: Optimization of conditions for deoxyvasicinone (4) formation.a
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-135-i18.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Solvent (0.2 M) | Catalyst (mol %) | Oxidant (equiv) | Additive (equiv) | Temp. (°C) | Time (h) | Yield of 4 (%) |
|---|---|---|---|---|---|---|---|
| 1 | DMSO | CuBr (20) | O2 | – | 100 | 2 | 21 |
| 2 | DMSO | CuBr (20) | O2 | DBU (0.4) | 100 | 17 | 25 |
| 3 | DMSO | CuBr (20) | O2 | DBU (2) | 100 | 3 | 22 |
| 4 | DMSO | CuBr (20) | O2 | – | 60 | 3 | 28 |
| 5 | MeCN | CuBr(10) | O2 | – | 80 | 24 | 43 |
| 6 | DMF | CuBr(10) | O2 | – | 80 | 24 | 42 |
| 7 | DMSO | CuI (20) | O2 | – | 60 | 3 | 29 |
| 8 | MeCN | CuCl2·2H2O (20) | O2 | – | 50 | 5 | 19 |
| 9 | MeCN | CuCl (10) | O2 |
DABCO (0.1),
TEMPO (0.05) |
80 | 12 | 50 |
| 10 | DMSO | CuCl (10) | O2 |
DABCO (0.1),
TEMPO (0.05) |
100 | 3 | 38 |
| 11 | PhMe | CuBr (20) | TBHP (5) | piperidineb (5) | rt | 0.5 | 61 |
| 12 | EtOH | KI (20) | TBHP (5) | piperidineb (5) | rt | 36 | 80 |
aReactions run on a 0.25 mmol scale. DBU = 1,8-Diazabicyclo[5.4.0]undec-7-ene. DABCO = 1,4-Diazabicyclo[2.2.2]octane. TEMPO = 2,2,6,6-Tetramethylpiperidine-1-oxy radical. bPiperidine was added at the end of the reaction and the reaction mixture was heated at 50 °C for 1 h.
Using the optimized conditions, a range of different quinazolinones were synthesized (Table 4). In general, yields were moderate to good for substrates with varying ring sizes. In this manner the natural products deoxyvasicine (4), mackinazolinone (5) and rutaecarpine (6) were prepared, in addition to the azepinoquinazolone 23, which has been demonstrated to be a more effective antitussive agent than codeine [48]. Dibromo-deoxyvasicinone analogue 25 was obtained in relatively high yield (88%) whereas the corresponding analogue of mackinazolinone (27) was obtained in only 50% yield.
Table 4: Scope of KI-catalyzed conversion of aminals to quinazolinones.a
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-135-i19.svg?max-width=637&scale=1.0)
|
|||
| Entry | Starting material | Product | Yield [%] |
|---|---|---|---|
| 1 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-135-i20.svg?max-width=637&scale=1.0)
7 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-135-i21.svg?max-width=637&scale=1.0)
4 |
84 |
| 2b |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-135-i22.svg?max-width=637&scale=1.0)
9 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-135-i23.svg?max-width=637&scale=1.0)
5 |
59 |
| 3b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-135-i24.svg?max-width=637&scale=1.0)
11 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-135-i25.svg?max-width=637&scale=1.0)
23 |
69 |
| 4 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-135-i26.svg?max-width=637&scale=1.0)
15 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-135-i27.svg?max-width=637&scale=1.0)
24 |
60 |
| 5 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-135-i28.svg?max-width=637&scale=1.0)
17 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-135-i29.svg?max-width=637&scale=1.0)
6 |
58 |
| 6 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-135-i30.svg?max-width=637&scale=1.0)
19 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-135-i31.svg?max-width=637&scale=1.0)
25 |
88 |
| 7 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-135-i32.svg?max-width=637&scale=1.0)
26 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-135-i33.svg?max-width=637&scale=1.0)
27 |
50 |
aReactions run on a 1 mmol scale. b0.5 mmol scale.
Interestingly, when quaternary aminal 28 was subjected to oxidative conditions in an attempt to prepare compound 29, deoxyvasicinone (4) was obtained as the major product in a process that involved demethylation (Scheme 1, reaction 1). The demethylation of aminals has been previously reported in cases where the product achieves aromaticity [49-51], which is presumably the driving force for this transformation. Aminal 30, which contains two tertiary amines and is readily obtainable by an acid-promoted hydride shift process [52-54], was also exposed to oxidative conditions (Scheme 1, reaction 2). We had hypothesized that quinazolinone 32 might be formed in this reaction by the debenzylation of an intermediate iminium ion. However, the major product from this reaction was identified to be 31, the apparent product of iminium hydrolysis.
![[1860-5397-9-135-i1]](/bjoc/content/inline/1860-5397-9-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Oxidation of other aminal systems.
Scheme 1: Oxidation of other aminal systems.
Conclusion
We have demonstrated that quinazoline alkaloids and their analogues can be synthesized from aminals by using Cu(OAc)2/O2/AcOH and KI/TBHP catalyst systems. The use of acetic acid in addition to oxygen and catalytic copper(II) salts was determined to prevent overoxidation of dihydroquinazolines, allowing access to these structures under mild conditions. A number of natural products and their analogues were obtainable by these methods, which should facilitate the preparation of novel materials for biological studies.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and 1H and 13C NMR spectra for all new compounds. | ||
| Format: PDF | Size: 733.7 KB | Download |
References
-
D’yakonov, A. L.; Telezhenetskaya, M. V. Chem. Nat. Compd. 1997, 33, 221. doi:10.1007/BF02234869
Return to citation in text: [1] -
Sheu, J.-R. Cardiovasc. Drug Rev. 1999, 17, 237. doi:10.1111/j.1527-3466.1999.tb00017.x
Return to citation in text: [1] -
Decker, M. Eur. J. Med. Chem. 2005, 40, 305. doi:10.1016/j.ejmech.2004.12.003
Return to citation in text: [1] [2] [3] [4] -
Rachana, S.; Ujata, B.; Mamta, P.; Priyanka, K. M.; Sonam, S. Indo Global J. Pharm. Sci. 2011, 1, 85.
Return to citation in text: [1] -
Nepali, K.; Sharma, S.; Ojha, R.; Dhar, K. L. Med. Chem. Res. 2013, 22, 1. doi:10.1007/s00044-012-0002-5
Return to citation in text: [1] [2] -
Jia, S.; Hu, C. Molecules 2010, 15, 1873. doi:10.3390/molecules15031873
Return to citation in text: [1] -
Dhankhar, S.; Kaur, R.; Ruhil, S.; Balhara, M.; Dhankhar, S.; Chhillar, A. K. Afr. J. Plant Sci. 2011, 5, 620.
Return to citation in text: [1] -
Corrêa, G. M.; Alcântara, A. F. de C. Rev. Bras. Farmacogn. 2012, 22, 220. doi:10.1590/S0102-695X2011005000196
Return to citation in text: [1] -
Asgarpanah, J.; Ramezanloo, F. Afr. J. Pharm. Pharmacol. 2012, 6, 1573.
Return to citation in text: [1] -
Hooper, D. Pharm. J. Trans. 1888, 18, 841.
Return to citation in text: [1] -
Bergman, J.; Bergman, S. Heterocycles 1981, 16, 347. doi:10.3987/R-1981-03-0347
Return to citation in text: [1] -
Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038. doi:10.1021/jo010727l
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S. Tetrahedron Lett. 2002, 43, 1905. doi:10.1016/S0040-4039(02)00135-1
Return to citation in text: [1] -
Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787. doi:10.1016/j.tet.2006.07.098
Return to citation in text: [1] -
Bowman, W. R.; Elsegood, M. R. J.; Stein, T.; Weaver, G. W. Org. Biomol. Chem. 2007, 5, 103. doi:10.1039/b614075k
Return to citation in text: [1] -
Zhou, J.; Fu, L.; Lv, M.; Liu, J.; Pei, D.; Ding, K. Synthesis 2008, 3974. doi:10.1055/s-0028-1083245
Return to citation in text: [1] -
Kraus, G. A.; Guo, H. J. Org. Chem. 2009, 74, 5337. doi:10.1021/jo900718g
Return to citation in text: [1] -
Giri, R.; Lam, J. K.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 686. doi:10.1021/ja9077705
Return to citation in text: [1] -
Kshirsagar, U. A.; Puranik, V. G.; Argade, N. P. J. Org. Chem. 2010, 75, 2702. doi:10.1021/jo100400z
Return to citation in text: [1] -
Dabiri, M.; Salehi, P.; Bahramnejad, M.; Alizadeh, M. Monatsh. Chem. 2010, 141, 877. doi:10.1007/s00706-010-0341-1
Return to citation in text: [1] -
Zeng, F.; Alper, H. Org. Lett. 2010, 12, 3642. doi:10.1021/ol101428v
Return to citation in text: [1] -
Kshirsagar, U. A.; Argade, N. P. Org. Lett. 2010, 12, 3716. doi:10.1021/ol101597p
Return to citation in text: [1] -
Tseng, M.-C.; Cheng, H.-T.; Shen, M.-J.; Chu, Y.-H. Org. Lett. 2011, 13, 4434. doi:10.1021/ol201793v
Return to citation in text: [1] -
Granger, B. A.; Kaneda, K.; Martin, S. F. Org. Lett. 2011, 13, 4542. doi:10.1021/ol201739u
Return to citation in text: [1] -
Fang, J.; Zhou, J. Org. Biomol. Chem. 2012, 10, 2389. doi:10.1039/c2ob07178a
Return to citation in text: [1] -
Murai, K.; Komatsu, H.; Nagao, R.; Fujioka, H. Org. Lett. 2012, 14, 772. doi:10.1021/ol203313n
Return to citation in text: [1] -
von Niementowski, S. J. Prakt. Chem. 1895, 51, 564. doi:10.1002/prac.18950510150
Return to citation in text: [1] -
Pachter, I.; Suld, G. J. Org. Chem. 1960, 25, 1680. doi:10.1021/jo01079a615
Return to citation in text: [1] -
D’yakonov, A. L.; Telezhenetskaya, M. V.; Yunusov, L. Yu. Chem. Nat. Compd. 1986, 22, 435. doi:10.1007/BF00579819
Return to citation in text: [1] -
Lee, E. S.; Park, J.-G.; Jahng, Y. Tetrahedron Lett. 2003, 44, 1883. doi:10.1016/S0040-4039(03)00080-7
Return to citation in text: [1] -
Zhang, C.; De, C. K.; Mal, R.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 416. doi:10.1021/ja077473r
Return to citation in text: [1] -
Dieckmann, A.; Richers, M. T.; Platonova, A. Yu.; Zhang, C.; Seidel, D.; Houk, K. N. J. Org. Chem. 2013, 78, 4132. doi:10.1021/jo400483h
Return to citation in text: [1] -
Zhang, C.; De, C. K.; Seidel, D. Org. Synth. 2012, 89, 274.
Return to citation in text: [1] -
Richers, M. T.; Deb, I.; Platonova, A. Y.; Zhang, C.; Seidel, D. Synthesis 2013, 45, 1730–1748. doi:10.1055/s-0033-1338852
Return to citation in text: [1] [2] -
Han, B.; Yang, X.-L.; Wang, C.; Bai, Y.-W.; Pan, T.-C.; Chen, X.; Yu, W. J. Org. Chem. 2012, 77, 1136. doi:10.1021/jo2020399
Return to citation in text: [1] [2] [3] -
Kumar, R. A.; Maheswari, C. U.; Ghantasala, S.; Jyothi, C.; Reddy, K. R. Adv. Synth. Catal. 2011, 353, 401. doi:10.1002/adsc.201000580
Return to citation in text: [1] [2] -
Kumar, R. A.; Saidulu, G.; Prasad, K. R.; Kumar, G. S.; Sridhar, B.; Reddy, K. R. Adv. Synth. Catal. 2012, 354, 2985. doi:10.1002/adsc.201200679
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n
Return to citation in text: [1] -
Punniyamurthy, T.; Rout, L. Coord. Chem. Rev. 2008, 252, 134. doi:10.1016/j.ccr.2007.04.003
Return to citation in text: [1] -
Wendlandt, A. E.; Suess, A. M.; Stahl, S. S. Angew. Chem., Int. Ed. 2011, 50, 11062. doi:10.1002/anie.201103945
Return to citation in text: [1] -
Hirano, K.; Miura, M. Chem. Commun. 2012, 48, 10704. doi:10.1039/c2cc34659a
Return to citation in text: [1] -
Zhang, C.; Tang, C.; Jiao, N. Chem. Soc. Rev. 2012, 41, 3464. doi:10.1039/c2cs15323h
Return to citation in text: [1] -
Jones, K. M.; Klussmann, M. Synlett 2012, 159. doi:10.1055/s-0031-1290117
Return to citation in text: [1] -
Ratnikov, M. O.; Doyle, M. P. J. Am. Chem. Soc. 2013, 135, 1549. doi:10.1021/ja3113559
Return to citation in text: [1] -
Mehta, D. R.; Naravane, J. S.; Desai, R. M. J. Org. Chem. 1963, 28, 445. doi:10.1021/jo01037a041
Return to citation in text: [1] -
Johns, S. R.; Lamberton, J. A.; Suares, H. Aust. J. Chem. 1985, 38, 1007. doi:10.1071/CH9851007
Return to citation in text: [1] -
Nakagawa, Y.; Stevens, R. V. J. Org. Chem. 1988, 53, 1873. doi:10.1021/jo00244a007
Return to citation in text: [1] -
Nepali, K.; Bande, M. S.; Sapra, S.; Garg, A.; Kumar, S.; Sharma, P.; Goyal, R.; Satti, N. K.; Suri, O. P.; Dhar, K. L. Med. Chem. Res. 2012, 21, 1271. doi:10.1007/s00044-011-9641-1
Return to citation in text: [1] -
Elderfield, R. C.; Kreysa, F. J.; Dunn, J. H.; Humphreys, D. D. J. Am. Chem. Soc. 1947, 69, 186. doi:10.1021/ja01193a506
Return to citation in text: [1] -
Yamazaki, C. J. Org. Chem. 1981, 46, 3956. doi:10.1021/jo00333a005
Return to citation in text: [1] -
Schulze, K.; Richter, C.; Ludwig, R. Tetrahedron Lett. 1989, 30, 2369. doi:10.1016/S0040-4039(01)80401-9
Return to citation in text: [1] -
Zhang, C.; Murarka, S.; Seidel, D. J. Org. Chem. 2009, 74, 419. doi:10.1021/jo802325x
Return to citation in text: [1] -
Mori, K.; Ohshima, Y.; Ehara, K.; Akiyama, T. Chem. Lett. 2009, 38, 524. doi:10.1246/cl.2009.524
Return to citation in text: [1] -
He, Y.-P.; Du, Y.-L.; Luo, S.-W.; Gong, L.-Z. Tetrahedron Lett. 2011, 52, 7064. doi:10.1016/j.tetlet.2011.10.062
Return to citation in text: [1]
| 49. | Elderfield, R. C.; Kreysa, F. J.; Dunn, J. H.; Humphreys, D. D. J. Am. Chem. Soc. 1947, 69, 186. doi:10.1021/ja01193a506 |
| 50. | Yamazaki, C. J. Org. Chem. 1981, 46, 3956. doi:10.1021/jo00333a005 |
| 51. | Schulze, K.; Richter, C.; Ludwig, R. Tetrahedron Lett. 1989, 30, 2369. doi:10.1016/S0040-4039(01)80401-9 |
| 36. | Kumar, R. A.; Maheswari, C. U.; Ghantasala, S.; Jyothi, C.; Reddy, K. R. Adv. Synth. Catal. 2011, 353, 401. doi:10.1002/adsc.201000580 |
| 48. | Nepali, K.; Bande, M. S.; Sapra, S.; Garg, A.; Kumar, S.; Sharma, P.; Goyal, R.; Satti, N. K.; Suri, O. P.; Dhar, K. L. Med. Chem. Res. 2012, 21, 1271. doi:10.1007/s00044-011-9641-1 |
| 1. | D’yakonov, A. L.; Telezhenetskaya, M. V. Chem. Nat. Compd. 1997, 33, 221. doi:10.1007/BF02234869 |
| 2. | Sheu, J.-R. Cardiovasc. Drug Rev. 1999, 17, 237. doi:10.1111/j.1527-3466.1999.tb00017.x |
| 3. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305. doi:10.1016/j.ejmech.2004.12.003 |
| 4. | Rachana, S.; Ujata, B.; Mamta, P.; Priyanka, K. M.; Sonam, S. Indo Global J. Pharm. Sci. 2011, 1, 85. |
| 5. | Nepali, K.; Sharma, S.; Ojha, R.; Dhar, K. L. Med. Chem. Res. 2013, 22, 1. doi:10.1007/s00044-012-0002-5 |
| 3. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305. doi:10.1016/j.ejmech.2004.12.003 |
| 27. | von Niementowski, S. J. Prakt. Chem. 1895, 51, 564. doi:10.1002/prac.18950510150 |
| 28. | Pachter, I.; Suld, G. J. Org. Chem. 1960, 25, 1680. doi:10.1021/jo01079a615 |
| 29. | D’yakonov, A. L.; Telezhenetskaya, M. V.; Yunusov, L. Yu. Chem. Nat. Compd. 1986, 22, 435. doi:10.1007/BF00579819 |
| 30. | Lee, E. S.; Park, J.-G.; Jahng, Y. Tetrahedron Lett. 2003, 44, 1883. doi:10.1016/S0040-4039(03)00080-7 |
| 5. | Nepali, K.; Sharma, S.; Ojha, R.; Dhar, K. L. Med. Chem. Res. 2013, 22, 1. doi:10.1007/s00044-012-0002-5 |
| 11. | Bergman, J.; Bergman, S. Heterocycles 1981, 16, 347. doi:10.3987/R-1981-03-0347 |
| 12. | Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038. doi:10.1021/jo010727l |
| 13. | Yadav, J. S.; Reddy, B. V. S. Tetrahedron Lett. 2002, 43, 1905. doi:10.1016/S0040-4039(02)00135-1 |
| 14. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787. doi:10.1016/j.tet.2006.07.098 |
| 15. | Bowman, W. R.; Elsegood, M. R. J.; Stein, T.; Weaver, G. W. Org. Biomol. Chem. 2007, 5, 103. doi:10.1039/b614075k |
| 16. | Zhou, J.; Fu, L.; Lv, M.; Liu, J.; Pei, D.; Ding, K. Synthesis 2008, 3974. doi:10.1055/s-0028-1083245 |
| 17. | Kraus, G. A.; Guo, H. J. Org. Chem. 2009, 74, 5337. doi:10.1021/jo900718g |
| 18. | Giri, R.; Lam, J. K.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 686. doi:10.1021/ja9077705 |
| 19. | Kshirsagar, U. A.; Puranik, V. G.; Argade, N. P. J. Org. Chem. 2010, 75, 2702. doi:10.1021/jo100400z |
| 20. | Dabiri, M.; Salehi, P.; Bahramnejad, M.; Alizadeh, M. Monatsh. Chem. 2010, 141, 877. doi:10.1007/s00706-010-0341-1 |
| 21. | Zeng, F.; Alper, H. Org. Lett. 2010, 12, 3642. doi:10.1021/ol101428v |
| 22. | Kshirsagar, U. A.; Argade, N. P. Org. Lett. 2010, 12, 3716. doi:10.1021/ol101597p |
| 23. | Tseng, M.-C.; Cheng, H.-T.; Shen, M.-J.; Chu, Y.-H. Org. Lett. 2011, 13, 4434. doi:10.1021/ol201793v |
| 24. | Granger, B. A.; Kaneda, K.; Martin, S. F. Org. Lett. 2011, 13, 4542. doi:10.1021/ol201739u |
| 25. | Fang, J.; Zhou, J. Org. Biomol. Chem. 2012, 10, 2389. doi:10.1039/c2ob07178a |
| 26. | Murai, K.; Komatsu, H.; Nagao, R.; Fujioka, H. Org. Lett. 2012, 14, 772. doi:10.1021/ol203313n |
| 35. | Han, B.; Yang, X.-L.; Wang, C.; Bai, Y.-W.; Pan, T.-C.; Chen, X.; Yu, W. J. Org. Chem. 2012, 77, 1136. doi:10.1021/jo2020399 |
| 3. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305. doi:10.1016/j.ejmech.2004.12.003 |
| 45. | Mehta, D. R.; Naravane, J. S.; Desai, R. M. J. Org. Chem. 1963, 28, 445. doi:10.1021/jo01037a041 |
| 46. | Johns, S. R.; Lamberton, J. A.; Suares, H. Aust. J. Chem. 1985, 38, 1007. doi:10.1071/CH9851007 |
| 47. | Nakagawa, Y.; Stevens, R. V. J. Org. Chem. 1988, 53, 1873. doi:10.1021/jo00244a007 |
| 6. | Jia, S.; Hu, C. Molecules 2010, 15, 1873. doi:10.3390/molecules15031873 |
| 7. | Dhankhar, S.; Kaur, R.; Ruhil, S.; Balhara, M.; Dhankhar, S.; Chhillar, A. K. Afr. J. Plant Sci. 2011, 5, 620. |
| 8. | Corrêa, G. M.; Alcântara, A. F. de C. Rev. Bras. Farmacogn. 2012, 22, 220. doi:10.1590/S0102-695X2011005000196 |
| 9. | Asgarpanah, J.; Ramezanloo, F. Afr. J. Pharm. Pharmacol. 2012, 6, 1573. |
| 34. | Richers, M. T.; Deb, I.; Platonova, A. Y.; Zhang, C.; Seidel, D. Synthesis 2013, 45, 1730–1748. doi:10.1055/s-0033-1338852 |
| 35. | Han, B.; Yang, X.-L.; Wang, C.; Bai, Y.-W.; Pan, T.-C.; Chen, X.; Yu, W. J. Org. Chem. 2012, 77, 1136. doi:10.1021/jo2020399 |
| 38. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n |
| 39. | Punniyamurthy, T.; Rout, L. Coord. Chem. Rev. 2008, 252, 134. doi:10.1016/j.ccr.2007.04.003 |
| 40. | Wendlandt, A. E.; Suess, A. M.; Stahl, S. S. Angew. Chem., Int. Ed. 2011, 50, 11062. doi:10.1002/anie.201103945 |
| 41. | Hirano, K.; Miura, M. Chem. Commun. 2012, 48, 10704. doi:10.1039/c2cc34659a |
| 42. | Zhang, C.; Tang, C.; Jiao, N. Chem. Soc. Rev. 2012, 41, 3464. doi:10.1039/c2cs15323h |
| 43. | Jones, K. M.; Klussmann, M. Synlett 2012, 159. doi:10.1055/s-0031-1290117 |
| 44. | Ratnikov, M. O.; Doyle, M. P. J. Am. Chem. Soc. 2013, 135, 1549. doi:10.1021/ja3113559 |
| 34. | Richers, M. T.; Deb, I.; Platonova, A. Y.; Zhang, C.; Seidel, D. Synthesis 2013, 45, 1730–1748. doi:10.1055/s-0033-1338852 |
| 35. | Han, B.; Yang, X.-L.; Wang, C.; Bai, Y.-W.; Pan, T.-C.; Chen, X.; Yu, W. J. Org. Chem. 2012, 77, 1136. doi:10.1021/jo2020399 |
| 52. | Zhang, C.; Murarka, S.; Seidel, D. J. Org. Chem. 2009, 74, 419. doi:10.1021/jo802325x |
| 53. | Mori, K.; Ohshima, Y.; Ehara, K.; Akiyama, T. Chem. Lett. 2009, 38, 524. doi:10.1246/cl.2009.524 |
| 54. | He, Y.-P.; Du, Y.-L.; Luo, S.-W.; Gong, L.-Z. Tetrahedron Lett. 2011, 52, 7064. doi:10.1016/j.tetlet.2011.10.062 |
| 31. | Zhang, C.; De, C. K.; Mal, R.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 416. doi:10.1021/ja077473r |
| 32. | Dieckmann, A.; Richers, M. T.; Platonova, A. Yu.; Zhang, C.; Seidel, D.; Houk, K. N. J. Org. Chem. 2013, 78, 4132. doi:10.1021/jo400483h |
| 36. | Kumar, R. A.; Maheswari, C. U.; Ghantasala, S.; Jyothi, C.; Reddy, K. R. Adv. Synth. Catal. 2011, 353, 401. doi:10.1002/adsc.201000580 |
| 37. | Kumar, R. A.; Saidulu, G.; Prasad, K. R.; Kumar, G. S.; Sridhar, B.; Reddy, K. R. Adv. Synth. Catal. 2012, 354, 2985. doi:10.1002/adsc.201200679 |
© 2013 Richers et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)