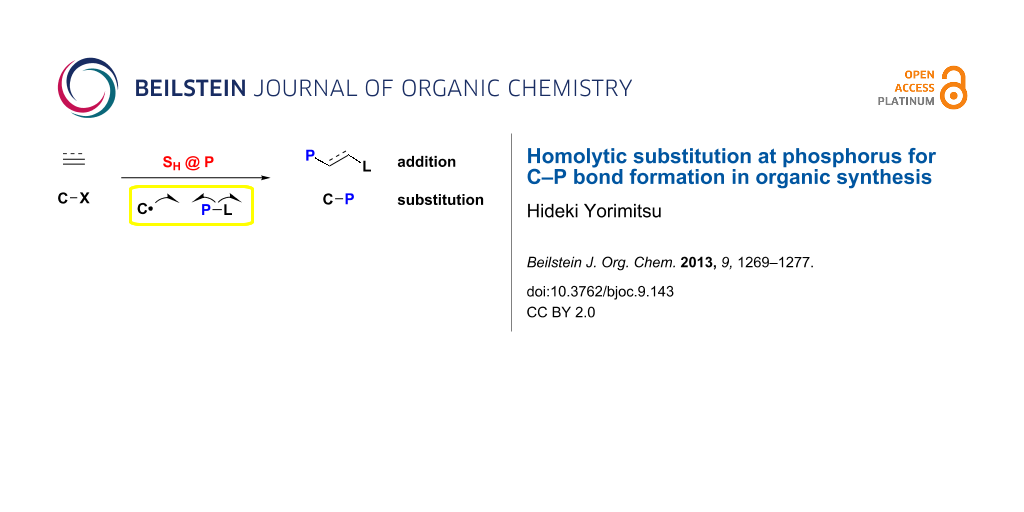
Abstract
Organophosphorus compounds are important in organic chemistry. This review article covers emerging, powerful synthetic approaches to organophosphorus compounds by homolytic substitution at phosphorus with a carbon-centered radical. Phosphination reagents include diphosphines, chalcogenophosphines and stannylphosphines, which bear a weak P–heteroatom bond for homolysis. This article deals with two transformations, radical phosphination by addition across unsaturated C–C bonds and substitution of organic halides.

Graphical Abstract
Introduction
Organophosphorus compounds constitute an important class of compounds in a wide range of applications in organic chemistry, as reagents, intermediates, ligands, bioactive agents, and functional materials [1-4]. The synthesis of organophosphorus compounds has therefore been extensively investigated (Scheme 1). Classical methods to form a C–P bond include ionic reactions such as nucleophilic substitution of P–X compounds with organometallic reagents, nucleophilic substitution of alkyl halides with phosphorus nucleophiles, and nucleophilic addition to polar unsaturated bonds. Recent advances in transition-metal catalysis have realized catalytic cross-coupling reactions of aryl halides with H–P compounds [5-7] and catalytic addition to nonpolar unsaturated carbon–carbon bonds [8-11]. In the field of radical chemistry, the addition of phosphorus radicals, mainly from H–P compounds, onto carbon–carbon multiple bonds [12-15] has held a special position since they provide transformations unattainable by polar reactions.
![[1860-5397-9-143-i1]](/bjoc/content/inline/1860-5397-9-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative C–P bond-forming reactions.
Scheme 1: Representative C–P bond-forming reactions.
Homolytic substitution is a reaction in which a radical (R•) attacks a saturated atom (X) in a molecule with the liberation of a leaving radical (L•) from the atom (Scheme 2). Homolytic substitution at halogen and chalcogen atoms is well known to proceed and hence has been widely used in organic synthesis [16-19]. In contrast, applications of homolytic substitution to C–P bond formation have been rarely explored. With the growing importance of organophosphorus compounds, increasing attention has been paid to homolytic substitution at phosphorus. The new tool for C–P bond formation has achieved interesting transformations that ionic reactions cannot. This review summarizes homolytic substitution at phosphorus for C–P bond formation in organic synthesis while the relevant mechanistic studies are found in the literature [19-21]. This review deals with two transformations, radical phosphination by addition across unsaturated C–C bonds and substitution of organic halides.
![[1860-5397-9-143-i2]](/bjoc/content/inline/1860-5397-9-143-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General equation of homolytic substitution.
Scheme 2: General equation of homolytic substitution.
Review
Radical addition of phosphination agents
Stannylphosphines of the type R3Sn–PR’2 are known to undergo radical addition to carbon–carbon unsaturated bonds. Schumann reported the addition of diphenyl(triphenylstannyl)phosphine to allyl chloride, styrene, and phenylacetylene (Scheme 3) [22,23]. The addition is most likely to proceed via a radical process as the absence of AIBN leads to considerable decreases in yield. Mitchell then reported that diphenyl(trimethylstannyl)phosphine reacts not only with terminal alkynes but also with internal alkynes and allenes (Scheme 4) [24,25]. It is noteworthy that the regioselectivity of the radical addition to propynamide is opposite to that of the relevant ionic Michael addition. Considering the regioselectivity, these addition reactions naturally involve C–P bond formation by homolytic substitution at phosphorus (Scheme 5). Studer recently reported similar silylphosphination of phenyl vinyl sulfone with Me3Si–PPh2 [26].
![[1860-5397-9-143-i3]](/bjoc/content/inline/1860-5397-9-143-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Addition of diphenyl(triphenylstannyl)phosphine.
Scheme 3: Addition of diphenyl(triphenylstannyl)phosphine.
![[1860-5397-9-143-i4]](/bjoc/content/inline/1860-5397-9-143-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Addition of diphenyl(trimethylstannyl)phosphine.
Scheme 4: Addition of diphenyl(trimethylstannyl)phosphine.
![[1860-5397-9-143-i5]](/bjoc/content/inline/1860-5397-9-143-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism of addition of R3Sn–PPh2.
Scheme 5: Plausible mechanism of addition of R3Sn–PPh2.
Tzschach reported that tetraorganodiphosphines R2P–PR2 add to phenylacetylene under UV irradiation or upon heating in the presence of AIBN (Scheme 6) [27]. The reaction consists of the addition of a diorganophosphanyl radical to phenylacetylene and the homolytic substitution of tetraorganodiphosphine with the resulting vinyl radical to afford the adduct and to regenerate the initial diorganophosphanyl radical (Scheme 7). The high E selectivity is attributable to kinetic control of the homolytic substitution, where R2P–PR2 preferentially approaches the vinyl radical from the roomier side. Although the transformation looks useful to construct an (E)-1,2-diphosphanylethene skeleton, the scope of alkyne is limited to phenylacetylene and the reactions result in unsatisfactory yields because of the instability of the products as well as the diphosphines in air.
![[1860-5397-9-143-i6]](/bjoc/content/inline/1860-5397-9-143-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Addition of tetraorganodiphosphines to phenylacetylene.
Scheme 6: Addition of tetraorganodiphosphines to phenylacetylene.
![[1860-5397-9-143-i7]](/bjoc/content/inline/1860-5397-9-143-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible mechanism of anti-diphosphination.
Scheme 7: Plausible mechanism of anti-diphosphination.
A more general, facile, and reliable method for diphosphination was later reported by Yorimitsu and Oshima, which utilizes diphosphines generated in situ from chlorophosphine and hydrophosphine in the presence of triethylamine [28]. A variety of terminal alkynes undergo the radical diphosphination (Table 1). The diphosphination was applicable to the synthesis of a new push–pull-type molecule that emits blue fluorescence (Scheme 8). The initially formed diphosphanylethylene derivatives are not very stable in air, and therefore sulfidation or oxidation was performed to accurately assess the efficiency of the diphosphination reactions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-143-i24.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-i8]](/bjoc/content/inline/1860-5397-9-143-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Radical diphosphination for synthesizing fluorescent material.
Scheme 8: Radical diphosphination for synthesizing fluorescent material.
Ogawa independently reported similar diphosphination under UV irradiation (Table 2) [29,30]. The reactions favor the formation of Z isomers, which results from photoinduced isomerization of initially formed E isomers. Ogawa’s diphosphination is thus potentially useful for the synthesis of (Z)-1,2-diphosphanyl-1-alkenes, which can serve as bidentate ligands.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-143-i25.svg?max-width=637&scale=1.0)
Morse developed photoinduced addition of tetrafluorodiphosphine to alkenes and alkynes in the gas phase (Table 3) [31-34]. The addition provides a series of intriguing bidentate phosphine ligands. The addition to alkynes yields 1:1 mixtures of E/Z isomers. Due to the high reactivity of a difluorophosphanyl radical, considerable polymerization takes place unless substrates or olefinic products are reasonably inert.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-143-i26.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-143-i27.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-143-i28.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-143-i29.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-143-i30.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-143-i31.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-143-i32.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-143-i33.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-143-i34.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-143-i35.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-143-i36.svg?max-width=637&scale=1.0)
Yorimitsu and Oshima reported radical addition of a P–S bond across alkyne by using diphenyl(organosulfanyl)phosphine (Table 4) [35]. The addition proceeds mainly in an anti fashion to afford the adducts bearing a sulfanyl group at the terminal carbon and a phosphanyl group at the internal carbon. The reaction mechanism is similar to that in Scheme 7 (Scheme 9). The regioselective outcome suggests that the homolytic substitution occurs exclusively at phosphorus, not at sulfur. A sulfanyl radical is liberated to add the terminal carbon of alkyne. To reverse the regioselectivity in radical addition of a P–S bond, S-thiophosphinyl O-ethyl dithiocarbonates were created, although the reversed addition excludes homolytic substitution at phosphorus (Scheme 10) [36].
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-143-i37.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-i9]](/bjoc/content/inline/1860-5397-9-143-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Mechanism of thiophosphination with diphenyl(organosulfanyl)phosphine.
Scheme 9: Mechanism of thiophosphination with diphenyl(organosulfanyl)phosphine.
![[1860-5397-9-143-i10]](/bjoc/content/inline/1860-5397-9-143-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Thiophosphination with S-thiophosphinyl O-ethyl dithiocarbonate.
Scheme 10: Thiophosphination with S-thiophosphinyl O-ethyl dithiocarbonate.
Kawaguchi, Nomoto, and Ogawa seminally studied the photoinduced radical chalcogenophosphination of alkynes and allenes by means of PhCh–ChPh/Ph2P–PPh2 binary systems (Ch = S, Se, Te) [30,37-39]. The regioselective outcome of the photoinduced thio- and selenophosphination of terminal alkynes (Table 5) is similar to that of the thermal thiophosphination (Scheme 9). Detailed mechanistic studies revealed that comproportionation between PhSe–SePh and Ph2P–PPh2 occurs smoothly to generate PhSe–PPh2 as the actual reactive species. Selenophosphination of terminal allene affords (2-phenylselenyl-2-alkenyl)diphenylphosphine regioselectively (Scheme 11). Notably, the sense of the regioselectivity of tellurophosphination by a PhTe–TePh/Ph2P–PPh2 system is opposite to those of the thio- and selenophosphination (Scheme 12). This reversal indicates that homolytic substitution at tellurium overwhelms that at phosphorus and that a diphenylphosphanyl radical is more reactive than a phenyltelluranyl radical.
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-143-i38.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-i11]](/bjoc/content/inline/1860-5397-9-143-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Photoinduced selenophosphination of allenes.
Scheme 11: Photoinduced selenophosphination of allenes.
![[1860-5397-9-143-i12]](/bjoc/content/inline/1860-5397-9-143-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Photoinduced tellurophosphination.
Scheme 12: Photoinduced tellurophosphination.
Substitution of halides (X), carboxys (COOR), or carboxylates (OCOR) with phosphorus
After scattered research efforts into the uncontrolled radical C–H phosphination under harsh reaction conditions [40], Barton elegantly devised radical decarboxylative phosphorylation of carboxylic thiohydroxamic mixed anhydrides (Scheme 13) [41]. Radical addition of a phenylsulfanyl radical to the thiocarbonyl generates the corresponding alkyl radical R•, which undergoes homolytic substitution at the phosphorus of P(SPh)3 to furnish (PhS)2P–R as the initial product (Scheme 14). Oxidative addition of the disulfide byproduct to the initial product furnishes a pentavalent phosphorus species that is eventually hydrolyzed to an S,S-diphenyl dithiophosphonate upon workup.
![[1860-5397-9-143-i13]](/bjoc/content/inline/1860-5397-9-143-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Decarboxylative phosphorylation of carboxylic acid derivatives.
Scheme 13: Decarboxylative phosphorylation of carboxylic acid derivatives.
![[1860-5397-9-143-i14]](/bjoc/content/inline/1860-5397-9-143-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Plausible mechanism of decarboxylative phosphorylation.
Scheme 14: Plausible mechanism of decarboxylative phosphorylation.
Barton also reported that white phosphorus reacts with N-acyloxythiopyridones, so-called Barton PTOC esters (Scheme 15) [42]. Photolysis of the esters in the presence of white phosphorus followed by oxidation with hydrogen peroxide yields alkylphosphonic acid. The efficient phosphination would stem from the highly strained structure and the weak P–P bonds of white phosphorus.
![[1860-5397-9-143-i15]](/bjoc/content/inline/1860-5397-9-143-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Radical phosphination of PTOC esters with white phosphorus.
Scheme 15: Radical phosphination of PTOC esters with white phosphorus.
After 13 years of silence, radical substitution reactions of organic halides and related compounds with phosphination agents have now been rapidly developing since 2006. Yorimitsu and Oshima invented radical phosphination of organic halides with tetraphenyldiphosphine (Table 6) [43]. Tetraphenyldiphosphine is generated in situ by radical reduction of chlorodiphenylphosphine with tris(trimethylsilyl)silane followed by condensation of the resulting diphenylphosphine with the remaining chlorophosphine (Scheme 16, equation 1 and 2). An aryl radical reacts with tetraphenyldiphosphine to liberate a diphenylphosphanyl radical, which abstracts hydrogen from tris(trimethylsilyl)silane to sustain the chain propagation (Scheme 16, equation 3–5). The in situ formations of diphenylphosphine and of tetraphenyldiphosphine can exclude the handling of pyrophoric diphenylphosphine and air-sensitive tetraphenyldiphosphine. The user-friendly conditions are also suitable for dicyclohexylphosphination with ClP(c-C6H11)2.
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-143-i39.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-i16]](/bjoc/content/inline/1860-5397-9-143-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Plausible mechanism of radical phosphination (Si = (Me3Si)3Si).
Scheme 16: Plausible mechanism of radical phosphination (Si = (Me3Si)3Si).
Phosphination of alkyl halides as substrates results in unsatisfactory yields. Instead, Barton’s alkyl imidazole-1-carbothioates are good substrates for this radical phosphination (Table 7). Conversion of an optically pure cis-carbothioate leads to trans-aminophosphine of potential use as a ligand (Scheme 17). Diphosphine approaches the resulting radical from the opposite side of the NHBoc group to invert the original stereochemistry.
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-143-i40.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-i17]](/bjoc/content/inline/1860-5397-9-143-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Stereoselective phosphination leading to (S,S)-aminophosphine derivative.
Scheme 17: Stereoselective phosphination leading to (S,S)-aminophosphine derivative.
Studer developed in 2007 new elegant reagents Me3Sn–PPh2 and Me3Si–PPh2 for radical phosphination [44]. The scope of his phosphination with Me3Sn–PPh2 is wide as summarized in Table 8. Although the low toxicity of Me3Si–PPh2 is advantageous, phosphination with Me3Si–PPh2 is limited to alkyl halides or imidazole-1-carbothioate. Density functional theory calculations have clarified the homolytic substitution process is a two-step mechanism via a tetracoordinated phosphorus atom (Figure 1). The spin density in the tetracoordinated phosphorus intermediate is localized mostly on the Sn atom while the remaining spin density is found in the equatorial position of the distorted trigonal prismatic P atom.
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-143-i41.svg?max-width=637&scale=1.0)
![[1860-5397-9-143-1]](/bjoc/content/figures/1860-5397-9-143-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Calculated reaction profile of homolytic substitution between Ph• and Me3Sn–PPh2 at the B2-PLYP-D/TZVVP//PBE-D/TZVP level. Gray lobes indicate major spin densities.
Figure 1: Calculated reaction profile of homolytic substitution between Ph• and Me3Sn–PPh2 at the B2-PLYP-D/T...
The rate constant for phosphination of an aryl radical with Me3Sn–PPh2 is calculated to be ca. 9 × 108 M−1s−1 by competition kinetics with Bu3SnH reduction [45]. This large rate constant allows for stereospecific trapping of axially chiral acyl radicals with Me3Sn–PPh2 (Scheme 18). Chemodivergent trapping of diastereomers of an N-(2-cyclohexenyl)acetanilide derivative is interesting (Scheme 19). One isomer undergoes direct phosphination while the other cyclizes prior to phosphination.
![[1860-5397-9-143-i18]](/bjoc/content/inline/1860-5397-9-143-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Phosphination with retention of axial chirality.
Scheme 18: Phosphination with retention of axial chirality.
![[1860-5397-9-143-i19]](/bjoc/content/inline/1860-5397-9-143-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Intermolecular phosphinative radical addition of alkyl iodides to activated alkenes proceeds in the presence of Me3M–PPh2 (M = Sn, Si) and V-40 (Table 9) [26]. Secondary and tertiary alkyl iodides participate in the addition reaction while primary alkyl iodide results in direct phosphination prior to the expected addition. Not only acrylate ester but also acrylamide, vinyl sulfone, and acrylonitrile are good radical acceptors in this addition.
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-143-i42.svg?max-width=637&scale=1.0)
Studer’s stannylphosphine technology is reliable enough to be applied to the construction of interesting π-conjugated frameworks. In collaboration with Yamaguchi, Studer invented a new radical reagent (Me3Sn)2PPh for the synthesis of highly strained bis(phosphoryl)-bridged biphenyls (Scheme 20) [46]. Subsequently, Liu reported an efficient synthesis of bis(phosphoryl)-bridged ladder triphenylene by means of the radical clipping with (Me3Sn)2PPh (Scheme 21) [47]. In light of the increasing importance of phosphoryl-bridged π-conjugated skeletons in organic material sciences, (Me3Sn)2PPh will serve as a key reagent.
![[1860-5397-9-143-i20]](/bjoc/content/inline/1860-5397-9-143-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Bis(phosphoryl)-bridged biphenyls by radical phosphination.
Scheme 20: Bis(phosphoryl)-bridged biphenyls by radical phosphination.
![[1860-5397-9-143-i21]](/bjoc/content/inline/1860-5397-9-143-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Bis(phosphoryl)-bridged ladder triphenylene by radical phosphination.
Scheme 21: Bis(phosphoryl)-bridged ladder triphenylene by radical phosphination.
Ogawa developed photoinduced phosphination of perfluoroalkyl iodides with tetraorganodiphosphines (Scheme 22) [48]. Remarkably, the phosphination proceeds quantitatively. The phosphine ligands thus synthesized are fluorophilic. Particularly, two molecules of perfluorodecyldiphenylphosphine coordinate to palladium dichloride to form a catalytically active palladium complex that is useful for a fluorous/organic biphasic system.
![[1860-5397-9-143-i22]](/bjoc/content/inline/1860-5397-9-143-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Photoinduced phosphination of perfluoroalkyl iodides with tetraphenyldiphosphine.
Scheme 22: Photoinduced phosphination of perfluoroalkyl iodides with tetraphenyldiphosphine.
Cummins devised radical phosphination of bromobenzene or bromocyclohexane with white phosphorus by means of a trivalent titanium complex (Scheme 23) [49]. This represents a unique direct method for preparing triorganophosphine without recourse to any trivalent phosphorus sources such as PCl3.
![[1860-5397-9-143-i23]](/bjoc/content/inline/1860-5397-9-143-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Ti(III)-mediated radical phosphination of organic bromides with white phosphorus.
Scheme 23: Ti(III)-mediated radical phosphination of organic bromides with white phosphorus.
Conclusion
Introduction of a phosphorus atom by a radical process has offered an intriguing tool for the synthesis of organophosphorus compounds. Radical addition of a phosphorus-centered radical has been representative so far. A recent dramatic growth in reports of homolytic substitution at phosphorus in organic synthesis has changed the landscape of radical phosphination. Radical addition that involves homolytic substitution at phosphorus always culminates in difunctionalization of a multiple bond. Therefore this methodology will find application in the synthesis of complex phosphines including bidentate ones. Radical substitution of halogen in organic halide with phosphorus will be an alternative to classical ionic substitution. Advantageously, it requires neither highly basic conditions nor transition metals. Homolytic substitution at phosphorus is still in its infancy. In light of the rich chemistry of organophosphorus compounds, it will find wider application in organic synthesis in the future.
Acknowledgements
Preparation of this article and parts of synthetic chemistry in this article were supported by JSPS and MEXT (Grants-in-Aid for Scientific Research, Nos. 24685007, 23655037, 22106523, and 24106721 “Reaction Integration”) and by The Uehara Memorial Foundation, NOVARTIS Foundation for the Promotion of Science, Kinki Invention Center, and Takeda Science Foundation. Special thanks are given to those listed as the coauthors in our papers cited herein, particularly to Dr. Akinori Sato, Dr. Azusa Kondoh, and Professor Koichiro Oshima for their invaluable contributions.
References
-
Organophosphorus Chemistry; Royal Society of Chemistry: Cambridge, U.K.; Vol. 1–40.
Return to citation in text: [1] -
New Aspects in Phosphorus Chemistry; Springer: Berlin, Germany; Vol. 1–5.
Return to citation in text: [1] -
Mathey, F., Ed. Science of Synthesis: Houben-Weyl Methods of Molecular Transformation; Thieme: Stuttgart, Germany, 2008; Vol. 42.
Return to citation in text: [1] -
Murphy, P. J., Ed. Organophosphorus Reagents; Oxford University Press: Oxford, U.K., 2004.
Return to citation in text: [1] -
Beletskaya, I. P.; Kazankova, M. A. Russ. J. Org. Chem. 2002, 38, 1391–1430. doi:10.1023/A:1022685801622
Return to citation in text: [1] -
Schwan, A. L. Chem. Soc. Rev. 2004, 33, 218–224. doi:10.1039/b307538a
Return to citation in text: [1] -
Tappe, F. M. J.; Trepohl, V. T.; Oestreich, M. Synthesis 2010, 3037–3062. doi:10.1055/s-0030-1257960
Return to citation in text: [1] -
Wicht, D. K.; Glueck, D. S. Hydrophosphination and Related Reactions. In Catalytic Heterofunctionalization; Togni, A.; Grützmacher, H., Eds.; Wiley: Weinheim, Germany, 2001. doi:10.1002/3527600159.ch5
Return to citation in text: [1] -
Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3159. doi:10.1021/cr0201068
Return to citation in text: [1] -
Tanaka, M. Top. Curr. Chem. 2004, 232, 25–54. doi:10.1007/b13778
Return to citation in text: [1] -
Delacroix, O.; Gaumont, A. C. Curr. Org. Chem. 2005, 9, 1851–1882. doi:10.2174/138527205774913079
Return to citation in text: [1] -
Baralle, A.; Baroudi, A.; Daniel, M.; Fensterbank, L.; Goddard, J.-P.; Lacôte, E.; Larraufie, M.-H.; Maestri, G.; Malacria, M.; Olivier, C. Main-Group Elements in Radical Chemistry. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley: Weinheim, Germany, 2012; Vol. 2, Chapter 28. doi:10.1002/9781119953678.rad023
Return to citation in text: [1] -
Leca, D.; Fensterbank, L.; Lacôte, E.; Malacria, M. Chem. Soc. Rev. 2005, 34, 858–865. doi:10.1039/b500511f
Return to citation in text: [1] -
Walling, C.; Pearson, M. S. Top. Phosphorus Chem. 1966, 3, 1–56.
Return to citation in text: [1] -
Stacy, F. W.; Harris, J. F. Org. React. 1963, 13, 150–376.
Return to citation in text: [1] -
Kyne, S. H.; Schiesser, C. H. Intramolecular Homolytic Substitution in Synthesis. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; Wiley: Weinheim, Germany, 2012; Vol. 2, Chapter 24. doi:10.1002/9781119953678.rad018
Return to citation in text: [1] -
Schiesser, C. H.; Wild, L. M. Tetrahedron 1996, 52, 13265–13314. doi:10.1016/0040-4020(96)00809-5
Return to citation in text: [1] -
Crich, D. Helv. Chim. Acta 2006, 89, 2167–2182. doi:10.1002/hlca.200690204
Return to citation in text: [1] -
Walton, J. C. Acc. Chem. Res. 1998, 31, 99–107. doi:10.1021/ar970259v
Return to citation in text: [1] [2] -
Bentrude, W. G. Acc. Chem. Res. 1982, 15, 117–125. doi:10.1021/ar00076a004
Return to citation in text: [1] -
Bentrude, W. G. Chapter 22: Phosphorus Radicals. In Free Radicals; Kochi, J. K., Ed.; Wiley: Weinheim, Germany, 1973; Vol. 2.
Return to citation in text: [1] -
Schumann, H.; Jutzi, P.; Schmidt, M. Angew. Chem., Int. Ed. Engl. 1965, 4, 869. doi:10.1002/anie.196508692
Return to citation in text: [1] -
Schumann, H. Angew. Chem., Int. Ed. Engl. 1969, 8, 937–950. doi:10.1002/anie.196909371
Return to citation in text: [1] -
Mitchell, T. N.; Belt, H.-J. J. Organomet. Chem. 1988, 345, C28–C30. doi:10.1016/0022-328X(88)80105-0
Return to citation in text: [1] -
Mitchell, T. N.; Belt, H.-J. J. Organomet. Chem. 1990, 386, 167–176. doi:10.1016/0022-328X(90)85241-P
Return to citation in text: [1] -
Lamas, M.-C.; Studer, A. Org. Lett. 2011, 13, 2236–2239. doi:10.1021/ol200483p
Return to citation in text: [1] [2] -
Tzschach, A.; Baensch, S. J. Prakt. Chem. 1971, 313, 254–258. doi:10.1002/prac.19713130209
Return to citation in text: [1] -
Sato, A.; Yorimitsu, A.; Oshima, K. Angew. Chem., Int. Ed. 2005, 44, 1694–1696. doi:10.1002/anie.200462603
Return to citation in text: [1] -
Kawaguchi, S.-i.; Nagata, S.; Shirai, T.; Tsuchii, K.; Nomoto, A.; Ogawa, A. Tetrahedron Lett. 2006, 47, 3919–3922. doi:10.1016/j.tetlet.2006.03.165
Return to citation in text: [1] -
Kawaguchi, S.-i.; Ogawa, A. J. Synth. Org. Chem., Jpn. 2010, 68, 705–717. doi:10.5059/yukigoseikyokaishi.68.705
Return to citation in text: [1] [2] -
Morse, K. W.; Morse, J. G. J. Am. Chem. Soc. 1973, 95, 8469–8470. doi:10.1021/ja00806a057
Return to citation in text: [1] -
Morse, J. G.; Morse, K. W. Inorg. Chem. 1975, 14, 565–569. doi:10.1021/ic50145a024
Return to citation in text: [1] -
Glanville, W. K.; Morse, K. W.; Morse, J. G. J. Fluorine Chem. 1976, 7, 153–158. doi:10.1016/S0022-1139(00)83992-5
Return to citation in text: [1] -
Morse, J. G.; Mielcarek, J. J. J. Fluorine Chem. 1988, 40, 41–49. doi:10.1016/S0022-1139(00)81060-X
Return to citation in text: [1] -
Wada, T.; Kondoh, A.; Yorimitsu, A.; Oshima, K. Org. Lett. 2008, 10, 1155–1157. doi:10.1021/ol800059n
Return to citation in text: [1] -
Sato, A.; Yorimitsu, A.; Oshima, K. Tetrahedron 2009, 65, 1553–1558. doi:10.1016/j.tet.2008.12.071
Return to citation in text: [1] -
Shirai, T.; Kawaguchi, S.-i.; Nomoto, A.; Ogawa, A. Tetrahedron Lett. 2008, 49, 4043–4046. doi:10.1016/j.tetlet.2008.04.068
Return to citation in text: [1] -
Kawaguchi, S.-i.; Shirai, T.; Ohe, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. J. Org. Chem. 2009, 74, 1751–1754. doi:10.1021/jo8020067
Return to citation in text: [1] -
Kawaguchi, S.-i.; Ohe, T.; Shirai, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Organometallics 2010, 29, 312–316. doi:10.1021/om9008982
Return to citation in text: [1] -
Sakurai, H.; Okamoto, Y. J. Synth. Org. Chem., Jpn. 1976, 34, 203–214.
Return to citation in text: [1] -
Barton, D. H. R.; Bridon, D.; Zard, S. Z. Tetrahedron Lett. 1986, 27, 4309–4312. doi:10.1016/S0040-4039(00)94261-8
Return to citation in text: [1] -
Barton, D. H. R.; Zhu, J. J. Am. Chem. Soc. 1993, 115, 2071–2072. doi:10.1021/ja00058a082
Return to citation in text: [1] -
Sato, A.; Yorimitsu, A.; Oshima, K. J. Am. Chem. Soc. 2006, 128, 4240–4241. doi:10.1021/ja058783h
Return to citation in text: [1] -
Vaillard, S. E.; Mück-Lichtenfeld, C.; Grimme, S.; Studer, A. Angew. Chem., Int. Ed. 2007, 46, 6533–6536. doi:10.1002/anie.200701650
Return to citation in text: [1] -
Bruch, A.; Ambrosius, A.; Fröhlich, R.; Studer, A.; Guthrie, D. B.; Zhang, H.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 11452–11454. doi:10.1021/ja105070k
Return to citation in text: [1] -
Bruch, A.; Fukazawa, A.; Yamaguchi, E.; Yamaguchi, S.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 12094–12098. doi:10.1002/anie.201104114
Return to citation in text: [1] -
Hanifi, D.; Pun, A.; Liu, Y. Chem.–Asian J. 2012, 7, 2615–2620. doi:10.1002/asia.201200631
Return to citation in text: [1] -
Kawaguchi, S.-i.; Minamida, Y.; Ohe, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Angew. Chem., Int. Ed. 2013, 52, 1748–1752. doi:10.1002/anie.201207383
Return to citation in text: [1] -
Cossairt, B. M.; Cummins, C. C. New J. Chem. 2010, 34, 1533–1536. doi:10.1039/c0nj00124d
Return to citation in text: [1]
| 1. | Organophosphorus Chemistry; Royal Society of Chemistry: Cambridge, U.K.; Vol. 1–40. |
| 2. | New Aspects in Phosphorus Chemistry; Springer: Berlin, Germany; Vol. 1–5. |
| 3. | Mathey, F., Ed. Science of Synthesis: Houben-Weyl Methods of Molecular Transformation; Thieme: Stuttgart, Germany, 2008; Vol. 42. |
| 4. | Murphy, P. J., Ed. Organophosphorus Reagents; Oxford University Press: Oxford, U.K., 2004. |
| 16. | Kyne, S. H.; Schiesser, C. H. Intramolecular Homolytic Substitution in Synthesis. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; Wiley: Weinheim, Germany, 2012; Vol. 2, Chapter 24. doi:10.1002/9781119953678.rad018 |
| 17. | Schiesser, C. H.; Wild, L. M. Tetrahedron 1996, 52, 13265–13314. doi:10.1016/0040-4020(96)00809-5 |
| 18. | Crich, D. Helv. Chim. Acta 2006, 89, 2167–2182. doi:10.1002/hlca.200690204 |
| 19. | Walton, J. C. Acc. Chem. Res. 1998, 31, 99–107. doi:10.1021/ar970259v |
| 36. | Sato, A.; Yorimitsu, A.; Oshima, K. Tetrahedron 2009, 65, 1553–1558. doi:10.1016/j.tet.2008.12.071 |
| 12. | Baralle, A.; Baroudi, A.; Daniel, M.; Fensterbank, L.; Goddard, J.-P.; Lacôte, E.; Larraufie, M.-H.; Maestri, G.; Malacria, M.; Olivier, C. Main-Group Elements in Radical Chemistry. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley: Weinheim, Germany, 2012; Vol. 2, Chapter 28. doi:10.1002/9781119953678.rad023 |
| 13. | Leca, D.; Fensterbank, L.; Lacôte, E.; Malacria, M. Chem. Soc. Rev. 2005, 34, 858–865. doi:10.1039/b500511f |
| 14. | Walling, C.; Pearson, M. S. Top. Phosphorus Chem. 1966, 3, 1–56. |
| 15. | Stacy, F. W.; Harris, J. F. Org. React. 1963, 13, 150–376. |
| 30. | Kawaguchi, S.-i.; Ogawa, A. J. Synth. Org. Chem., Jpn. 2010, 68, 705–717. doi:10.5059/yukigoseikyokaishi.68.705 |
| 37. | Shirai, T.; Kawaguchi, S.-i.; Nomoto, A.; Ogawa, A. Tetrahedron Lett. 2008, 49, 4043–4046. doi:10.1016/j.tetlet.2008.04.068 |
| 38. | Kawaguchi, S.-i.; Shirai, T.; Ohe, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. J. Org. Chem. 2009, 74, 1751–1754. doi:10.1021/jo8020067 |
| 39. | Kawaguchi, S.-i.; Ohe, T.; Shirai, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Organometallics 2010, 29, 312–316. doi:10.1021/om9008982 |
| 8. | Wicht, D. K.; Glueck, D. S. Hydrophosphination and Related Reactions. In Catalytic Heterofunctionalization; Togni, A.; Grützmacher, H., Eds.; Wiley: Weinheim, Germany, 2001. doi:10.1002/3527600159.ch5 |
| 9. | Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3159. doi:10.1021/cr0201068 |
| 10. | Tanaka, M. Top. Curr. Chem. 2004, 232, 25–54. doi:10.1007/b13778 |
| 11. | Delacroix, O.; Gaumont, A. C. Curr. Org. Chem. 2005, 9, 1851–1882. doi:10.2174/138527205774913079 |
| 31. | Morse, K. W.; Morse, J. G. J. Am. Chem. Soc. 1973, 95, 8469–8470. doi:10.1021/ja00806a057 |
| 32. | Morse, J. G.; Morse, K. W. Inorg. Chem. 1975, 14, 565–569. doi:10.1021/ic50145a024 |
| 33. | Glanville, W. K.; Morse, K. W.; Morse, J. G. J. Fluorine Chem. 1976, 7, 153–158. doi:10.1016/S0022-1139(00)83992-5 |
| 34. | Morse, J. G.; Mielcarek, J. J. J. Fluorine Chem. 1988, 40, 41–49. doi:10.1016/S0022-1139(00)81060-X |
| 5. | Beletskaya, I. P.; Kazankova, M. A. Russ. J. Org. Chem. 2002, 38, 1391–1430. doi:10.1023/A:1022685801622 |
| 6. | Schwan, A. L. Chem. Soc. Rev. 2004, 33, 218–224. doi:10.1039/b307538a |
| 7. | Tappe, F. M. J.; Trepohl, V. T.; Oestreich, M. Synthesis 2010, 3037–3062. doi:10.1055/s-0030-1257960 |
| 35. | Wada, T.; Kondoh, A.; Yorimitsu, A.; Oshima, K. Org. Lett. 2008, 10, 1155–1157. doi:10.1021/ol800059n |
| 26. | Lamas, M.-C.; Studer, A. Org. Lett. 2011, 13, 2236–2239. doi:10.1021/ol200483p |
| 28. | Sato, A.; Yorimitsu, A.; Oshima, K. Angew. Chem., Int. Ed. 2005, 44, 1694–1696. doi:10.1002/anie.200462603 |
| 24. | Mitchell, T. N.; Belt, H.-J. J. Organomet. Chem. 1988, 345, C28–C30. doi:10.1016/0022-328X(88)80105-0 |
| 25. | Mitchell, T. N.; Belt, H.-J. J. Organomet. Chem. 1990, 386, 167–176. doi:10.1016/0022-328X(90)85241-P |
| 29. | Kawaguchi, S.-i.; Nagata, S.; Shirai, T.; Tsuchii, K.; Nomoto, A.; Ogawa, A. Tetrahedron Lett. 2006, 47, 3919–3922. doi:10.1016/j.tetlet.2006.03.165 |
| 30. | Kawaguchi, S.-i.; Ogawa, A. J. Synth. Org. Chem., Jpn. 2010, 68, 705–717. doi:10.5059/yukigoseikyokaishi.68.705 |
| 22. | Schumann, H.; Jutzi, P.; Schmidt, M. Angew. Chem., Int. Ed. Engl. 1965, 4, 869. doi:10.1002/anie.196508692 |
| 23. | Schumann, H. Angew. Chem., Int. Ed. Engl. 1969, 8, 937–950. doi:10.1002/anie.196909371 |
| 19. | Walton, J. C. Acc. Chem. Res. 1998, 31, 99–107. doi:10.1021/ar970259v |
| 20. | Bentrude, W. G. Acc. Chem. Res. 1982, 15, 117–125. doi:10.1021/ar00076a004 |
| 21. | Bentrude, W. G. Chapter 22: Phosphorus Radicals. In Free Radicals; Kochi, J. K., Ed.; Wiley: Weinheim, Germany, 1973; Vol. 2. |
| 27. | Tzschach, A.; Baensch, S. J. Prakt. Chem. 1971, 313, 254–258. doi:10.1002/prac.19713130209 |
| 42. | Barton, D. H. R.; Zhu, J. J. Am. Chem. Soc. 1993, 115, 2071–2072. doi:10.1021/ja00058a082 |
| 41. | Barton, D. H. R.; Bridon, D.; Zard, S. Z. Tetrahedron Lett. 1986, 27, 4309–4312. doi:10.1016/S0040-4039(00)94261-8 |
| 48. | Kawaguchi, S.-i.; Minamida, Y.; Ohe, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Angew. Chem., Int. Ed. 2013, 52, 1748–1752. doi:10.1002/anie.201207383 |
| 49. | Cossairt, B. M.; Cummins, C. C. New J. Chem. 2010, 34, 1533–1536. doi:10.1039/c0nj00124d |
| 46. | Bruch, A.; Fukazawa, A.; Yamaguchi, E.; Yamaguchi, S.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 12094–12098. doi:10.1002/anie.201104114 |
| 47. | Hanifi, D.; Pun, A.; Liu, Y. Chem.–Asian J. 2012, 7, 2615–2620. doi:10.1002/asia.201200631 |
| 45. | Bruch, A.; Ambrosius, A.; Fröhlich, R.; Studer, A.; Guthrie, D. B.; Zhang, H.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 11452–11454. doi:10.1021/ja105070k |
| 26. | Lamas, M.-C.; Studer, A. Org. Lett. 2011, 13, 2236–2239. doi:10.1021/ol200483p |
| 43. | Sato, A.; Yorimitsu, A.; Oshima, K. J. Am. Chem. Soc. 2006, 128, 4240–4241. doi:10.1021/ja058783h |
| 44. | Vaillard, S. E.; Mück-Lichtenfeld, C.; Grimme, S.; Studer, A. Angew. Chem., Int. Ed. 2007, 46, 6533–6536. doi:10.1002/anie.200701650 |
© 2013 Yorimitsu; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)