Abstract
Although four-coordinate square-planar geometries, with a formally 16-electron counting, are absolutely dominant in isolated Pt(II) complexes, three-coordinate, 14-electron Pt(II) complexes are believed to be key intermediates in a number of platinum-mediated organometallic transformations. Although very few authenticated three-coordinate Pt(II) complexes have been characterized, a much larger number of complexes can be described as operationally three-coordinate in a kinetic sense. In these compounds, which we have called masked T-shaped complexes, the fourth position is occupied by a very weak ligand (agostic bond, solvent molecule or counteranion), which can be easily displaced. This review summarizes the structural features of the true and masked T-shaped Pt(II) complexes reported so far and describes synthetic strategies employed for their formation. Moreover, recent experimental and theoretical reports are analyzed, which suggest the involvement of such intermediates in reaction mechanisms, particularly C–H bond-activation processes.

Graphical Abstract
Review
Scope of this review
Reaction intermediates are transient species able to undergo transformations along chemical processes. Electron deficient transition-metal complexes with vacant coordination sites are well-suited to play such a role. Coordinatively and electronically unsaturated species have often been invoked as crucial intermediates in reactions involving late transition-metal complexes. Ligand dissociation, forming intermediates with open coordination sites, has been proposed as the initial step in many reactions involving square-planar d8 organometallic complexes [1]. Four-coordinate square-planar structures, with a formally 16-electron counting, are absolutely dominant in isolated Pt(II) complexes. However, three-coordinate, 14-electron Pt(II) complexes are believed to be key intermediates in a number of reactions, e.g., β-hydrogen elimination, thermal decomposition of dialkyls, insertion of olefins into M–H bonds, electrophilic attack at Pt–C bonds, and ligand cycloplatination [2]. Likewise, related Pd(II) complexes [3,4] are relevant in cross-coupling reactions and C–H bond-activation processes. The accessibility of three-coordinate Pd(II) species have been recently discussed [5].
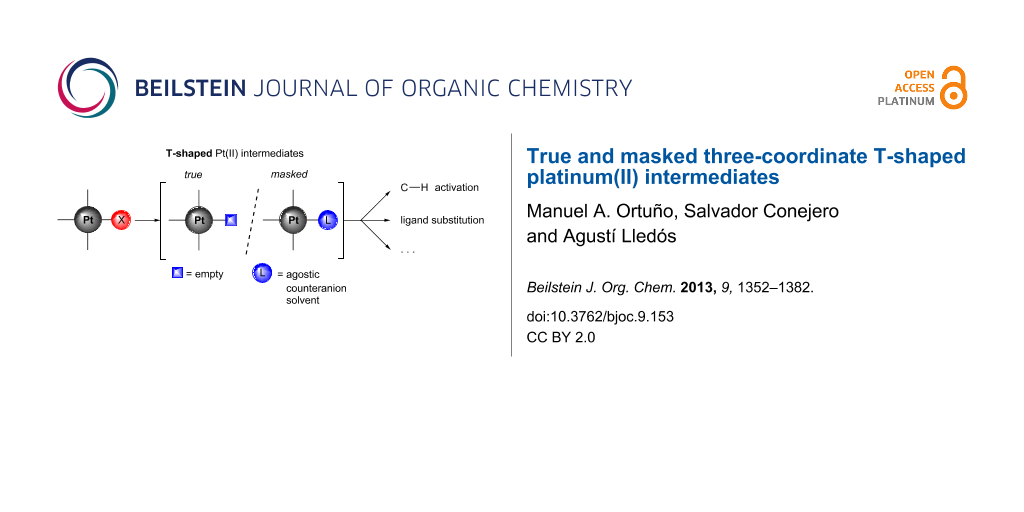
Three-coordinate Pt(II) intermediates are the focus of this review. Despite the kinetic perception of the intermediacy of these coordinatively unsaturated species in important organometallic processes, direct proofs of 14-electron Pt(II) complexes have been difficult to find. The strong readiness to alleviate the unsaturation makes them very reactive species but hampers their isolation. Low-coordinate Pt(II) complexes have been generated in gas-phase experiments [6,7] but have remained elusive in solution. With a few exceptions the fourth coordination site is occupied by a weak ligand, e.g., an agostic interaction, a counteranion or a solvent molecule. However, if this additional interaction is weak and labile enough, the three-coordinate species is very accessible and the complex can be considered as “operationally unsaturated” in a kinetic sense [8]. We will name these compounds “masked” three-coordinate complexes to distinguish them from the true low-coordinate complexes. In this review, we will summarize recent advances in true and masked three-coordinate Pt(II) complexes, highlighting both their structural features and their possible participation as reaction intermediates. Computational studies have become an invaluable tool for the investigation of short-lived elusive intermediates and will be quoted throughout the article. The review is organized as follows. First, a general picture of the electronic and geometrical structure of three-coordinate Pt(II) complexes will be presented. Then, the structural features of the main families of these compounds will be summarized. The next sections will be devoted to the spectroscopic tools for their detection and the synthetic strategies employed to their formation. Afterwards, the rearrangement processes exhibited by the low-coordinate complexes in solution will be discussed. Finally, participation of three-coordinate Pt(II) intermediates in reactions, mainly C–H bond-activation processes and ligand exchanges, will be analyzed. A thorough review on the bonding and stereochemistry of three-coordinate transition-metal compounds was published several years ago [9].
Electronic and geometrical structure
As we will discuss later on, three-coordinate, 14-electron Pt(II) d8 complexes display a T-shaped structure, i.e., a structure with two ligands mutually trans and the third ligand trans to the vacant position. A qualitative molecular orbital scheme of the d block of a T-shaped d8 metal complex can be easily derived from that of a square-planar complex by removing one of the ligands (Figure 1) [10,11]. The three nonbonding orbitals in square-planar complexes (dxz, dxy and dyz) are not affected by the removal of a ligand. The dz2 orbital is slightly stabilized due to the disappearance of a small antibonding interaction. However, the most striking difference is observed in the lowest unoccupied dx2−y2 orbital; its energy strongly decreases by removing an antibonding interaction with one ligand and its shape changes by mixing with the py orbital. This hybridization takes place so that the orbital is directed away from the three ligands toward the empty coordination site, making this position suitable for the approach of an incoming ligand. This simple picture has been corroborated by DFT calculations on T-shaped [Pt(alkyl)(PMe3)2]+ complexes [2].
The splitting of the d-block orbitals favors a singlet ground state for T-shaped d8 complexes and these species usually exhibit a low-spin reactivity. Recent DFT calculations on the 14-electron [Pt(FPNP)]+ complex (FPNP = (4-F-2-(iPr2P)C6H3)2N) predict a relatively small (10–12 kcal mol−1) separation between the singlet and the triplet states of this intermediate [12]. It is worth pointing out that a d8 C2v ML3 fragment is isolobal with CH2, although the ordering of the two valence orbitals a1 and b2 differs. For a singlet 14-electron T-shaped ML3, the two electrons fill the b2 level (dyz), while for a singlet CH2 fragment they occupy the a1 orbital (empty dx2−y2 in ML3, Figure 1) [13].
![[1860-5397-9-153-1]](/bjoc/content/figures/1860-5397-9-153-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Qualitative orbital diagram for a d8 metal in ML4 square-planar and ML3 T-shaped complexes.
Figure 1: Qualitative orbital diagram for a d8 metal in ML4 square-planar and ML3 T-shaped complexes.
Steric reasons should favor trigonal-planar D3h-like structures for three-coordinate Pt(II) complexes instead of the sterically unfavorable T-shaped structure. The preference of these Pt(II) compounds (with a low spin d8 configuration) for T-shaped structures is due to electronic effects and can be understood from the Walsh diagram shown in Figure 2. The Walsh diagram describes the variation of the L–M–L angle from a T-shaped structure through D3h to a Y-shaped structure. A d8, 14-electron ML3 complex would have the lowest four levels filled in Figure 2. In a D3h geometry, the degeneracy of e’ orbitals will promote a Jahn–Teller distortion towards T or Y geometries [10,14].
![[1860-5397-9-153-2]](/bjoc/content/figures/1860-5397-9-153-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Walsh diagram for the d-block of a d8 ML3 complex upon bending of one L–M–L angle.
Figure 2: Walsh diagram for the d-block of a d8 ML3 complex upon bending of one L–M–L angle.
DFT calculations on asymmetric cis-[Pt(alkyl)(PMe3)2]+ show that, in spite of the reduced symmetry, the bending energy profile maintains the same basic features: the T-shaped configurations of the 14-electron species are energy minima, and their cis-like to trans-like interconversion occurs via transition states with Y-shaped configurations [2].
The X-ray structure of the recently reported three-coordinate platinum complex [Pt(SiMe2Ph)2(IPr)] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) shows a unique Y-shaped geometry in which the Si–Pt–Si angle is very acute (80.9°) and far from the ideal values for both trigonal-planar and T-shaped structures (Y1, Figure 3) [15]. Computations on non-sterically demanding models [Pt(R)2(Im)] (R = SiMe3, Me; Im = imidazol-2-ylidene) appealed to the trans influence of both NHC and silyl ligands to explain the structure. However, a recent DFT investigation concluded that Y1 is better described as a Pt(0) σ-disilane complex [16] than as a Pt(II) disilyl species. Thorough geometrical and electronic analyses support a Pt(0)···disilane coordination via donation and back-donation interactions. This suggestion also explains the experimentally observed 195Pt NMR chemical shift, which is closer to the Pt(0) rather than the Pt(II) species.
![[1860-5397-9-153-3]](/bjoc/content/figures/1860-5397-9-153-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Neutral Y-shaped Pt complex Y1 [15]. Angles are given in degrees.
Figure 3: Neutral Y-shaped Pt complex Y1 [15]. Angles are given in degrees.
The nonequivalence of axial and equatorial positions in ML3 complexes raises the question of which positions are preferred by the ligands in such compounds. Although in most of the experimentally characterized systems the steric demands dictate the disposition of the ligands, electronic effects are also at work, and they were analyzed for simple model systems with theoretical methods. An early study based on perturbation theory concluded that in T-shaped ML3 d8 complexes both axial and equatorial bonds should have similar strength and that the most electronegative atoms will substitute axially [17]. DFT calculations on [PtXY(PH3)] (X, Y = Cl, CH3, SiH3, Si(OH)3) demonstrated the importance of the trans influence in governing the stability of T-shaped isomers. In the most stable isomer, the ligand with the smaller trans influence is located trans to the PH3 ligand [18].
Structurally characterized compounds
The T-shaped structure is easily recognized by the absence of one ligand in a square-planar disposition. Nevertheless, the number of well-characterized three-coordinate Pt(II) complexes is very low. To achieve a true T-shaped structure, the vacancy at the metal center must be blocked to avoid intra- and intermolecular interactions, such as agostic bonds and counteranion or solvent coordination. These interactions mask the T-shaped structure, but due to their potential labile nature, the three-coordinate species are still accessible.
In this section we will describe the main families of T-shaped Pt(II) complexes, which are structurally characterized (Figure 4). First, true T-shaped structures with no stabilization at the vacant site are compiled. Then, complexes involving agostic, counteranion and solvent interactions will be summarized. From now on, true T-shaped complexes are labeled as T complexes, while agostic, counteranion and solvent-stabilized complexes are designated as A, C and S complexes, respectively.
![[1860-5397-9-153-4]](/bjoc/content/figures/1860-5397-9-153-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: General classification of T-shaped Pt(II) structures according to the fourth coordination site.
Figure 4: General classification of T-shaped Pt(II) structures according to the fourth coordination site.
True T-shaped
The isolation and characterization of such highly electrophilic species is challenging. Accordingly, the number of well-characterized true T-shaped Pt(II) complexes is scarce. The empty site should be blocked somehow to prevent undesirable contacts, and in most cases, bulky ligands are required to protect the empty coordination site. Strong electron-donor ligands also help stabilizing the formally 14-electron compounds. It is worthy of note that almost all of them are cationic.
The first true T-shaped complexes were prepared by using phosphine ligands (Figure 5). In the 80s, Goel et al. proposed, from nuclear magnetic resonance (NMR) data, a T-shaped structure for the cationic hydride trans-[(t-Bu3P)2Pt(H)]+ T1 containing bulky phosphine ligands [19]. However, it was not until 2005 when Braunschweig et al. successfully characterized a true 14-electron T-shaped Pt(II) boryl complex T2a by means of X-ray studies [20]. By halide abstraction, the cationic trans-[Pt(B(Fc)Br)(PCy3)2]+ T2a (Fc = ferrocenyl; Cy = cyclohexyl) could be obtained, in which the boryl ligand is located trans to the empty site. No agostic interactions were detected, the shortest Pt–H and Pt–C distances being 2.542 Å and 3.117 Å, respectively. Inspired by this chemical template, the synthesis of Pt–boryl derivatives of the type trans-[Pt(BRR’)(PCy3)2]+ has been extended [21]. Even at extreme electronic conditions in dicationic trans-[Pt(BR(4-picoline))(PCy3)2]2+ T3, the complex remains truly T-shaped, although small Lewis donors such as CO and acetonitrile can coordinate to the open coordination site of T3 [22]. DFT-based electron localization function (ELF) analyses [20] and geometry optimizations [22] supported the lack of agostic interactions in these complexes. This absence has been attributed to the strong trans influence exerted by the ligand in trans position with respect to the vacant site [23,24].
![[1860-5397-9-153-5]](/bjoc/content/figures/1860-5397-9-153-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Hydride, boryl and borylene true T-shaped Pt(II) complexes.
Figure 5: Hydride, boryl and borylene true T-shaped Pt(II) complexes.
Moreover, in the past few years the synthesis of 14-electron Pt(II) complexes was extended by using N-heterocyclic carbene (NHC) ligands (Figure 6), which have been proven to be useful stabilizing electron-deficient transition-metal species [25-27]. In this regard, recent studies state that the use of IMes* (4,5-dimethyl-1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) and IMes (1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) ligands in [Pt(Me)(NHC)2]+ T4 and [Pt(NHC’)(NHC)]+ T5 (NHC = IMes*, IMes; NHC’ = cyclometalated ligand) provides pure T-shaped species with no agostic stabilization [28]. Additionally, the resulting [Pt(Ar)(IMes*)2]+ T6 formed after C–H bond activation has also proven to be a three-coordinate species with no agostic interactions according to the X-ray structure of the derivative T6d, in which the closest Pt–H contact is located at 3.117 Å. The absence of agostic interactions has been attributed to geometrical constraints, the limited flexibility of the mesityl groups in IMes* and IMes hampering the approach of the CH bond to the metal center. Once again, Lewis acids as acetonitrile can access the empty site, which means that a potential reactant molecule can coordinate to the metal for further reactions.
![[1860-5397-9-153-6]](/bjoc/content/figures/1860-5397-9-153-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: NHC-based true T-shaped Pt(II) complexes.
Figure 6: NHC-based true T-shaped Pt(II) complexes.
Masked T-shaped via agostic interaction
The agostic interaction [29,30] is usually explained as an intramolecular 3-center-2-electron interaction between a metal M and a C–H bond. This type of contact is a recurrent event in unsaturated transition-metal complexes [31] and it can be characterized by structural and spectroscopic techniques [29,30] together with computational tools [32,33]. The agostic interaction shows different behaviors. It can be a transient species prior to the C–H bond breaking or stabilize low electron count situations. In this section we will focus on the latter aspect. Since this intramolecular interaction is difficult to avoid, even in the presence of bulky ligands, it is the most common motif for the stabilization of three-coordinate complexes.
Early works on T-shaped Pt(II) species are related to complexes stabilized with phosphine ligands (Figure 7). Pioneering studies on alkyl complexes from Orpen’s and Spencer’s research groups reported the synthesis of cationic [Pt(norbornyl)(P–P)]+ complexes A1a–e (P–P = bidentate phosphine ligand) [34,35]. Further studies involved other alkyl ligands such as ethyl (A2b–c), 3,3-dimethylbutyl (A3b–c) and 2,3,3-trimethylbutyl (A4) [36-38]. NMR spectroscopic data and X-ray structures of A1a [34] and A2b [36] demonstrate that these compounds display a β-agostic interaction filling the fourth coordination site. This interaction is strong enough to cleave the C–H bond, creating a chemical equilibrium with the corresponding hydrido–alkene derivatives. Indeed, the detected ground state of the compounds in brackets (A2a, A2d, A2e and A3a in Figure 7) is the hydrido–alkene isomer instead of the agostic–alkyl one. Later, Baratta et al. succeeded in the isolation of 14-electron Pt(II) complexes bearing bulky phosphine ligands [39]. The usage of trans-[Pt(Me)Cl(PR3)2] as a starting material, followed by halide removal and the release of methane by intramolecular C–H bond activation provided the cationic T-shaped cyclometalated complexes A5a–b. The X-ray structure of A5b shows that the platinum atom exhibits a δ-agostic interaction with one hydrogen atom of the methyl group of the non-cyclometalated phosphine ligand. Subsequent hydrogenation generates the corresponding hydride complexes A6a–b. On the basis of NMR studies (1JPt,H values of ca. 2000 Hz, see next section), a weak interaction trans to the hydride ligand, typically an agostic contact, is supposed to exist. Carmona and co-workers also prepared quite similar complexes, A5c and A6c, where R labels represent isopropyl groups [40]. Weller and co-workers reported the formation of complex trans-[Pt(Me)(PiPr3)2]+ A7 in which a γ-agostic interaction is located in the fourth coordination site [41]. The addition of tetrahydrofuran (THF) rapidly forms the corresponding adduct. Braunschweig et al. obtained the agostic structure A8 by employing BCat as a ligand (BCat = catecholatoboryl) in three-coordinate Pt(II) complexes [21]. Unlike the previous pure T-shaped boryl complexes T2 (Figure 5), the weaker trans influence of the BCat ligand allows the formation of an agostic contact. Treatment of A8 with Lewis bases also removes the agostic interaction, generating the adduct complex.
![[1860-5397-9-153-7]](/bjoc/content/figures/1860-5397-9-153-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Phosphine-based agostic T-shaped Pt(II) complexes. Compounds in brackets correspond with hydrido–alkene ground states.
Figure 7: Phosphine-based agostic T-shaped Pt(II) complexes. Compounds in brackets correspond with hydrido–al...
Natural Bond Orbital (NBO) and Atoms In Molecules (AIM) calculations on complexes A1, A2b, A5b and A7 classified the Pt···H–C contacts as agostic interactions for all the species with the only exception being A7, in which a H-bond character can also be ascribed [42].
Concerning other types of ligands (Figure 8), the phenylpyridyl complex A9 reported by Rourke and co-workers exhibits a bifurcated δ-agostic interaction that has been determined by X-ray studies [43]. This is one of the rare neutral T-shaped Pt(II) complexes. Referring to NHC ligands, Rivada-Wheelaghan et al. isolated the three-coordinate methyl [Pt(Me)(IPr)2]+ (A10) and cyclometalated [Pt(NHC’)NHC]+ complexes (NHC = IPr A11a, It-Bu (1,3-bis(tert-butyl)imidazol-2-ylidene) A11b; NHC’ = cyclometalated ligand) [44]. In sharp contrast with the analogous T5 and T6 (Figure 6) [28], δ- and ζ-agostic interactions at the fourth coordination site were detected by X-ray and NMR studies for complexes containing IPr (A11a) and It-Bu (A11b), respectively.
![[1860-5397-9-153-8]](/bjoc/content/figures/1860-5397-9-153-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Phenylpyridine and NHC-based agostic T-shaped Pt(II) complexes.
Figure 8: Phenylpyridine and NHC-based agostic T-shaped Pt(II) complexes.
Selected Pt···H–C parameters from the X-ray structures of agostic compounds are collected in Table 1. As it can be expected, short Pt–H and Pt–C distances are observed for the β-agostic interactions in A1a and A2b. In addition, Pt–H–C angles larger than 100° correlate with large Pt–C distances as shown in A7, A8 and A11a. Although a remote contact is described in A11a, the largest distances correspond with the γ-agostic A8. In other words, the strength of the agostic interaction is not only governed by geometrical constraints and the surroundings of the CH group, but also by the trans influence of the ligand in trans position with respect to the agostic interaction [23,24]. In this case, the boryl ligand in A8 exhibits a higher trans influence than the alkyl group in A11a [45,46], and therefore the agostic interaction in the former is weaker.
Table 1: Selected geometrical parameters of X-ray characterized agostic T-shaped Pt(II) structures.
aNot available. bBifurcated agostic interaction.
Masked T-shaped via counteranion interaction
Most of the T-shaped Pt(II) complexes are cationic. Thus, there is the possibility of stabilizing the unsaturated structure by nonbulky, weakly coordinating counteranions. Triflate (OTf = SO3CF3) and tetrafluoroborate are the best candidates to play such a role (Figure 9). All the well-characterized compounds bear a coordinated triflate anion. In the 80s, Whitesides et al. described the synthesis of trans-[Pt(Np)(OTf)(PMe3)2] (Np = neopentyl) C1a. This compound reacts with benzene forming trans-[Pt(Ph)(OTf)(PMe3)2] C1b through a mechanism that involves trans-[Pt(Np)(PMe3)2]+ as a reactive intermediate [47]. The labile character of the coordinated triflate has been alleged to justify the significant difference between the 1JPt,P found for [Pt(Me)(OTf)(dmpe)] C2 (dmpe = 1,2-bis(dimethylphosphino)ethane) in CD2Cl2 (4572 Hz) and in acetone (4305 Hz). In the latter media one solvent molecule is proposed to displace the triflate ligand, forming [Pt(Me)(acetone)(dmpe)][OTf] [48]. The complex [Pt(Me)(OTf)(dbbipy)] C3 (dbbipy = 4,4'-di-tert-butyl-2,2'-bipyridine) was prepared by treatment of [PtCl(Me)(dbbipy)] with AgOTf [49]. Regarding tridentate ligands, the X-ray structure of the triflate complex [Pt(3,3'-iPr2-BQA)(OTf)] C4b (BQA = bis(8-quinolinyl)amine) shows a coordinated OTf ligand (Pt–O = 2.097 Å) in trans position to the amido N-donor of the pincer-like amido ligand. In solution, the labile triflate ligand can be displaced, allowing a reaction with benzene [50]. Milstein and co-workers have reported a series of pincer-type Pt(II) complexes C5–8 containing an XCX ligand core (X = N, P), incorporating the anions OTf− and BF4− [51-53]. The structure of C5b was determined by X-ray crystallography [51]. The Pt–O bond distance in this compound, in which the triflate anion is coordinated to platinum in trans position with respect to the aromatic ring, is considerably longer than in C4b (C4b: 2.097 Å [50]; C5b: 2.249 Å [51]). The weakly coordinating character of triflate accounts for its sensitivity to the trans influence.
![[1860-5397-9-153-9]](/bjoc/content/figures/1860-5397-9-153-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Counteranion coordination in T-shaped Pt(II) complexes.
Figure 9: Counteranion coordination in T-shaped Pt(II) complexes.
Masked T-shaped via solvent coordination
In solution, a solvent molecule can occupy the vacant position of a T-shaped three-coordinate Pt(II) compound via σ-interaction. A number of phosphine-based complexes with different solvent molecules coordinated to the T-shaped frame have been characterized (Figure 10). In this regard, Kubas and co-workers prepared a set of hydride trans-[PtH(PiPr3)2(solv)]+ compounds where “solv” stands for η1-ClCH2Cl (S1a), OEt2 (S1b) and THF (S1c) solvent molecules [54]. Interestingly, the closely related species T1 (Figure 5) does not include any solvent molecule filling the vacancy [19]. Moreover, the structures of S1a and S1c were fully confirmed by X-ray studies. The similar dichloromethane adduct S1d was later detected by NMR spectroscopy [55]. This work was extended to methyl complexes with the formula trans-[Pt(Me)(PR3)2(solv)] (S2) [56,57]. Similar structures have been obtained by the use of chelating phosphine ligands in diethylether (S3) [58]. In this line, Peters and co-workers developed several bidentate phosphine ligands to yield complexes S4 in which one THF molecule is coordinated to the metal center [59,60]. Romeo’s group was actively working on solvento Pt(II) complexes S5. For instance, they employed an extended series of phosphine ligands to obtain cis and trans-solvento complexes containing methanol [61]. The formation of acetonitrile-d3 adducts with triethylphosphine ligands was also reported [2,62].
![[1860-5397-9-153-10]](/bjoc/content/figures/1860-5397-9-153-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Phosphine-based solvento Pt(II) complexes.
Figure 10: Phosphine-based solvento Pt(II) complexes.
Several examples involving chelating nitrogen-based ligands have also been reported (Figure 11). Bercaw and co-workers developed cationic solvento Pt(II) complexes S6 bearing tetramethylethylenediamine (tmeda) [63]. X-ray crystallography of S6a·BArF and S6c·BArF verified the solvent coordination [64]. Diimine analogues, valuable in C–H bond-activation processes, have been extensively reported elsewhere (S7) [65-72]. Later, Tilset’s group prepared and characterized a set of solvento tolyl complexes of the type [Pt(Tol)(N–N)(CH3CN)] S8 by means of 1H and 19F NMR [73]. The formation of p-xylene derivates S9 containing a molecule of the weakly coordinating 2,2,2-trifluoroethanol (TFE) has also been reported [69,74]. Unsaturated bipyridyl compounds S10 have been isolated in which the presence of solvent molecules has been detected [75,76]. The stabilizing role of the solvent molecule is quite relevant. For instance, the removal of the THF molecule in S10b provokes the decomposition of the complex [75]. The facile displacement of the solvent ligand allows the use of these compounds as catalysts in hydrophenylation reactions [76]. Other related complexes have been prepared elsewhere [77-81].
![[1860-5397-9-153-11]](/bjoc/content/figures/1860-5397-9-153-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Nitrogen-based solvento Pt(II) complexes.
Figure 11: Nitrogen-based solvento Pt(II) complexes.
Pincer ligands [82,83] naturally define a three-coordinate environment and, in this way, they are well-suited as ligands in T-shaped compounds. Accordingly, a number of T-shaped Pt(II) pincer complexes with an additional solvent molecule as a fourth ligand have been reported (S11–19, Figure 12). Van Eldik and co-workers studied the aquo complexes S11 by using several tridentate nitrogen-based ligands (NNN) [84]. Peters and co-workers isolated [Pt(OTf)(NNN)] species, though NMR data were collected in acetonitrile-d3 solution, in which the solvento adduct S12 is favored [50]. Extensive work carried out by van Koten and co-workers [85,86] concerned NCN aquo compounds such as S13. Unsaturated complexes bearing NCP ligands were characterized by Milstein and co-workers, including water (S14a) [51] and acetone (S14b) [87] solvent molecules. Isolation of compound S15 in acetone/pyridine solution provided suitable crystals for X-ray studies, which showed that a pyridine molecule has been added [87]. Other investigations have been devoted to complexes with PCP ligands. For instance, Bullock and co-workers prepared a dihydrogen adduct of the type [PtH2(PCP)]+ in dichloromethane, but NMR data suggest a mixture of the former compound and the corresponding solvento complex S16a [88]. Milstein et al. proposed that the closely related complex S16b exists in THF solution, since an interaction with the BF4− counteranion is not observed, although the elemental analysis evidences the lack of the THF molecule in the solid state [89]. Contrarily, the naphthyl derivative S17 was found together with the BF4− adduct C8 (Figure 9) [53]. For silicon-related ligands, Turculet and co-workers successfully crystallized S18 in a diethylether solution at low temperature, showing one diethylether molecule directly coordinated to the platinum center [90]. A set of dicationic complexes based on carbene groups have been developed by Limbach and co-workers, forming pyridine S19a and acetonitrile S19b,c adducts [91].
![[1860-5397-9-153-12]](/bjoc/content/figures/1860-5397-9-153-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Pincer-based solvento Pt(II) complexes.
Figure 12: Pincer-based solvento Pt(II) complexes.
The relevance of solvent-stabilized T-shaped Pt(II) complexes extends beyond the organometallic field. Their presence in cisplatin–protein adducts has also been proposed concerning the bovine Cu, Zn superoxide dismutase [92] and the hen egg white lysozyme [93]. The corresponding crystallographic structures show the platinum coordinated to a histidine residue of the protein and two ligands (Cl− [92] or NH3 [93]). However, in both situations the fourth ligand was not fully detectable, so that the authors suggested the participation of a coordinating water molecule. Indeed, quantum mechanics/molecular mechanics (QM/MM) calculations on the cisplatin–hen egg white lysozyme adduct confirmed the facile inclusion of a solvent water molecule in the first coordination shell of the platinum complex (Figure 13) [94].
![[1860-5397-9-153-13]](/bjoc/content/figures/1860-5397-9-153-13.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: Structure of the QM/MM optimized cisplatin–protein adduct [94].
Figure 13: Structure of the QM/MM optimized cisplatin–protein adduct [94].
NMR coupling constants to 195Pt as sensitive probes for coordinatively unsaturated Pt(II) complexes
NMR spectroscopy has provided additional evidence for the existence of low electron-count Pt(II) complexes not only through the observation (in some cases) of NMR signals for the C–H bonds involved in agostic interactions, but also by the magnitude of the coupling constant of some of the ligands around the metal center with the NMR active 195Pt nuclei (33.7% natural abundance). This is actually not the case with 31P NMR, for which the JPt,P values seem to be virtually insensitive to the nature of the complex. As an example, in the boryl derivatives reported by Braunschweig et al. [21] the JPt,P coupling constant is almost the same both in the starting material, [Pt(BR2)Br(PCy3)2], and in the three-coordinate Pt(II) species, [Pt(BR2)(PCy3)2][BArF] T2. On the other hand, the coupling constants of the proton and carbon atoms of alkyl and hydride ligands with 195Pt are very sensitive to the presence of coordinating ligands trans to them (Figure 14).
![[1860-5397-9-153-14]](/bjoc/content/figures/1860-5397-9-153-14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: NMR coupling constants used for the characterization of three-coordinate Pt(II) species.
Figure 14: NMR coupling constants used for the characterization of three-coordinate Pt(II) species.
A nice example is illustrated in the hydride, T-shaped Pt(II) complex trans-[PtH(Pt-Bu3)2][X] T1 (X = PF6, BF4, ClO4, SO3CF3) reported by Goel and Srivastava [19]. The magnitude of the 1JPt,H coupling constant of the hydride ligand in its precursor trans-[PtHCl(Pt-Bu3)2] is 1070 Hz, but in the 14-electron derivative trans-[PtH(Pt-Bu3)2][X] T1 it increases to ca. 2600 Hz, the largest 1JPt,H reported in the literature. A decrease of this coupling constant to 2050–2070 Hz was observed in the related hydride Pt(II) complexes trans-[PtH(PR2(2,6-Me2C6H3))2]+ (Figure 7, R = Ph A6a, Cy A6b, iPr A6c), which are stabilized by agostic interactions involving the methyl groups of the aryl fragments. In the same vein, weakly coordinating ligands such as the solvent stabilized Pt(II) derivatives trans-[PtH(ClCH2Cl)(PR3)2][BArF] (Figure 10, R = iPr S1a, Cy S1d) exhibit a smaller JPt,H of 1480 (R = Cy) or 1852 Hz (R = iPr) in agreement with the presence of a ligand trans to the hydride. With regard to Pt–H two-bond and Pt–C one-bond coupling constants in Pt–Calkyl complexes (Figure 15 and Table 2), Weller et al. reported that the methyl group in the complex trans-[Pt(Me)(PiPr3)2]+ A7 exhibits a 2JPt,H value of 106 Hz. This value is almost identical to those reported for the NHC derivatives trans-[Pt(Me)(NHC)2]+ (NHC = IMes* T4a, IMes T4b; 110 and 103 Hz, respectively). The 1JPt,C is also very large for these methyl derivatives, ranging from 755 to 780 Hz (Table 2). Cyclometalated phosphine [39,40] and NHC [28,44] Pt(II) compounds were also shown to have very large 2JPt,H coupling constants (slightly larger than those for the non-cyclometalated versions) spanning from 107 Hz to ca. 135 Hz (average) and 1JPt,C values between 805 and 975 Hz.
![[1860-5397-9-153-15]](/bjoc/content/figures/1860-5397-9-153-15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: The chemical formula of the complexes discussed in Table 2.
Figure 15: The chemical formula of the complexes discussed in Table 2.
In those cases where solvent molecules (THF, acetonitrile) have been reported to coordinate to some of the complexes shown in Figure 15, smaller values of both the 2JPt,H and the 1JPt,C coupling constant are observed. For example, the 2JPt,H of the methyl ligand decreases to 98 Hz in the THF adduct of derivative A7, whereas the acetonitrile adduct of complex A11b (A11b·NCMe) shows resonances for the Pt–CH2 fragment with 2JPt,H and 1JPt,C values of 87 and 798 Hz, respectively [28].
Synthetic routes to stable, solvent-stabilized and transient 14-electron Pt(II) species
True and agostic T-shaped Pt(II) complexes
Several methods have been described to prepare coordinatively unsaturated Pt(II) complexes with a T-shaped geometry. Although the number of species that have been authenticated by crystallographic or spectroscopic methods is still very limited, the best and most general method for obtaining them is by removing a halogen ligand (Cl, Br, I) from the platinum coordination center in 1 by using a halogen abstractor with a poor coordinating anion, such as tetrakis[3,5-(trifluoromethyl)phenyl]borate or hexafluoroantimonate (Scheme 1) [19-21,28,39,44].
![[1860-5397-9-153-i1]](/bjoc/content/inline/1860-5397-9-153-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
This procedure has been successfully employed for the generation of complexes stabilized by two phosphine PR3 or two NHC ligands, in which the third coordination site is occupied by alkyl, hydride or boryl ligands. Therefore, high trans-influence ligands are present in all these cases, favoring the dissociation of the Pt–X bond. The generality of this method is so powerful that it has provided access to an intriguing dicationic borylene Pt(II) complex T3 which is not stabilized by agostic interactions (Scheme 2) [22].
![[1860-5397-9-153-i2]](/bjoc/content/inline/1860-5397-9-153-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Halogen abstraction from 2 forming the dicationic complex T3 [22].
Scheme 2: Halogen abstraction from 2 forming the dicationic complex T3 [22].
A stable T-shaped structure trans-[Pt(Me)(PiPr3)2]+ (A7) has also been generated [41] by methide abstraction from the neutral derivative cis-[Pt(Me)2(PiPr3)2] by using highly electrophilic Lewis acids such as B(C6F5)3 or [CPh3][1-H-closo-CB11Me11] (with the concomitant formation of MeB(C6F5)3− or MeCPh3, respectively). This synthetic route uses the same procedure described by Goldberg et al. for the generation of a transient, neutral Pt(II) derivative [Pt(Me)(κ2-TpMe2)] from the anionic Pt(II) complex K[Pt(Me)2(κ2-TpMe2)] [95]. Alternatively, the cationic compound [Pt(Me)(PiPr3)2]+ was synthesized by homolytic cleavage of the Pt–Me bond by using the neutral radical [1-H-closo-CB11Me11]• as a reagent.
H2 addition across Pt–Calkyl bonds can also lead to electron-deficient Pt(II) species stabilized by agostic interactions. Baratta and co-workers studied the addition of H2 to preformed T-shaped Pt(II) complexes A5a,b to prepare Pt(II) hydrides A6a,b (Scheme 3) [39].
![[1860-5397-9-153-i3]](/bjoc/content/inline/1860-5397-9-153-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Hydrogenation of complexes A5a and A5b [39].
Scheme 3: Hydrogenation of complexes A5a and A5b [39].
Alternatively, Carmona and co-workers have recently described that the hydrogenation of the 16-electron Pt(II) carbene 3 bearing a cyclometalated phosphine ligand (Scheme 4) resulted in the formation of the cyclometalated complex A5c, which can be further hydrogenated to give the Pt(II) hydride A6c [40], similar to those reported by Baratta [39].
![[1860-5397-9-153-i4]](/bjoc/content/inline/1860-5397-9-153-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Hydrogenation of complexes 3 and A5c [40].
Scheme 4: Hydrogenation of complexes 3 and A5c [40].
Low electron-count species can also be prepared by C–H bond-activation reactions from coordinatively unsaturated compounds in a way similar to the addition of H2 to Pt–Calkyl bonds mentioned above. The cyclometalated complex T5a shown in Scheme 5 reacts with aromatic compounds to yield the corresponding aryl complexes while keeping the unsaturated nature at the platinum atom. The complexes thus synthesized are not stabilized by agostic interactions according to spectroscopic (NMR), crystallographic and theoretical methods [28].
![[1860-5397-9-153-i5]](/bjoc/content/inline/1860-5397-9-153-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Intermolecular C–H bond activation from T5a [28].
Scheme 5: Intermolecular C–H bond activation from T5a [28].
Spencer et al. reported that the formation of stable electron-deficient Pt(II) complexes stabilized by strong β-agostic interactions can be accessible by protonation of electron-rich alkene Pt(0) compounds 4 (Scheme 6) [35,36].
![[1860-5397-9-153-i6]](/bjoc/content/inline/1860-5397-9-153-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Protonation of complexes 4 [35,36].
Scheme 6: Protonation of complexes 4 [35,36].
Finally, Rourke et al. isolated a neutral Pt(II) derivative A9 by a direct and simple approach that involves a cyclometalation process of the 2-substituted bulky pyridine 5 when it is reacted with the platinum salt K2PtCl4 (Scheme 7) [43].
![[1860-5397-9-153-i7]](/bjoc/content/inline/1860-5397-9-153-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Pt(II) complexes stabilized by solvent molecules
Although some of these compounds have been prepared by halogen [58] or methide [74] abstraction as described above, the vast majority of the Pt(II) derivatives stabilized by solvent molecules have been obtained by protonation of neutral alkyl, aryl or hydride Pt(II), as shown in Scheme 8 (see for example: [55,59,60,63,67,69,72,77-81]), or Pt(IV) complexes [96-98]. Nonetheless, this method has not yet been exploited for the preparation of true 14-electron species or agostic stabilized derivatives.
![[1860-5397-9-153-i8]](/bjoc/content/inline/1860-5397-9-153-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Transient electron-deficient Pt(II) complexes from six- and five-coordinate Pt(IV) derivatives
In some cases, transient, highly reactive 14-electron Pt(II) compounds have been generated by thermal decomposition of trimethyl, five-coordinate Pt(IV) complexes 7. These latter compounds are in some cases sufficiently stable to be isolated and characterized by X-ray diffraction studies, but thermally unstable at moderate to high temperatures releasing ethane and a three-coordinate Pt(II) intermediate [Pt(Me)L2]+ (Scheme 9). The coordinatively unsaturated complexes [Pt(Me)L2]+ thus generated undergo subsequent reactivity with appropriate reagents [99-102].
![[1860-5397-9-153-i9]](/bjoc/content/inline/1860-5397-9-153-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reductive elimination of ethane from 7.
Scheme 9: Reductive elimination of ethane from 7.
Similarly, methane reductive elimination from Pt(IV) complexes [PtH(Me)(R)(η3-N3)]n+ 9 (R = Me, H; N3 = tris-pyridine[2.1.1]-(2,6)-pyridinophane or tris(pyrazolyl)borate; n = 0, 1) produces a transient three-coordinate Pt(II) complex [Pt(R)(η2-N3)]n+ 10 (Scheme 10), which is able to activate C–H bonds of hydrocarbons or form solvent adducts [103-105].
![[1860-5397-9-153-i10]](/bjoc/content/inline/1860-5397-9-153-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Reductive elimination of methane from six-coordinate Pt(IV) complexes.
Scheme 10: Reductive elimination of methane from six-coordinate Pt(IV) complexes.
Solution behavior
In masked three-coordinate d8 complexes, the fourth coordination site is occupied by a weakly bound ligand, which can be easily displaced. In this way, in solution the ligands of these compounds are opened to conformational events that will be discussed in this section. The empty site can also be available for incoming substrates, allowing the participation of these complexes as reaction intermediates. This aspect will be discussed in the next section.
Most of the reports of fluxional processes correspond to agostic bonded complexes. The equivalence of 1H and 13C chemical shifts observed in solution for the γ-, δ- and remote-agostic contacts is a common feature of the complexes collected in Figure 15 and arises from the rapid intramolecular exchange of the C–H bond involved in the agostic interaction. In several cases, such as A11a [44], the overlapping of the proton signals and signal averaging of the ligands avoid drawing a conclusion about the nature of the agostic interaction at work in solution. Recently, ab initio molecular dynamics (AIMD) simulations of some representative T-shaped Pt(II) complexes (T5b, A2b and A11a), performed in explicit dichloromethane solvent, have provided a detailed description of the mechanism by which the equivalence of signals takes place [106]. Simulations showed that the dynamics of the agostic interaction in solution strongly depends on the complex. Contingent upon the strength of the agostic interaction and the flexibility of the ligand, several events related with the occupation of the fourth coordination site by an agostic bond could happen: (i) the same C–H bond maintains the agostic interaction with the platinum atom for the entire simulation (A2b); (ii) an agostic interaction is present throughout the simulation, though the C–H bond changes by a rotation of alkyl groups (A11b); (iii) the agostic interaction moves on and off (A11a) [106].
There are indirect evidences that agostic bonds, solvent and weakly coordinating counteranions could easily exchange their role of stabilizing the unsaturated complex by placement at the vacant site. Although no kinetic studies on exchanges of these kinds of ligands have been reported, the detection of the agostic and solvent complexes for the same system and, for pincer complexes, of the counteranion and solvent forms, suggests that the exchange can take place. For instance, in trans-[Pt(Me)(PiPr3)2]+ (A7) a γ-agostic interaction can be displaced by addition of THF, forming the corresponding adduct [41]. NMR data in CD3CN of the triflate complex [Pt(BQA)(OTf)] (C4a) leads to its formulation as [Pt(BQA)(NCCD3)][OTf] (S12) [50]. Similar behavior has been observed for other pincer complexes. The naphthyl-based PCP–Pt(II) complex was found in THF as a 1:2 mixture of the counteranion (BF4−, C8) and solvent (S17) forms [53].
Energetically accessible T-shaped species can also be intermediates in the site exchange of bidentate ligands of square-planar complexes. Fluxional motions of the 2,9-dimethyl-1,10-phenanthroline ligand (dmphen) have been observed in cationic complexes as [Pt(Me)(NN)(L)]+ (NN = dmphen; L = SOR(R’), PR3) [107-111]. The driving force of these flipping processes is the distortion of the dmphen ligand with respect to the coordination plane, which is caused by the methyl groups. Indeed, the fluxional motion is not detected when the unhindered 1,10-phenanthroline ligand is used. Interestingly, the mechanism can be switchable between associative and dissociative [107-109]. For the latter scenario, 14-electron T-shaped species involving phosphine ligands can be envisaged as feasible intermediates. The dissociative pathway fully prevails when overcrowded PR3 ligands are employed, where R stands for o-methoxyphenyl 11 [110] and o-tolyl 12 [111]. The detailed mechanism (Scheme 11) is described as follows: (i) dissociative nitrogen decomplexation, (ii) isomerization between two nonequivalent exchanging sites, and (iii) nitrogen coordination recovering the initial chelating situation. The fluxional motion of dmphen is not affected by the counteranion, solvent, or the presence of weak nucleophiles deliberately added. In addition, for 11a and 12a both flipping of dmphen and phosphine rotation motions are synchronized, behaving as molecular gears. The calculated activation barriers are ca. 13.3 and 16.4 kcal mol−1 for the motions involving 11 and 12, respectively.
![[1860-5397-9-153-i11]](/bjoc/content/inline/1860-5397-9-153-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Proposed dissociative mechanism for the fluxional motion of dmphen in [Pt(Me)(dmphen)(PR3)]+ complexes.
Scheme 11: Proposed dissociative mechanism for the fluxional motion of dmphen in [Pt(Me)(dmphen)(PR3)]+ comple...
Concerning 14-electron intermediates b and b’, the assistance of methoxy groups in 11b,b’ [110] as well as the presence of agostic interactions in 12b,b’ [111] can be postulated (Figure 16). Indeed, 12a easily undergoes a cyclometalation process, probably assisted by an agostic contact via intermediate 12b.
![[1860-5397-9-153-16]](/bjoc/content/figures/1860-5397-9-153-16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 16: Feasible interactions for unsaturated intermediates 11b (left) and 12b (right) during fluxional motions [110,111].
Figure 16: Feasible interactions for unsaturated intermediates 11b (left) and 12b (right) during fluxional mot...
Three-coordinate Pt(II) species as reaction intermediates
Coordinatively unsaturated Pt(II) species are considered as intermediates in many organometallic processes. Early studies by Whitesides and co-workers dealing with the thermal decomposition of [Pt(R)2L2] complexes demonstrated that the dissociation of a phosphine ligand is a preliminary requisite for the reaction to occur [112]. Since then, T-shaped three-coordinate 14-electron intermediates have been proposed in a large number of reaction mechanisms. A review concerning dissociative pathways in Pt(II) complexes was published in 1990 [113].
Due to both the low electron count and the presence of a vacant site in the coordination sphere, T-shaped Pt(II) species are suitable intermediates in ligand-exchange and bond-breaking processes of unreactive bonds, such as C–H bond activations. A selection of results from the past few years regarding these two issues are collected in this section to illustrate the growing importance of three-coordinate Pt(II) species as reaction intermediates.
Intramolecular C–H bond activation
Intramolecular C–H bond activation is a common reaction of unsaturated Pt(II) complexes. Cyclometalation processes, sometimes involving agostic situations [114], have been thoroughly reviewed recently [115].
According to Baratta and co-workers, the abstraction of Cl from 13a,b with Na[BArF] directly generates the cyclometalated products A5a,b and methane (Scheme 12) [39]. Unsaturated intermediates such as [Pt(Me)(PR3)2]+, probably stabilized by agostic contacts, might be considered.
![[1860-5397-9-153-i12]](/bjoc/content/inline/1860-5397-9-153-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Halogen abstraction from 13a,b and subsequent cyclometalation to yield complexes A5a,b [39].
Scheme 12: Halogen abstraction from 13a,b and subsequent cyclometalation to yield complexes A5a,b [39].
Weller et al. reported some reactivity features of complex A7 [41]. Although it shows an agostic interaction, no cyclometalation via C–H bond activation is observed in CD2Cl2 (40 °C, 7 days). However, the addition of THF rapidly traps the T-shaped complex forming the adduct 14 (Scheme 13). This species does undergo cyclometalation in the presence of trace amounts of acids to yield 16 [116]. The unsaturated agostic species 15 has been proposed as a reaction intermediate.
![[1860-5397-9-153-i13]](/bjoc/content/inline/1860-5397-9-153-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Proposed mechanism for the acid-catalyzed cyclometalation of 14 via intermediate 15 [41].
Scheme 13: Proposed mechanism for the acid-catalyzed cyclometalation of 14 via intermediate 15 [41].
Goldberg and co-workers put forward the participation of T-shaped Pt(II) species in processes, implying the five-coordinate Pt(IV) complex 7a (Scheme 14) [102]. This compound undergoes reductive elimination in benzene-d6 solvent with the concomitant formation of ethane. The resulting three-coordinate 8a is supposed to be stabilized by an agostic interaction or solvent coordination. Indeed, two possible scenarios related to these interactions are envisaged. Firstly, 8a evolves to 17 through a cyclometalation process ((i) in Scheme 14) which involves one t-Bu group of the bidentate ligand. On the other hand, 8a can also activate the C–D bonds of benzene-d6 molecules forming 18 ((ii) in Scheme 14). Since both 17 and 18 exhibit potential empty coordination sites, benzene activation from 17 and cyclometalation from 18 are plausible scenarios. Nevertheless, the three-coordinate intermediate 17 is not isolable and reacts further with a molecule of the starting material 7a leading to the dinuclear species 19. The inclusion of deuterium atoms in the t-Bu groups of 19 suggests that both intra- and intermolecular C–H bond activations are competitive pathways. The transient formation of derivative 17 was confirmed by trapping experiments with ethylene.
![[1860-5397-9-153-i14]](/bjoc/content/inline/1860-5397-9-153-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Proposed mechanism for the formation of 19 [102].
Scheme 14: Proposed mechanism for the formation of 19 [102].
Examples of cyclometalation processes preceded by ligand dissociation have been described by Romeo and co-workers [111,117,118]. For instance, [Pt2(Hbph)4(μ-SEt2)2] 20 (Hbph = η1-biphenyl monoanion) undergoes cyclometalation to form [Pt2(bph)2(μ-SEt2)2] 22 and biphenyl H2bph (Scheme 15) [117]. Intramolecular C–H bond activation seems to be driven by thioether dissociation via 21, a process previously reported for the complex [Pt2(Me)4(μ-SMe2)2] [119].
![[1860-5397-9-153-i15]](/bjoc/content/inline/1860-5397-9-153-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Cyclometalation of 20 via thioether dissociation [117].
Scheme 15: Cyclometalation of 20 via thioether dissociation [117].
Marrone et al. computed the cyclometalation process of [Pt(Me)2(PR3)(DMSO)] 23 to yield [Pt(Me)(PR2R’)(DMSO)] 28 (R = o-tolyl, R’ = cyclometalated group; DMSO = dimethylsulfoxide) [120] as experimentally reported in [118]. They demonstrated that the reaction involves coordinatively unsaturated 14-electron T-shaped complexes through (i) DMSO dissociation, (ii) C–H bond activation, (iii) methane release, and (iv) DMSO association (Figure 17). Starting from 23, initial ligand dissociation generates the T-shaped intermediate 24, in which one o-tolyl group of the phosphine ligand establishes an agostic interaction with the platinum center. This agostic interaction weakens the C–H bond inducing an oxidative-addition (OA) process. The resulting five-coordinate Pt(IV) hydride complex 25, located at 26.0 kcal mol−1 above reactants, quickly undergoes reductive elimination (RE) providing the methane adduct 26. After methane releasing, the cyclometalated T-shaped structure 27 is obtained, once again stabilized by an agostic interaction. The corresponding transition states for OA and RE processes are isoenergetic with respect to 25. Finally, the agostic coordination mode in 27 is displaced by a DMSO molecule forming the cyclometalated product 28. Moreover, they showed that the reaction mechanism starting from the four-coordinate 16-electron complex 23 does not provide low-energy paths for OA, demonstrating the kinetic inertness of these types of compounds.
![[1860-5397-9-153-17]](/bjoc/content/figures/1860-5397-9-153-17.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 17: Gibbs energy profile (in chloroform solvent) for the cyclometalation of 23 [120].
Figure 17: Gibbs energy profile (in chloroform solvent) for the cyclometalation of 23 [120].
Three-coordinate species have been invoked by Nolan and co-workers to account for the reaction of cis-[Pt(NO3)2L2] 29 with tetramethylthiourea (tmtu) (Scheme 16) [121]. The sulfur atom of tmtu displaces one nitrate ligand of 29 forming 30, which eventually evolves to the cyclometalated product 33 via C–H bond activation. The increase of nitrate concentration as well as the addition of coordinating agents, such as triethylamine and triphenylphosphine, clearly retard the process. Therefore, a dissociative mechanism has been proposed to generate the unsaturated species 31. Then, one methyl group of the coordinated tmtu can stabilize the open coordination site via agostic interaction, 32, inducing an intramolecular C–H bond activation process to yield 33. It was also observed that less σ-donor phosphine ligands increase the reaction rate. Less-electron-donating ligands may not stabilize 31, favoring an agostic contact in 32, which, consequently, accelerates the cyclometalation reaction. Indeed, the presence of stronger σ-donors such as ICy ligand (1,3-biscyclohexylimidazol-2-ylidene) inhibits the process.
![[1860-5397-9-153-i16]](/bjoc/content/inline/1860-5397-9-153-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Coordination of tmtu to 29 and subsequent C–H bond activation via three-coordinate species 31 and 32 [121].
Scheme 16: Coordination of tmtu to 29 and subsequent C–H bond activation via three-coordinate species 31 and 32...
T-shaped Pt(II) complexes bearing NHC ligands can be prepared starting from the pertinent iodo-precursors by halide removal (Scheme 17) [44]. For IPr [44], IMes* and IMes ligands [28], the corresponding methyl complexes A10a and T4a,b can be isolated. Interestingly, cyclometalation involving a methyl group of the carbene arm was observed upon heating these methyl derivatives. It is noteworthy that the process barely depends on the nature of the fourth coordination site, either agostic A10a or pure empty site T4a,b. In the case of It-Bu ligand [44], a similar cyclometalation reaction is observed even at low temperatures (−70 °C), but the putative methyl intermediate could not be detected.
![[1860-5397-9-153-i17]](/bjoc/content/inline/1860-5397-9-153-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Cyclometalation process of NHC-based Pt(II) complexes [28,44].
Scheme 17: Cyclometalation process of NHC-based Pt(II) complexes [28,44].
The agostic complex A9 can exchange the site of cyclometalation by C(sp3)–H bond activation (Scheme 18) [43]. The addition of one equivalent of L (L = DMSO, PPh3 or pyridine) to A9 in chloroform or acetone at room temperature yields the products 37, in which one methyl group (previously agostic) has been cyclometalated, and L has entered into the coordination sphere of the platinum atom. The authors reasonably propose a σ-bond metathesis (σ-CAM) mechanism via intermediate 36 invoking the well-known capacity of agostic interactions to facilitate the C–H bond cleavage.
![[1860-5397-9-153-i18]](/bjoc/content/inline/1860-5397-9-153-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Cyclometalation process of complex A9 [43].
Scheme 18: Cyclometalation process of complex A9 [43].
The “rollover” process is a class of cyclometalation, in which a heteroaryl ligand undergoes decoordination and bond-rotation processes prior to C–H bond activation. A recent review on this topic collects the most important features of this reaction [122]. Early work by Young and co-workers [123] proposes the formation of coordinately unsaturated species 39 and 39’ as intermediates in the “rollover” reactions of 38, leading to polymeric species (Scheme 19).
![[1860-5397-9-153-i19]](/bjoc/content/inline/1860-5397-9-153-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: “Rollover” reaction of 38 and subsequent oligomerization [123].
Scheme 19: “Rollover” reaction of 38 and subsequent oligomerization [123].
Zucca and co-workers reported the synthesis of several cyclometalated compounds 44 by means of substitution and “rollover” processes (Scheme 20) [124]. Starting from derivative 41, DMSO displacement by 6-substituted 2,2’-bipyridines (NN ligands) yields the corresponding bidentate derivatives 42. Due to the steric congestion between the R group of the bipyridine and the methyl ligand 42 becomes unstable promoting the decoordination of the nitrogen atom. Subsequent C–H bond activation is proposed to take place through an agostic intermediate 43 generated by rotation around the 2,2’-C–C bond of the bipyridine ligand. After the release of methane, the vacant site is easily occupied by one DMSO ligand yielding 44. Interestingly, from 44 (R = t-Bu, Ph) the corresponding hydride compounds can be prepared [125]. It is worth pointing out that, depending on the ligand present in solution, both 16-electron and 14-electron species are obtained, though the latter are not stable, and only oligomers with bridging hydrides can be detected [125].
![[1860-5397-9-153-i20]](/bjoc/content/inline/1860-5397-9-153-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Proposed mechanism for the formation of cyclometalated species 44 [124].
Scheme 20: Proposed mechanism for the formation of cyclometalated species 44 [124].
Wang and co-workers disclosed the spontaneous self-assembling of [Pt(Me)2(NPA)] 45 by a “rollover” cyclometalation process (Scheme 21) [126]. The suggested mechanism begins with a C–N bond rotation by chelating ligand dissociation forming species 46, which is stabilized by solvent coordination. Eventually, an agostic interaction in 47 prior to the rate-determining C–H bond cleavage should displace the solvent molecule. An oxidative addition and reductive elimination (OA/RE) scenario via hydride Pt(IV) 48 and subsequent methane release yield the corresponding solvent adduct 49, from which self-association generates the cyclic tetramer 50. Indeed, when good coordinating agents such as acetonitrile are added, the reaction slows down.
![[1860-5397-9-153-i21]](/bjoc/content/inline/1860-5397-9-153-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Self-assembling process of 45 by “rollover” reaction [126].
Scheme 21: Self-assembling process of 45 by “rollover” reaction [126].
A “rollover” process has been observed for the already cyclometalated compound A9 in DMSO providing two zwitterionic products, cis- and trans-51 (Scheme 22) [127]. On the other hand, by switching the DMSO solvent to the less polar chloroform the expected cyclometalated product 37a (Scheme 18) is obtained. DFT calculations correctly explain the relative stabilities of 51 with respect to 37a depending on the polarity of the solvent [127].
![[1860-5397-9-153-i22]](/bjoc/content/inline/1860-5397-9-153-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: “Rollover” reaction of A9. Energies (solvent) in kcal mol−1 [127].
Scheme 22: “Rollover” reaction of A9. Energies (solvent) in kcal mol−1 [127].
This kind of reactivity has also attracted attention in gas-phase conditions [122]. The gas-phase behavior of cationic species 52 has been analyzed by means of combined experimental and computational studies (Scheme 23) [128]. DFT calculations discourage the initial loss of dimethylsulfide. Instead, decomplexation and C–C bond rotation processes starting from the four-coordinate complex 52 are favored. The resulting isomeric compounds exhibit an empty coordination site that is filled by an agostic interaction prior to the C–H bond activation. Intermediate 53 evolves through a σ-CAM process, whereas intermediate 54 undergoes oxidative addition and reductive elimination processes. The release of methane and dimethylsulfide yields 55. Further studies including labeling experiments support the reversibility of these “rollover” reactions. The highly unsaturated species 55 is still reactive and can coordinate and decompose XMe2 molecules (X = S [128] and O [129]) and dehydrogenate alkanes [130]. Finally, other cyclometalation processes including “rollover” reactions have also been observed for the complex [Pt(Me)L(SMe2)] bearing a diimine ligand instead of the ubiquitous bipyridyl backbone [131].
![[1860-5397-9-153-i23]](/bjoc/content/inline/1860-5397-9-153-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Proposed mechanisms for the “rollover” cyclometalation of 52 in gas-phase ion-molecule reactions [128].
Scheme 23: Proposed mechanisms for the “rollover” cyclometalation of 52 in gas-phase ion-molecule reactions [128].
β-H elimination
As previously noted, the agostic complexes A1–4 (Figure 7) are in equilibrium with the hydrido–alkene isomers [34-38]. Experimental evidence points out that substituted alkyls and large chelate ring-size diphosphine ligands favor the β-agostic isomer. As a representative example, Scheme 24 shows the β-H elimination for A1d together with the reverse 1,2-insertion for 56. Interestingly, upon crystallization in dichloromethane solvent, A1d eliminates norbornene from 56 and generates the dinuclear complex 57 [35]. The presence of chloride as a bridging ligand suggests that solvent molecules are involved in the reaction. Therefore, the participation of T-shaped intermediates, probably stabilized as solvento adducts, might be relevant in the overall process.
![[1860-5397-9-153-i24]](/bjoc/content/inline/1860-5397-9-153-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: β-H elimination and 1,2-insertion equilibrium involving A1d and the subsequent generation of 57 [35].
Scheme 24: β-H elimination and 1,2-insertion equilibrium involving A1d and the subsequent generation of 57 [35].
Although β-elimination should be easily accomplished, Goldberg and co-workers realized that other reactions can compete. The thermolysis of five-coordinate Pt(IV) complexes 7 containing nacnac ([{(o-iPr2C6H3)NC(CH3)}2CH]−, 7b) [99,101] and AnIM ([o-C6H4-{N(C6H3iPr2)}(CH=NC6H3iPr2)]−, 7c) [101] ligands produces D-59 in benzene-d6 (Scheme 25). The first step in this reaction seems to be the direct reductive elimination of 7 liberating ethane. The resulting intermediate 8 undergoes cyclometalation to give the complex 58. The subsequent β-H elimination process shall provide the expected hydride complex 59. However, solvent molecules come into play, so that the unsaturated intermediate 58 activates the C–D bond of benzene-d6 forming 60. Cyclometalation and subsequent β-H elimination processes generate D-59, which has fully incorporated the corresponding deuterium atoms. This latter evidence suggests that the intermolecular C–D bond activation of benzene-d6 is indeed faster than the β-elimination. On the other hand, when the reaction is conducted in a cyclohexane-d12 solution, D-59 is hardly obtained and 59 prevails. It means that, in sharp contrast to the arene solvent, the intermolecular C–D bond activation of alkanes becomes slower than the β-elimination.
![[1860-5397-9-153-i25]](/bjoc/content/inline/1860-5397-9-153-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Proposed mechanism for thermolysis of 7b and 7c in benzene-d6 and cyclohexane-d12 solvents [101].
Scheme 25: Proposed mechanism for thermolysis of 7b and 7c in benzene-d6 and cyclohexane-d12 solvents [101].
This type of reaction has also been observed for the agostic complex A11a [28], although the CH group of the agostic contact is not involved. Upon heating or under UV irradiation (Scheme 26), one hydrogen of the cyclometalated isopropyl group in A11a undergoes a β-H elimination process yielding 61, in which the alkene and the hydride ligands are located mutually trans.
![[1860-5397-9-153-i26]](/bjoc/content/inline/1860-5397-9-153-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: β-H elimination process of A11a [28].
Scheme 26: β-H elimination process of A11a [28].
Intermolecular C–H bond activation
Three-coordinate T-shaped Pt(II) complexes have been postulated in hydrocarbon C–H bond activations, particularly in the Shilov system for the functionalization of methane. A number of excellent reviews about C–H bond activation have been published [132-137].
Labile ligands in masked T-shaped compounds allow further reactivity. Although some solvent complexes exhibit associative pathways eluding 14-electron species [73,138], the participation of such coordinatively unsaturated intermediates should be taken into account for other systems [135,136]. A good example is the investigation reported by Wick and Goldberg (Scheme 27) [95]. From the anionic species [Pt(Me)2Tp’]− 62, they attempted to generate the unsaturated species 63 through the abstraction of one methyl ligand. Indeed, the treatment of 62 with B(C6F5)3 in benzene, cyclohexane and n-pentane provides the products 64 arising from the C–H bond activation of solvent molecules. Interestingly, the activation of pentane molecules only occurs at the primary carbon atom.
![[1860-5397-9-153-i27]](/bjoc/content/inline/1860-5397-9-153-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Intermolecular C–H bond activation from 62 [95].
Scheme 27: Intermolecular C–H bond activation from 62 [95].
A similar situation has been observed for the reductive elimination of methane in the complex 65 (Scheme 28) [105]. Experimental evidence is consistent with a dissociative methane loss from 66 as the rate-determining step. Therefore, as shown in Scheme 27, the unsaturated intermediate 63 is supposed to operate again. When the reaction is carried out in CD3CN/C6F6 mixtures, acetonitrile binds the transient 63 forming the corresponding adduct 67. On the other hand, when benzene-d6 and toluene-d8 are used as solvents, intermolecular C–D bond activations occur with the formation of 68. In cyclohexane-d12 solution, the observation of deuterated methane isotopomers indicates C–D bond-activation processes, though the corresponding alkyl Pt(IV) product could not be characterized.
![[1860-5397-9-153-i28]](/bjoc/content/inline/1860-5397-9-153-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Reductive elimination of methane from 65 followed by CD3CN coordination or C–D bond-activation processes [105].
Scheme 28: Reductive elimination of methane from 65 followed by CD3CN coordination or C–D bond-activation proc...
Nevertheless, the participation of one arm of the Tp’ ligand stabilizing the open coordination site in 63 should be considered. As pointed out by Keinan and co-workers [139], the intermediate [Pt(Me)Tp] can adopt two different structures; the bidentate κ2 coordination mode (69) provides a T-shaped structure (Figure 18 left) whereas the κ3-complex (69’) exhibits a see-saw geometry (Figure 18 right). The former is slightly favored by only 1.8 kcal mol−1.
![[1860-5397-9-153-18]](/bjoc/content/figures/1860-5397-9-153-18.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 18: DFT-optimized structures describing the κ2 (69, left) and κ3 (69’, right) coordination modes of [Pt(Me)Tp] [139]. Bond distances in angstroms, angles in degrees and Gibbs energies in kcal mol−1.
Figure 18: DFT-optimized structures describing the κ2 (69, left) and κ3 (69’, right) coordination modes of [Pt...
Recently, arene activation has been reported regarding true T-shaped species with NHC ligands [28]. The cyclometalated complexes A11a, A11b, T5a and T5b were tested toward C–H bond-activation processes by using benzene as a solvent (Scheme 29). No reaction was observed for A11a and A11b even under drastic conditions (high temperatures and long reaction times). On the other hand, T5a yields the phenyl product T6a, whereas T5b barely reacts.
![[1860-5397-9-153-i29]](/bjoc/content/inline/1860-5397-9-153-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Intermolecular arene C–H bond activation from NHC-based complexes [28].
Scheme 29: Intermolecular arene C–H bond activation from NHC-based complexes [28].
DFT calculations suggest an oxidative addition and reductive elimination scenario via Pt(IV) hydride intermediates 70 (Figure 19). The steric environment of the agostic complexes A11a and A11b complicates the reaction, which is reflected in the high energy barriers (more than 40 kcal mol−1, red line). In sharp contrast, the true T-shaped species T5a and T5b, with no agostic bonds, show lower energy profiles (ca. 30 kcal mol−1, blue line) and the reaction thermodynamics (ΔEr values, Figure 19) accounts for the observation of T6a and the poor detection of T7.
![[1860-5397-9-153-19]](/bjoc/content/figures/1860-5397-9-153-19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 19: Energy profiles (in benzene solvent) for the benzene C–H bond activation from A11a, A11b, T5a and T5b [28].
Figure 19: Energy profiles (in benzene solvent) for the benzene C–H bond activation from A11a, A11b, T5a and T...
Other arenes can also be activated by the use of T5a to afford T6 (Figure 6). In toluene solvent, only the two products m-T6b and p-T6b corresponding to meta- and para-site activations are observed in a 5:1 molar ratio. Neither ortho- nor benzylic C–H bond activations are detected. Once again, DFT calculations provide reasonable ΔE‡ for both meta and para-routes (ca. 30 kcal mol−1), and the thermodynamic effects, i.e., that only m-T6b and p-T6b are slightly more stable than T5a, explain the experimental evidence.
Some pincer complexes can indeed activate the C–H bonds of benzene, though the mechanism is not fully understood [50,140]. In this line, Ozerov and co-workers have attempted to access unsaturated species by means of abstraction of triflate ligand from 71 (Scheme 30) [12]. The presumable generation of the low electron-count intermediate 72, results in the intermolecular C–H bond activation of several arene solvents. Phenyl 73a and phenyl-d5 73b together with o-fluoro 73c and o-chlorophenyl 73d are obtained. The toluene solvent also reacts forming a mixture of o-, m- and p-tolyl complexes (8% o-73e and 92% m-73e and p-73e). Additional experiments proved that the reverse processes are not kinetically accessible. In some cases, C–X bond-activation processes involving PhBr and CH2Cl2 molecules have also been registered.
![[1860-5397-9-153-i30]](/bjoc/content/inline/1860-5397-9-153-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Intermolecular arene C–H bond activation from PNP-based complex 71 [12].
Scheme 30: Intermolecular arene C–H bond activation from PNP-based complex 71 [12].
Complexes of the type [Pt(Me)(NN)]+ 74 (Scheme 31) have shown reactivity toward the C–H bond activation of benzene in gas-phase ion-molecule reactions [7]. Suitable labeling experiments disclose the reversibility of the C–H bond activation. Interestingly, the relative rate constants reveal that 74d reacts slower than 74a–c. In the absence of solvent, both reactants 74a–c and phenyl products 76a–c are believed to exhibit true T-shaped structures. On the other hand, the lower reactivity of 74d has been attributed to an interaction of the empty site of the platinum with one ortho-chlorine atom of the (o,o’-Cl2C6H3)N=C(CH3)–C(CH3)=N(o,o’-Cl2C6H3) (MeDABDCP) ligand [141], which blocks the approach of a benzene molecule. Oxidative addition and reductive elimination (OA/RE) as well as σ-CAM scenarios have been evaluated for 74b, but unfortunately the results are functional-dependent. B3LYP cannot reproduce the experimental evidences, mPW1K and mPW1PW91 favor the σ-CAM mechanism, and M05-2X considers both of them [7]. The bipyridyl complex 74b can also activate toluene and methane molecules forming 74bb and 74bc, respectively [142]. Concerning the toluene activation, meta-, para- and benzylic positions can be activated forming m-76bb, p-76bb and Bn-76bb, respectively. BP86 calculations suggest that OA/RE and σ-CAM are competitive pathways.
![[1860-5397-9-153-i31]](/bjoc/content/inline/1860-5397-9-153-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Intermolecular C–H bond-activation by gas-phase ion-molecule reactions of 74 [7,142].
Scheme 31: Intermolecular C–H bond-activation by gas-phase ion-molecule reactions of 74 [7,142].
H2 activation
Some agostic complexes previously reported can activate small molecules such as H2 (Scheme 32). For instance, the complexes A5a and A5b react with dihydrogen (1 atm H2, 20 °C) in dichloromethane solution to yield the corresponding agostic hydride products A6a and A6b [39]. Both species were found in equilibrium, exhibiting A5/A6 ratios of 4:1 and 1:2 for species a and b, respectively. A similar behavior is observed for the analogous reaction of A5c (1 bar H2, −30 °C to room temperature), in which A6c is obtained in a 1:3 ratio [40]. These results suggest that hydride species seem to be favored according to the basicity of the phosphine ligand. In previous works, the dichloromethane molecule in the solvento complex S1a was labile enough to allow H2 activation but, unlike A6, the corresponding dihydrogen adduct 77 was observed (Scheme 32) [54]. The latter undergoes a fluxional process which can be slowed down by cooling at −60 °C.
![[1860-5397-9-153-i32]](/bjoc/content/inline/1860-5397-9-153-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Dihydrogen activation through complexes A5a, A5b [39], A5c [40] and S1a [54].
Scheme 32: Dihydrogen activation through complexes A5a, A5b [39], A5c [40] and S1a [54].
Weller and co-workers reported an alternative procedure to obtain 77 from A7 by addition of dihydrogen in CD2Cl2 solution (Scheme 33) [41]. The agostic interaction in A7 is supposed to be displaced by one dihydrogen molecule forming the putative intermediate 78. The concomitant release of methane may be the driving force of the reaction, thus the entropic factors together with the nature of the phosphine ligands should be taken into account. Additionally, the related compound 16 (Scheme 13) can also activate dihydrogen molecules; the cyclometalated ring is opened and the corresponding hydride complex S1c is obtained [54].
![[1860-5397-9-153-i33]](/bjoc/content/inline/1860-5397-9-153-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Dihydrogen activation through complexes A7 and 16 [41]. For a: see Scheme 13.
Scheme 33: Dihydrogen activation through complexes A7 and 16 [41]. For a: see Scheme 13.
X2 activation
The cyclometalated compounds A11a and T5a can activate X2 molecules (X = Br, I) in dichloromethane solution affording 79 and 80 in which one C(sp3)–X bond is constructed (Scheme 34) [143]. It is noteworthy that the reaction occurs in the presence (A11a) or the absence (T5a) of agostic interactions in the starting materials.
![[1860-5397-9-153-i34]](/bjoc/content/inline/1860-5397-9-153-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Br2 and I2 bond activations through complexes A11a and T5a [143].
Scheme 34: Br2 and I2 bond activations through complexes A11a and T5a [143].
Interestingly, during the reaction of A11a with Br2 at low temperature (−78 °C) a paramagnetic, see-saw Pt(III) alkyl intermediate 81a could be isolated and characterized (Scheme 35), although the iodo-analogue 81b could not. DFT calculations correctly predicted the feasibility of 81a (−9.5 kcal mol−1 below reactants) and explained the nondetection of 81b (+4.5 kcal mol−1 above reactants) in terms of higher relative energies.
![[1860-5397-9-153-i35]](/bjoc/content/inline/1860-5397-9-153-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Detection and isolation of the Pt(III) complex 81a [143].
Scheme 35: Detection and isolation of the Pt(III) complex 81a [143].
A related Cl2 activation process involving unsaturated species has been recently reported by Rourke and co-workers (Scheme 36) [144]. Under certain conditions, the reaction of 82 with PhI·Cl2 yields 83. Although suitable crystals of 83 could not be obtained, the structure was elucidated by NMR studies showing the formation of one C(sp3)–Cl bond. Similarly to the agostic species A9, the resulting CH2Cl group is interacting with the open coordination site. Further reaction of 83 with PhI·Cl2 forms the oxidation product 84.
![[1860-5397-9-153-i36]](/bjoc/content/inline/1860-5397-9-153-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Cl2 bond activation through complexes 82 and 83 [144].
Scheme 36: Cl2 bond activation through complexes 82 and 83 [144].
cis–trans Isomerization
The cis–trans isomerization of several solvento species S5 has been studied and the formation of low electron-count Pt(II) complexes 85 through dissociative pathways accounts for the experimental observations [2,61]. The general mechanism is depicted as follows: (i) rate-determining dissociation step of a solvent molecule, (ii) isomerization process, and (iii) fast association of a solvent molecule (Scheme 37).
![[1860-5397-9-153-i37]](/bjoc/content/inline/1860-5397-9-153-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: cis–trans Isomerization mechanism of the solvento Pt(II) complexes S5 [2,61].
Scheme 37: cis–trans Isomerization mechanism of the solvento Pt(II) complexes S5 [2,61].
As shown in Scheme 37, the 14-electron structures 85 are involved in the process. Due to their high tendency to fulfill the empty coordination site, these species strongly favor intramolecular contacts. Indeed, it has been experimentally observed that the complexes S5 bearing R = Et, n-Pr and n-Bu groups react much faster than other derivatives with R groups without β-hydrogen atoms [61]. This phenomenon has been called the β-hydrogen kinetic effect [2]. It is defined by an incipient β-agostic interaction that can stabilize transient T-shaped intermediates and transition states, and therefore, an increase of the reaction rate would be expected. Further computational studies were performed on complexes [Pt(R)(PMe3)2(solv)]+ 85 describing the isomerization process in different solvents [2]. The β-hydrogen kinetic effect can be detected during the first steps of the isomerization energy profiles (Figure 20) for both the methyl (85a, red line) and the ethyl (85b, blue line) complexes in acetonitrile. Note that the agostic contact in cis-85b decreases the overall energy requirement with respect to cis-85a, but at the same time, it increases the relative energy barrier to reach the Y-shaped transition state TS85, i.e., 4.3 kcal mol−1 for the ethyl (TS85b) and 2.3 kcal mol−1 for the methyl (TS85a) derivatives. By removal of the agostic interaction in cis-85b (through a rotation of the C–C bond of the ethyl ligand), this energy stabilization was estimated to be 5.9 kcal mol−1. Other solvents such as methanol, dimethylsulfoxide and benzene were also considered. The donor ability strongly affects the dissociation step, although it has a little impact on the strength of the agostic interaction.
![[1860-5397-9-153-20]](/bjoc/content/figures/1860-5397-9-153-20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 20: Energy profiles for the isomerization of complexes [Pt(R)(PMe3)2(NCMe)]+ where R means Me (85a, red line) and Et (85b, blue line) [2].
Figure 20: Energy profiles for the isomerization of complexes [Pt(R)(PMe3)2(NCMe)]+ where R means Me (85a, red...
In the presence of benzyl ligands, a similar cis–trans isomerization via the unsaturated species [Pt(R)(PEt3)2]+ (Scheme 37) has been claimed [62]. DFT calculations on the model derivative [Pt(Bn)(PMe3)2]+ predict a Pt···η2-benzyl coordination mode in 86 involving the ipso-carbon of the benzyl ligand (Figure 21). This stabilization accounts for 7.6 kcal mol−1, i.e., 1.7 kcal mol−1 stronger than the above-mentioned agostic contact for the same family of compounds (5.9 kcal mol−1, [2]). The interaction is favored by electron-donating groups on the phenyl ring and, as a consequence, the reaction rate is enhanced.
![[1860-5397-9-153-21]](/bjoc/content/figures/1860-5397-9-153-21.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 21: DFT-optimized structure of intermediate 86 [62]. Bond distances in angstrom and angles in degrees.
Figure 21: DFT-optimized structure of intermediate 86 [62]. Bond distances in angstrom and angles in degrees.
Dissociative ligand substitution
Experimental evidences obtained in poorly coordinating solvents strongly support that neutral complexes of the type cis-[Pt(R)2S2] (87) undergo ligand substitutions through dissociative pathways as depicted in Scheme 38: (i) sulfur ligand dissociation/reassociation via transient 14-electron structures (88), (ii) subsequent ligand addition (89), and (iii) displacement of the sulfur ligand (90) [117,119]. The electron-rich metal, the Pt–S bond weakening due to the trans-influence of the R groups, and the stabilization of T-shaped intermediates favor this dissociative pathway. Interestingly, for the complex cis-[Pt(Ph)2(CO)(SMe2)], in which one thioether ligand has been replaced by CO, the operating mechanism becomes associative [145]. Steric and β-hydrogen kinetic effects [2] have been invoked to explain the fast reaction of [Pt(Hbph)2(DMSO)2] (Hbph = η1-biphenyl monoanion) compared to species containing other R groups [117].
![[1860-5397-9-153-i38]](/bjoc/content/inline/1860-5397-9-153-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Proposed dissociative ligand-substitution mechanism of cis-[Pt(R)2S2] complexes (87) [117].
Scheme 38: Proposed dissociative ligand-substitution mechanism of cis-[Pt(R)2S2] complexes (87) [117].
Puddephatt and co-workers have demonstrated that, depending on the nature of the R group, dissociative mechanisms can also facilitate the substitution of dimethylsulfide by phthalazine in the dinuclear species 91 (Scheme 39) [146]. When alkyl derivatives are employed, kinetic evidence mainly points to an associative mechanism via 92, although a minor part of the reaction has been related to a dissociative pathway. The inclusion of phenyl and p-tolyl ligands in 91 completely changes the mechanism. A first-order reaction is observed and large negative entropies of activation are collected. The proposed mechanism involves the dissociation of dimethylsulfide generating the putative unsaturated intermediate 93. This species is supposed to be stabilized by a donor–aceptor Pt–Pt bond, i.e., the electron-rich four-coordinate platinum atom interacts with the electron-deficient three-coordinate counterpart. Consequently, the electron density of the former platinum atom decreases, thereby favoring the subsequent attack of the N–N ligand to yield 94.
![[1860-5397-9-153-i39]](/bjoc/content/inline/1860-5397-9-153-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Proposed mechanisms for the ligand substitution of the dinuclear species 91 [146].
Scheme 39: Proposed mechanisms for the ligand substitution of the dinuclear species 91 [146].
Conclusion
There are only a very few authenticated three-coordinate Pt(II) complexes. Yet, T-shaped intermediates play an important role in a number of organometallic transformations. Given the electronic and coordinative unsaturated nature of such compounds, they can only be isolated when the vacant coordination site is blocked to avoid intra- and intermolecular interactions. Steric effects are important in preventing the coordination of a fourth external ligand. The use of bulky species, which hamper the entry of ligands into the platinum coordination sphere, is a good strategy toward this goal. The presence of shorter or less flexible alkyl chains in bulky ligands could avoid agostic interactions. Electronic effects, as well as the involvement of strong electron-donating ligands, also have a stabilizing impact. Overall, the number of true T-shaped complexes that are well-characterized is still very low. Nevertheless, a much larger number of Pt(II) complexes can be described as operationally three-coordinate in a kinetic sense. This happens when the fourth position in a square-planar complex is occupied by a very weak ligand, which can be easily displaced. We have named these compounds masked T-shaped complexes. An intramolecular agostic interaction, a weakly coordinated solvent molecule or a counteranion can play such a role. Whether or not the interaction of the platinum atom with the fourth ligand is very weak, the three-coordinate complex should be very close in energy to the square-planar ground state. Consequently, in practice the masked T-shaped complexes can be devised as a resting state of the three-coordinate species.
The accessibility of three-coordinate complexes makes them suitable intermediates in Pt(II) chemistry. In spite of the difficulty in detecting these intermediates, this review gathers a number of recent experimental and theoretical reports, which claim the involvement of T-shaped Pt(II) intermediates in reaction mechanisms. In the near future, the acquired knowledge on the synthetic routes and structural features, as well as the advances in the detection techniques and computational methods, should lead to an improved design of such compounds and to a wider recognition of the role they play in chemical transformations.
Acknowledgements
Financial support from the Spanish Ministerio de Economia y Competitividad (DGICYT, Projects CTQ2010-17476, CTQ2011-23336 and ORFEO Consolider-Ingenio 2010 CSD2007-00006) and the Junta de Andalucía (project No. FQM-3151) is acknowledged. M. A. O thanks the Spanish MECD for a research grant.
References
-
Hartwig, J. F. Organotransition metal chemistry: from bonding to catalysis; University Science: Sausalito, CA, USA, 2010.
Return to citation in text: [1] -
Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Yamashita, M.; Hartwig, J. F. J. Am. Chem. Soc. 2004, 126, 5344–5345. doi:10.1021/ja0315107
Return to citation in text: [1] -
Walter, M. D.; White, P. S.; Brookhart, M. New J. Chem. 2013, 37, 1128–1133. doi:10.1039/c3nj41145a
Return to citation in text: [1] -
Moncho, S.; Ujaque, G.; Lledós, A.; Espinet, P. Chem.–Eur. J. 2008, 14, 8986–8994. doi:10.1002/chem.200800423
Return to citation in text: [1] -
Gerdes, G.; Chen, P. Organometallics 2003, 22, 2217–2225. doi:10.1021/om0209982
Return to citation in text: [1] -
Butschke, B.; Schröder, D.; Schwarz, H. Organometallics 2009, 28, 4340–43496. doi:10.1021/om900388k
Return to citation in text: [1] [2] [3] [4] -
Ozerov, O. V.; Watson, L. A.; Pink, M.; Caulton, K. G. J. Am. Chem. Soc. 2007, 129, 6003–6016. doi:10.1021/ja062327r
Return to citation in text: [1] -
Alvarez, S. Coord. Chem. Rev. 1999, 193–195, 13–41. doi:10.1016/S0010-8545(99)00085-5
Return to citation in text: [1] -
Albright, T. A.; Burdett, J. K.; Whango, M. H. Orbital interactions in chemistry; Wiley: New York, 1985.
Return to citation in text: [1] [2] -
Jean, Y. Molecular orbitals of transition metal complexes; Oxford University Press: New York, 2005.
Return to citation in text: [1] -
DeMott, J. C.; Bhuvanesh, N.; Ozerov, O. V. Chem. Sci. 2013, 4, 642–649. doi:10.1039/c2sc21385k
Return to citation in text: [1] [2] [3] -
Su, M.-D.; Chu, S.-Y. J. Phys. Chem. A 1998, 102, 10159–10166. doi:10.1021/jp982861o
Return to citation in text: [1] -
Komiya, S.; Albright, T. A.; Hoffmann, R.; Kochi, J. K. J. Am. Chem. Soc. 1976, 98, 7255–7265. doi:10.1021/ja00439a024
Return to citation in text: [1] -
Berthon-Gelloz, G.; de Bruin, B.; Tinant, B.; Markó, I. E. Angew. Chem. 2009, 121, 3207–3210. doi:10.1002/ange.200900435
Angew. Chem. Int. Ed. 2009, 48, 3161–3164. doi:10.1002/anie.200900435
Return to citation in text: [1] [2] -
Takagi, N.; Sakaki, S. J. Am. Chem. Soc. 2012, 134, 11749–11759. doi:10.1021/ja304110h
Return to citation in text: [1] -
Su, M.-D. Mol. Phys. 1993, 80, 1223–1251. doi:10.1080/00268979300102991
Return to citation in text: [1] -
Sturmayr, D.; Schubert, U. Monatsh. Chem. 2003, 134, 791–795. doi:10.1007/s00706-002-0568-6
Return to citation in text: [1] -
Goel, R. G.; Srivastava, R. C. Can. J. Chem. 1983, 61, 1352–1359. doi:10.1139/v83-238
Return to citation in text: [1] [2] [3] [4] -
Braunschweig, H.; Radacki, K.; Rais, D.; Scheschkewitz, D. Angew. Chem. 2005, 117, 5796–5799. doi:10.1002/ange.200501588
Angew. Chem. Int. Ed. 2005, 44, 5651–5654. doi:10.1002/anie.200501588
Return to citation in text: [1] [2] [3] -
Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879
Return to citation in text: [1] [2] [3] [4] [5] -
Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Jimenez-Halla, J. O. C.; Kupfer, T.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2013, 125, 3055–3058. doi:10.1002/anie.201209717
Return to citation in text: [1] [2] [3] [4] -
Lam, W. H.; Lam, K. C.; Lin, Z.; Shimada, S.; Perutz, R. N.; Marder, T. B. Dalton Trans. 2004, 1556–1562. doi:10.1039/b402632b
Return to citation in text: [1] [2] -
Toner, A.; Matthes, J.; Gründemann, S.; Limbach, H.-H.; Chaudret, B.; Clot, E.; Sabo-Etienne, S. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6945–6950. doi:10.1073/pnas.0608979104
Return to citation in text: [1] [2] -
Dorta, R.; Stevens, E. D.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2003, 125, 10490–10491. doi:10.1021/ja0362151
Return to citation in text: [1] -
Lavallo, V.; Canac, Y.; De Hope, A.; Donnadieu, B.; Bertrand, G. Angew. Chem. 2005, 117, 7402–7405. doi:10.1002/ange.200502566
Angew. Chem., Int. Ed. 2005, 44, 7236–7239 doi:10.1002/anie.200502566
Return to citation in text: [1] -
Clavier, H.; Nolan, S. P. Chem. Commun. 2010, 46, 841–861. doi:10.1039/b922984a
Return to citation in text: [1] -
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] -
Brookhart, M.; Green, M. L. H. J. Organomet. Chem. 1983, 250, 395–408. doi:10.1016/0022-328X(83)85065-7
Return to citation in text: [1] [2] -
Brookhart, M.; Green, M. L. H.; Wong, L. L. Carbon-Hydrogen-Transition Metal Bonds. In Progress in Inorganic Chemistry; Lippard, S. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; Vol. 36, pp 1–124. doi:10.1002/9780470166376.ch1
Return to citation in text: [1] [2] -
Brookhart, M.; Green, M. L. H.; Parkin, G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6908–6914. doi:10.1073/pnas.0610747104
Return to citation in text: [1] -
Clot, E.; Eisenstein, O. Struct. Bond. 2004, 113, 1–36. doi:10.1007/b97940
Return to citation in text: [1] -
Lein, M. Coord. Chem. Rev. 2009, 253, 625–634. doi:10.1016/j.ccr.2008.07.007
Return to citation in text: [1] -
Carr, N.; Dunne, B. J.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Chem. Commun. 1988, 926–928. doi:10.1039/C39880000926
Return to citation in text: [1] [2] [3] [4] -
Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026
Return to citation in text: [1] [2] [3] [4] [5] -
Carr, N.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1992, 2653–2662. doi:10.1039/DT9920002653
Return to citation in text: [1] [2] [3] -
Spencer, J. L.; Mhinzi, G. S. J. Chem. Soc., Dalton Trans. 1995, 3819–3824. doi:10.1039/DT9950003819
Return to citation in text: [1] [2] -
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] -
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Thakur, T. S.; Desiraju, G. R. J. Mol. Struct.: THEOCHEM 2007, 810, 143–154. doi:10.1016/j.theochem.2007.02.012
Return to citation in text: [1] [2] -
Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n
Return to citation in text: [1] [2] [3] [4] [5] -
Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] -
Zhu, J.; Lin, Z. Inorg. Chem. 2005, 44, 9384–9390. doi:10.1021/ic0513641
Return to citation in text: [1] -
Braunschweig, H.; Brenner, P.; Müller, A.; Radacki, K.; Rais, D.; Uttinger, K. Chem.–Eur. J. 2007, 13, 7171–7176. doi:10.1002/chem.200700539
Return to citation in text: [1] -
Brainard, R. L.; Nutt, W. R.; Lee, T. R.; Whitesides, G. M. Organometallics 1988, 7, 2379–2386. doi:10.1021/om00101a019
Return to citation in text: [1] -
Peters, R. G.; White, S.; Roddick, D. M. Organometallics 1998, 17, 4493–4499. doi:10.1021/om980334l
Return to citation in text: [1] -
Hill, G. S.; Rendina, L. M.; Puddephatt, R. J. J. Chem. Soc., Dalton Trans. 1996, 1809–1813. doi:10.1039/DT9960001809
Return to citation in text: [1] -
Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z
Return to citation in text: [1] [2] [3] [4] [5] -
Poverenov, E.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Ben-David, Y.; Milstein, D. Organometallics 2005, 24, 1082–1090. doi:10.1021/om049182m
Return to citation in text: [1] [2] [3] [4] -
Poverenov, E.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Organometallics 2005, 24, 5937–5944. doi:10.1021/om050637x
Return to citation in text: [1] -
Schwartsburd, L.; Poverenov, E.; Shimon, L. J. W.; Milstein, D. Organometallics 2007, 26, 2931–2936. doi:10.1021/om0700720
Return to citation in text: [1] [2] [3] -
Butts, M. D.; Scott, B. L.; Kubas, G. K. J. Am. Chem. Soc. 1996, 118, 11831–11843. doi:10.1021/ja961836y
Return to citation in text: [1] [2] [3] [4] -
Stahl, S. S.; Labinger, J. A.; Bercaw, J. E. Inorg. Chem. 1998, 37, 2422–2431. doi:10.1021/ic970944y
Return to citation in text: [1] [2] -
Holtcamp, M. W.; Labinger, J. A.; Bercaw, J. E. Inorg. Chim. Acta 1997, 265, 117–125. doi:10.1016/S0020-1693(97)05675-2
Return to citation in text: [1] -
Konze, W. V.; Scott, B. L.; Kubas, G. J. Chem. Commun. 1999, 1807–1808. doi:10.1039/a902274k
Return to citation in text: [1] -
Konze, W. V.; Scott, B. L.; Kubas, G. J. J. Am. Chem. Soc. 2002, 124, 12550–12556. doi:10.1021/ja020798h
Return to citation in text: [1] [2] -
Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2001, 123, 5100–5101. doi:10.1021/ja0058987
Return to citation in text: [1] [2] -
Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2003, 125, 8870–8888. doi:10.1021/ja0296071
Return to citation in text: [1] [2] -
Romeo, R.; Alibrandi, G. Inorg. Chem. 1997, 36, 4822–4830. doi:10.1021/ic970210l
Return to citation in text: [1] [2] [3] [4] -
Guido, E.; D’Amico, G.; Russo, N.; Sicilia, E.; Rizzato, S.; Albinati, A.; Romeo, A.; Plutino, M. R.; Romeo, R. Inorg. Chem. 2011, 50, 2224–2239. doi:10.1021/ic101879s
Return to citation in text: [1] [2] [3] -
Holtcamp, M. W.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 1997, 119, 848–849. doi:10.1021/ja9620595
Return to citation in text: [1] [2] -
Holtcamp, M. W.; Henling, L. M.; Day, M. W.; Labinger, J. A.; Bercaw, J. E. Inorg. Chim. Acta 1998, 270, 467–478. doi:10.1016/S0020-1693(97)06102-1
Return to citation in text: [1] -
Johansson, L.; Ryan, O. B.; Rømming, C.; Tilset, M. Organometallics 1998, 17, 3957–3966. doi:10.1021/om9801498
Return to citation in text: [1] -
Johansson, L.; Ryan, O. B.; Tilset, M. J. Am. Chem. Soc. 1999, 121, 1974–1975. doi:10.1021/ja984028a
Return to citation in text: [1] -
Johansson, L.; Tilset, M.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2000, 122, 10846–10855. doi:10.1021/ja0017460
Return to citation in text: [1] [2] -
Procelewska, J.; Zahl, A.; van Eldik, R.; Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. Inorg. Chem. 2002, 41, 2808–2810. doi:10.1021/ic0201710
Return to citation in text: [1] -
Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2002, 124, 1378–1399. doi:10.1021/ja011189x
Return to citation in text: [1] [2] [3] -
Heyduk, A. F.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2003, 125, 6366–6367. doi:10.1021/ja029099v
Return to citation in text: [1] -
Owen, J. S.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2006, 128, 2005–2016. doi:10.1021/ja056387t
Return to citation in text: [1] -
Chen, G. S.; Labinger, J. A.; Bercaw, J. E. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6915–6920. doi:10.1073/pnas.0610981104
Return to citation in text: [1] [2] -
Johansson, L.; Ryan, O. B.; Rømming, C.; Tilset, M. J. Am. Chem. Soc. 2001, 123, 6579–6590. doi:10.1021/ja010277e
Return to citation in text: [1] [2] -
Heyduk, A. F.; Driver, T. G.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2004, 126, 15034–15035. doi:10.1021/ja045078k
Return to citation in text: [1] [2] -
McKeown, B. A.; Foley, N. A.; Lee, J. P.; Gunnoe, T. B. Organometallics 2008, 27, 4031–4033. doi:10.1021/om8006008
Return to citation in text: [1] [2] -
McKeown, B. A.; Gonzalez, H. E.; Friedfeld, M. R.; Gunnoe, T. B.; Cundari, T. R.; Sabat, M. J. Am. Chem. Soc. 2011, 133, 19131–19152. doi:10.1021/ja206064v
Return to citation in text: [1] [2] -
Thomas, C. M.; Peters, J. C. Organometallics 2005, 24, 5858–5867. doi:10.1021/om050538j
Return to citation in text: [1] [2] -
Zhang, F.; Prokopchuk, E. M.; Broczkowski, M. E.; Jennings, M. C.; Puddepthatt, R. J. Organometallics 2006, 25, 1583–1591. doi:10.1021/om050982m
Return to citation in text: [1] [2] -
Karshtedt, D.; McBee, J. L.; Bell, A. T.; Tilley, T. D. Organometallics 2006, 25, 1801–1811. doi:10.1021/om0600902
Return to citation in text: [1] [2] -
Britovsek, G. J. P.; Taylor, R. A.; Sunley, G. J.; Law, D. J.; White, A. J. P. Organometallics 2006, 25, 2074–2079. doi:10.1021/om060036l
Return to citation in text: [1] [2] -
Zhao, S.-B.; Wu, G.; Wang, S. Organometallics 2006, 25, 5979–5989. doi:10.1021/om0608861
Return to citation in text: [1] [2] -
Albrecht, M.; van Koten, G. Angew. Chem. 2001, 113, 3866–3898. doi:10.1002/1521-3773(20011015)113:20<3866::AID-ANGE3866>3.0.CO;2-Y
Angew. Chem., Int. Ed. 2001, 40, 3750–3781 doi:10.1002/1521-3773(20011015)40:20<3750::AID-ANIE3750>3.3.CO;2-Y
Return to citation in text: [1] -
van der Boom, M. E.; Milstein, D. Chem. Rev. 2003, 103, 1759–1792. doi:10.1021/cr960118r
Return to citation in text: [1] -
Hofmann, A.; Jaganyi, D.; Munro, O. Q.; Liehr, G.; van Eldik, R. Inorg. Chem. 2003, 42, 1688–1700. doi:10.1021/ic020605r
Return to citation in text: [1] -
Grove, D. M.; van Koten, G.; Louwen, J. N.; Noltes, J. G.; Spek, A. L.; Ubbels, H. J. C. J. Am. Chem. Soc. 1982, 104, 6609–6616. doi:10.1021/ja00388a022
Return to citation in text: [1] -
Terheijden, J.; van Koten, G.; Vinke, I. C.; Spek, A. L. J. Am. Chem. Soc. 1985, 107, 2891–2898. doi:10.1021/ja00296a010
Return to citation in text: [1] -
Poverenov, E.; Efremenko, I.; Frenkel, A. I.; Ben-David, Y.; Shimon, L. J. W.; Leitus, G.; Konstantinovski, L.; Martin, J. M. L.; Milstein, D. Nature 2008, 455, 1093–1096. doi:10.1038/nature07356
Return to citation in text: [1] [2] -
Kimmich, B. F. M.; Bullock, R. M. Organometallics 2002, 21, 1504–1507. doi:10.1021/om0108651
Return to citation in text: [1] -
Vuzman, D.; Poverenov, E.; Diskin-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Dalton Trans. 2007, 5692–5700. doi:10.1039/B711444C
Return to citation in text: [1] -
Mitton, S. J.; McDonald, R.; Turculet, L. Organometallics 2009, 28, 5122–5136. doi:10.1021/om9003863
Return to citation in text: [1] -
Serra, D.; Cao, P.; Cabrera, J.; Padilla, R.; Rominger, F.; Limbach, M. Organometallics 2011, 30, 1885–1895. doi:10.1021/om101128f
Return to citation in text: [1] -
Calderone, V.; Casini, A.; Mangani, S.; Messori, L.; Orioli, P. L. Angew. Chem. 2006, 118, 1289–1291. doi:10.1002/ange.200502599
Angew. Chem., Int. Ed. 2006, 45, 1267–1269 doi:10.1002/anie.200502599
Return to citation in text: [1] [2] -
Casini, A.; Mastrobuoni, G.; Temperini, C.; Gabbiani, C.; Francese, S.; Moneti, G.; Supuran, C. T.; Scozzafava, A.; Messori, L. Chem. Commun. 2007, 156–158. doi:10.1039/b611122j
Return to citation in text: [1] [2] -
Ortega-Carrasco, E.; Cossío, F. P.; Lledós, A.; Maréchal, J.-D. J. Inorg. Biochem. 2012, 117, 230–236. doi:10.1016/j.jinorgbio.2012.09.020
Return to citation in text: [1] [2] -
Wick, D. D.; Goldberg, K. I. J. Am. Chem. Soc. 1997, 119, 10235–10236. doi:10.1021/ja971952g
Return to citation in text: [1] [2] [3] -
Reinartz, S.; White, P. S.; Brookhart, M.; Templeton, J. L. Organometallics 2000, 19, 3854–3866. doi:10.1021/om000440z
Return to citation in text: [1] -
Reinartz, S.; White, P. S.; Brookhart, M.; Templeton, J. L. J. Am. Chem. Soc. 2001, 123, 6425–6426. doi:10.1021/ja0104047
Return to citation in text: [1] -
Reinartz, S.; Baik, M.-H.; White, P. S.; Brookhart, M.; Templeton, J. L. Inorg. Chem. 2001, 40, 4726–4732. doi:10.1021/ic010099q
Return to citation in text: [1] -
Fekl, U.; Goldberg, K. I. J. Am. Chem. Soc. 2002, 124, 6804–6805. doi:10.1021/ja0258677
Return to citation in text: [1] [2] -
Fekl, U.; Kaminsky, W.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 15286–15287. doi:10.1021/ja037781z
Return to citation in text: [1] -
Kloek, S. M.; Goldberg, K. I. J. Am. Chem. Soc. 2007, 129, 3460–3461. doi:10.1021/ja0669629
Return to citation in text: [1] [2] [3] [4] -
Luedtke, A. T.; Goldberg, K. I. Inorg. Chem. 2007, 46, 8496–8498. doi:10.1021/ic701504z
Return to citation in text: [1] [2] [3] -
Vedernikov, A. N.; Caulton, K. G. Angew. Chem. 2002, 114, 4276–4278. doi:10.1002/1521-3757(20021104)114:21<4276::AID-ANGE4276>3.0.CO;2-8
Angew. Chem. Int. Ed. 2002, 41, 4102–4104. doi:10.1002/1521-3773(20021104)41:21<4102::AID-ANIE4102>3.0.CO;2-#
Return to citation in text: [1] -
Vedernikov, A. N.; Huffman, J. C.; Caulton, K. G. New J. Chem. 2003, 27, 665–667. doi:10.1039/b302055j
Return to citation in text: [1] -
Jensen, M. P.; Wick, D. D.; Reinartz, S.; White, P. S.; Templeton, J. L.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 8614–8624. doi:10.1021/ja028477t
Return to citation in text: [1] [2] [3] -
Lledós, A.; Ortuño, M. A.; Vidossich, P.; Ujaque, G.; Conejero, S. Dalton Trans. 2013. doi:10.1039/c3dt50761k
Return to citation in text: [1] [2] -
Romeo, R.; Fenech, L.; Scolaro, L. M.; Albinati, A.; Macchioni, A.; Zuccaccia, C. Inorg. Chem. 2001, 40, 3293–3302. doi:10.1021/ic0014080
Return to citation in text: [1] [2] -
Romeo, R.; Fenech, L.; Carnabuci, S.; Plutino, M. R.; Romeo, A. Inorg. Chem. 2002, 41, 2839–2847. doi:10.1021/ic011206j
Return to citation in text: [1] [2] -
Romeo, R.; Carnabuci, S.; Plutino, M. R.; Romeo, A.; Rizzato, S.; Albinati, A. Inorg. Chem. 2005, 44, 1248–1262. doi:10.1021/ic0485920
Return to citation in text: [1] [2] -
Romeo, R.; Carnabuci, S.; Fenech, L.; Plutino, M. R.; Albinati, A. Angew. Chem. 2006, 118, 4606–4610. doi:10.1002/ange.200600827
Angew. Chem., Int. Ed. 2006, 45, 4494–4498. doi:10.1002/anie.200600827
Return to citation in text: [1] [2] [3] [4] -
Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j
Return to citation in text: [1] [2] [3] [4] [5] -
Foley, P.; DiCosimo, R.; Whitesides, G. M. J. Am. Chem. Soc. 1980, 102, 6713–6725. doi:10.1021/ja00542a009
Return to citation in text: [1] -
Romeo, R. Comments Inorg. Chem. 1990, 11, 21–57. doi:10.1080/02603599008035817
Return to citation in text: [1] -
Omae, I. J. Organomet. Chem. 2011, 696, 1128–1145. doi:10.1016/j.jorganchem.2010.11.023
Return to citation in text: [1] -
Albrecht, M. Chem. Rev. 2010, 110, 576–623. doi:10.1021/cr900279a
Return to citation in text: [1] -
Thorn, D. L. Organometallics 1998, 17, 348–352. doi:10.1021/om970759s
Return to citation in text: [1] -
Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Romeo, R.; Plutino, M. R.; Romeo, A. Helv. Chim. Acta 2005, 88, 507–522. doi:10.1002/hlca.200590035
Return to citation in text: [1] [2] -
Scott, J. D.; Puddephatt, R. J. Organometallics 1983, 2, 1643–1648. doi:10.1021/om50005a028
Return to citation in text: [1] [2] -
Marrone, A.; Re, N.; Romeo, R. Organometallics 2008, 27, 2215–2222. doi:10.1021/om701070f
Return to citation in text: [1] [2] -
Fantasia, S.; Pasini, A.; Nolan, S. P. Dalton Trans. 2009, 8107–8110. doi:10.1039/b911164f
Return to citation in text: [1] [2] -
Butschke, B.; Schwarz, H. Chem. Sci. 2012, 3, 308–326. doi:10.1039/c1sc00651g
Return to citation in text: [1] [2] -
Skapski, A. C.; Sutcliffe, V. F.; Young, G. B. J. Chem. Soc., Chem. Commun. 1985, 609–611. doi:10.1039/C39850000609
Return to citation in text: [1] [2] -
Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Soro, B.; Zucca, A. Organometallics 2003, 22, 4770–4777. doi:10.1021/om0301583
Return to citation in text: [1] [2] -
Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Petretto, G. L.; Zucca, A. Organometallics 2008, 27, 3415–3421. doi:10.1021/om800027g
Return to citation in text: [1] [2] -
Zhao, S.-B.; Wang, R.-Y.; Wang, S. J. Am. Chem. Soc. 2007, 129, 3092–3093. doi:10.1021/ja0702770
Return to citation in text: [1] [2] -
Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. Organometallics 2011, 30, 3603–3609. doi:10.1021/om200293e
Return to citation in text: [1] [2] [3] -
Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Chem.–Eur. J. 2008, 14, 11050–11060. doi:10.1002/chem.200801658
Return to citation in text: [1] [2] [3] -
Butschke, B.; Tabrizi, S. G.; Schwarz, H. Chem.–Eur. J. 2010, 16, 3962–3969. doi:10.1002/chem.200902742
Return to citation in text: [1] -
Butschke, B.; Schwarz, H. Chem.–Eur. J. 2012, 18, 14055–14062. doi:10.1002/chem.201201652
Return to citation in text: [1] -
Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Helv. Chim. Acta 2008, 91, 1902–1915. doi:10.1002/hlca.200890204
Return to citation in text: [1] -
Shilov, A. E.; Shul’pin, G. B. Chem. Rev. 1997, 97, 2879–2932. doi:10.1021/cr9411886
Return to citation in text: [1] -
Periana, R. A.; Taube, D. J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Science 1998, 280, 560–564. doi:10.1126/science.280.5363.560
Return to citation in text: [1] -
Labinger, J. A.; Bercaw, J. E. Nature 2002, 417, 507–514. doi:10.1038/417507a
Return to citation in text: [1] -
Fekl, U.; Goldberg, K. I. Adv. Inorg. Chem. 2003, 54, 259–320. doi:10.1016/S0898-8838(03)54005-3
Return to citation in text: [1] [2] -
Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471–2526. doi:10.1021/cr030710y
Return to citation in text: [1] [2] -
Balcells, D.; Clot, E.; Eisenstein, O. Chem. Rev. 2010, 110, 749–823. doi:10.1021/cr900315k
Return to citation in text: [1] -
Johansson, L.; Tilset, M. J. Am. Chem. Soc. 2001, 123, 739–740. doi:10.1021/ja002505v
Return to citation in text: [1] -
Iron, M. A.; Lo, H. C.; Martin, J. M. L.; Keinan, E. J. Am. Chem. Soc. 2002, 124, 7041–7054. doi:10.1021/ja025667v
Return to citation in text: [1] [2] -
Liang, L.-C.; Lin, J.-M.; Lee, W.-Y. Chem. Commun. 2005, 2462–2464. doi:10.1039/b501520k
Return to citation in text: [1] -
Moret, M.-E.; Chen, P. Organometallics 2007, 26, 1523–1530. doi:10.1021/om0610039
Return to citation in text: [1] -
Butschke, B.; Schwarz, H. Organometallics 2011, 30, 1588–1598. doi:10.1021/om101138d
Return to citation in text: [1] [2] -
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; García-Garrido, S. E.; Maya, C.; Lledós, A.; Conejero, S. J. Am. Chem. Soc. 2012, 134, 15261–15264. doi:10.1021/ja307139p
Return to citation in text: [1] [2] [3] -
Crosby, S. H.; Thomas, H. R.; Clarkson, G. J.; Rourke, J. P. Chem. Commun. 2012, 48, 5775–5777. doi:10.1039/c2cc31572f
Return to citation in text: [1] [2] -
Romeo, R.; Grassi, A.; Scolaro, L. M. Inorg. Chem. 1992, 31, 4383–4390. doi:10.1021/ic00047a028
Return to citation in text: [1] -
Rashidi, M.; Nabavizadeh, S. M.; Zare, A.; Jamali, S.; Puddephatt, R. J. Inorg. Chem. 2010, 49, 8435–8443. doi:10.1021/ic1010067
Return to citation in text: [1] [2]
| 69. | Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2002, 124, 1378–1399. doi:10.1021/ja011189x |
| 74. | Heyduk, A. F.; Driver, T. G.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2004, 126, 15034–15035. doi:10.1021/ja045078k |
| 75. | McKeown, B. A.; Foley, N. A.; Lee, J. P.; Gunnoe, T. B. Organometallics 2008, 27, 4031–4033. doi:10.1021/om8006008 |
| 76. | McKeown, B. A.; Gonzalez, H. E.; Friedfeld, M. R.; Gunnoe, T. B.; Cundari, T. R.; Sabat, M. J. Am. Chem. Soc. 2011, 133, 19131–19152. doi:10.1021/ja206064v |
| 65. | Johansson, L.; Ryan, O. B.; Rømming, C.; Tilset, M. Organometallics 1998, 17, 3957–3966. doi:10.1021/om9801498 |
| 66. | Johansson, L.; Ryan, O. B.; Tilset, M. J. Am. Chem. Soc. 1999, 121, 1974–1975. doi:10.1021/ja984028a |
| 67. | Johansson, L.; Tilset, M.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2000, 122, 10846–10855. doi:10.1021/ja0017460 |
| 68. | Procelewska, J.; Zahl, A.; van Eldik, R.; Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. Inorg. Chem. 2002, 41, 2808–2810. doi:10.1021/ic0201710 |
| 69. | Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2002, 124, 1378–1399. doi:10.1021/ja011189x |
| 70. | Heyduk, A. F.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2003, 125, 6366–6367. doi:10.1021/ja029099v |
| 71. | Owen, J. S.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2006, 128, 2005–2016. doi:10.1021/ja056387t |
| 72. | Chen, G. S.; Labinger, J. A.; Bercaw, J. E. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6915–6920. doi:10.1073/pnas.0610981104 |
| 73. | Johansson, L.; Ryan, O. B.; Rømming, C.; Tilset, M. J. Am. Chem. Soc. 2001, 123, 6579–6590. doi:10.1021/ja010277e |
| 125. | Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Petretto, G. L.; Zucca, A. Organometallics 2008, 27, 3415–3421. doi:10.1021/om800027g |
| 64. | Holtcamp, M. W.; Henling, L. M.; Day, M. W.; Labinger, J. A.; Bercaw, J. E. Inorg. Chim. Acta 1998, 270, 467–478. doi:10.1016/S0020-1693(97)06102-1 |
| 125. | Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Petretto, G. L.; Zucca, A. Organometallics 2008, 27, 3415–3421. doi:10.1021/om800027g |
| 123. | Skapski, A. C.; Sutcliffe, V. F.; Young, G. B. J. Chem. Soc., Chem. Commun. 1985, 609–611. doi:10.1039/C39850000609 |
| 122. | Butschke, B.; Schwarz, H. Chem. Sci. 2012, 3, 308–326. doi:10.1039/c1sc00651g |
| 84. | Hofmann, A.; Jaganyi, D.; Munro, O. Q.; Liehr, G.; van Eldik, R. Inorg. Chem. 2003, 42, 1688–1700. doi:10.1021/ic020605r |
| 124. | Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Soro, B.; Zucca, A. Organometallics 2003, 22, 4770–4777. doi:10.1021/om0301583 |
| 123. | Skapski, A. C.; Sutcliffe, V. F.; Young, G. B. J. Chem. Soc., Chem. Commun. 1985, 609–611. doi:10.1039/C39850000609 |
| 77. | Thomas, C. M.; Peters, J. C. Organometallics 2005, 24, 5858–5867. doi:10.1021/om050538j |
| 78. | Zhang, F.; Prokopchuk, E. M.; Broczkowski, M. E.; Jennings, M. C.; Puddepthatt, R. J. Organometallics 2006, 25, 1583–1591. doi:10.1021/om050982m |
| 79. | Karshtedt, D.; McBee, J. L.; Bell, A. T.; Tilley, T. D. Organometallics 2006, 25, 1801–1811. doi:10.1021/om0600902 |
| 80. | Britovsek, G. J. P.; Taylor, R. A.; Sunley, G. J.; Law, D. J.; White, A. J. P. Organometallics 2006, 25, 2074–2079. doi:10.1021/om060036l |
| 81. | Zhao, S.-B.; Wu, G.; Wang, S. Organometallics 2006, 25, 5979–5989. doi:10.1021/om0608861 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 82. |
Albrecht, M.; van Koten, G. Angew. Chem. 2001, 113, 3866–3898. doi:10.1002/1521-3773(20011015)113:20<3866::AID-ANGE3866>3.0.CO;2-Y
Angew. Chem., Int. Ed. 2001, 40, 3750–3781 doi:10.1002/1521-3773(20011015)40:20<3750::AID-ANIE3750>3.3.CO;2-Y |
| 83. | van der Boom, M. E.; Milstein, D. Chem. Rev. 2003, 103, 1759–1792. doi:10.1021/cr960118r |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 75. | McKeown, B. A.; Foley, N. A.; Lee, J. P.; Gunnoe, T. B. Organometallics 2008, 27, 4031–4033. doi:10.1021/om8006008 |
| 43. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n |
| 76. | McKeown, B. A.; Gonzalez, H. E.; Friedfeld, M. R.; Gunnoe, T. B.; Cundari, T. R.; Sabat, M. J. Am. Chem. Soc. 2011, 133, 19131–19152. doi:10.1021/ja206064v |
| 43. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n |
| 124. | Minghetti, G.; Stoccoro, S.; Cinellu, M. A.; Soro, B.; Zucca, A. Organometallics 2003, 22, 4770–4777. doi:10.1021/om0301583 |
| 87. | Poverenov, E.; Efremenko, I.; Frenkel, A. I.; Ben-David, Y.; Shimon, L. J. W.; Leitus, G.; Konstantinovski, L.; Martin, J. M. L.; Milstein, D. Nature 2008, 455, 1093–1096. doi:10.1038/nature07356 |
| 88. | Kimmich, B. F. M.; Bullock, R. M. Organometallics 2002, 21, 1504–1507. doi:10.1021/om0108651 |
| 51. | Poverenov, E.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Ben-David, Y.; Milstein, D. Organometallics 2005, 24, 1082–1090. doi:10.1021/om049182m |
| 87. | Poverenov, E.; Efremenko, I.; Frenkel, A. I.; Ben-David, Y.; Shimon, L. J. W.; Leitus, G.; Konstantinovski, L.; Martin, J. M. L.; Milstein, D. Nature 2008, 455, 1093–1096. doi:10.1038/nature07356 |
| 50. | Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z |
| 85. | Grove, D. M.; van Koten, G.; Louwen, J. N.; Noltes, J. G.; Spek, A. L.; Ubbels, H. J. C. J. Am. Chem. Soc. 1982, 104, 6609–6616. doi:10.1021/ja00388a022 |
| 86. | Terheijden, J.; van Koten, G.; Vinke, I. C.; Spek, A. L. J. Am. Chem. Soc. 1985, 107, 2891–2898. doi:10.1021/ja00296a010 |
| 129. | Butschke, B.; Tabrizi, S. G.; Schwarz, H. Chem.–Eur. J. 2010, 16, 3962–3969. doi:10.1002/chem.200902742 |
| 122. | Butschke, B.; Schwarz, H. Chem. Sci. 2012, 3, 308–326. doi:10.1039/c1sc00651g |
| 127. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. Organometallics 2011, 30, 3603–3609. doi:10.1021/om200293e |
| 128. | Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Chem.–Eur. J. 2008, 14, 11050–11060. doi:10.1002/chem.200801658 |
| 128. | Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Chem.–Eur. J. 2008, 14, 11050–11060. doi:10.1002/chem.200801658 |
| 90. | Mitton, S. J.; McDonald, R.; Turculet, L. Organometallics 2009, 28, 5122–5136. doi:10.1021/om9003863 |
| 126. | Zhao, S.-B.; Wang, R.-Y.; Wang, S. J. Am. Chem. Soc. 2007, 129, 3092–3093. doi:10.1021/ja0702770 |
| 91. | Serra, D.; Cao, P.; Cabrera, J.; Padilla, R.; Rominger, F.; Limbach, M. Organometallics 2011, 30, 1885–1895. doi:10.1021/om101128f |
| 126. | Zhao, S.-B.; Wang, R.-Y.; Wang, S. J. Am. Chem. Soc. 2007, 129, 3092–3093. doi:10.1021/ja0702770 |
| 89. | Vuzman, D.; Poverenov, E.; Diskin-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Dalton Trans. 2007, 5692–5700. doi:10.1039/B711444C |
| 127. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. Organometallics 2011, 30, 3603–3609. doi:10.1021/om200293e |
| 53. | Schwartsburd, L.; Poverenov, E.; Shimon, L. J. W.; Milstein, D. Organometallics 2007, 26, 2931–2936. doi:10.1021/om0700720 |
| 127. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. Organometallics 2011, 30, 3603–3609. doi:10.1021/om200293e |
| 131. | Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Helv. Chim. Acta 2008, 91, 1902–1915. doi:10.1002/hlca.200890204 |
| 130. | Butschke, B.; Schwarz, H. Chem.–Eur. J. 2012, 18, 14055–14062. doi:10.1002/chem.201201652 |
| 42. | Thakur, T. S.; Desiraju, G. R. J. Mol. Struct.: THEOCHEM 2007, 810, 143–154. doi:10.1016/j.theochem.2007.02.012 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 21. | Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879 |
| 50. | Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z |
| 101. | Kloek, S. M.; Goldberg, K. I. J. Am. Chem. Soc. 2007, 129, 3460–3461. doi:10.1021/ja0669629 |
| 99. | Fekl, U.; Goldberg, K. I. J. Am. Chem. Soc. 2002, 124, 6804–6805. doi:10.1021/ja0258677 |
| 101. | Kloek, S. M.; Goldberg, K. I. J. Am. Chem. Soc. 2007, 129, 3460–3461. doi:10.1021/ja0669629 |
| 51. | Poverenov, E.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Ben-David, Y.; Milstein, D. Organometallics 2005, 24, 1082–1090. doi:10.1021/om049182m |
| 52. | Poverenov, E.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Organometallics 2005, 24, 5937–5944. doi:10.1021/om050637x |
| 53. | Schwartsburd, L.; Poverenov, E.; Shimon, L. J. W.; Milstein, D. Organometallics 2007, 26, 2931–2936. doi:10.1021/om0700720 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 51. | Poverenov, E.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Ben-David, Y.; Milstein, D. Organometallics 2005, 24, 1082–1090. doi:10.1021/om049182m |
| 101. | Kloek, S. M.; Goldberg, K. I. J. Am. Chem. Soc. 2007, 129, 3460–3461. doi:10.1021/ja0669629 |
| 49. | Hill, G. S.; Rendina, L. M.; Puddephatt, R. J. J. Chem. Soc., Dalton Trans. 1996, 1809–1813. doi:10.1039/DT9960001809 |
| 34. | Carr, N.; Dunne, B. J.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Chem. Commun. 1988, 926–928. doi:10.1039/C39880000926 |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 36. | Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026 |
| 37. | Carr, N.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1992, 2653–2662. doi:10.1039/DT9920002653 |
| 38. | Spencer, J. L.; Mhinzi, G. S. J. Chem. Soc., Dalton Trans. 1995, 3819–3824. doi:10.1039/DT9950003819 |
| 50. | Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z |
| 128. | Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Chem.–Eur. J. 2008, 14, 11050–11060. doi:10.1002/chem.200801658 |
| 47. | Brainard, R. L.; Nutt, W. R.; Lee, T. R.; Whitesides, G. M. Organometallics 1988, 7, 2379–2386. doi:10.1021/om00101a019 |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 48. | Peters, R. G.; White, S.; Roddick, D. M. Organometallics 1998, 17, 4493–4499. doi:10.1021/om980334l |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 19. | Goel, R. G.; Srivastava, R. C. Can. J. Chem. 1983, 61, 1352–1359. doi:10.1139/v83-238 |
| 55. | Stahl, S. S.; Labinger, J. A.; Bercaw, J. E. Inorg. Chem. 1998, 37, 2422–2431. doi:10.1021/ic970944y |
| 51. | Poverenov, E.; Gandelman, M.; Shimon, L. J. W.; Rozenberg, H.; Ben-David, Y.; Milstein, D. Organometallics 2005, 24, 1082–1090. doi:10.1021/om049182m |
| 54. | Butts, M. D.; Scott, B. L.; Kubas, G. K. J. Am. Chem. Soc. 1996, 118, 11831–11843. doi:10.1021/ja961836y |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 62. | Guido, E.; D’Amico, G.; Russo, N.; Sicilia, E.; Rizzato, S.; Albinati, A.; Romeo, A.; Plutino, M. R.; Romeo, R. Inorg. Chem. 2011, 50, 2224–2239. doi:10.1021/ic101879s |
| 63. | Holtcamp, M. W.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 1997, 119, 848–849. doi:10.1021/ja9620595 |
| 59. | Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2001, 123, 5100–5101. doi:10.1021/ja0058987 |
| 60. | Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2003, 125, 8870–8888. doi:10.1021/ja0296071 |
| 61. | Romeo, R.; Alibrandi, G. Inorg. Chem. 1997, 36, 4822–4830. doi:10.1021/ic970210l |
| 56. | Holtcamp, M. W.; Labinger, J. A.; Bercaw, J. E. Inorg. Chim. Acta 1997, 265, 117–125. doi:10.1016/S0020-1693(97)05675-2 |
| 57. | Konze, W. V.; Scott, B. L.; Kubas, G. J. Chem. Commun. 1999, 1807–1808. doi:10.1039/a902274k |
| 58. | Konze, W. V.; Scott, B. L.; Kubas, G. J. J. Am. Chem. Soc. 2002, 124, 12550–12556. doi:10.1021/ja020798h |
| 53. | Schwartsburd, L.; Poverenov, E.; Shimon, L. J. W.; Milstein, D. Organometallics 2007, 26, 2931–2936. doi:10.1021/om0700720 |
| 50. | Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z |
| 106. | Lledós, A.; Ortuño, M. A.; Vidossich, P.; Ujaque, G.; Conejero, S. Dalton Trans. 2013. doi:10.1039/c3dt50761k |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 106. | Lledós, A.; Ortuño, M. A.; Vidossich, P.; Ujaque, G.; Conejero, S. Dalton Trans. 2013. doi:10.1039/c3dt50761k |
| 96. | Reinartz, S.; White, P. S.; Brookhart, M.; Templeton, J. L. Organometallics 2000, 19, 3854–3866. doi:10.1021/om000440z |
| 97. | Reinartz, S.; White, P. S.; Brookhart, M.; Templeton, J. L. J. Am. Chem. Soc. 2001, 123, 6425–6426. doi:10.1021/ja0104047 |
| 98. | Reinartz, S.; Baik, M.-H.; White, P. S.; Brookhart, M.; Templeton, J. L. Inorg. Chem. 2001, 40, 4726–4732. doi:10.1021/ic010099q |
| 55. | Stahl, S. S.; Labinger, J. A.; Bercaw, J. E. Inorg. Chem. 1998, 37, 2422–2431. doi:10.1021/ic970944y |
| 59. | Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2001, 123, 5100–5101. doi:10.1021/ja0058987 |
| 60. | Thomas, J. C.; Peters, J. C. J. Am. Chem. Soc. 2003, 125, 8870–8888. doi:10.1021/ja0296071 |
| 63. | Holtcamp, M. W.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 1997, 119, 848–849. doi:10.1021/ja9620595 |
| 67. | Johansson, L.; Tilset, M.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2000, 122, 10846–10855. doi:10.1021/ja0017460 |
| 69. | Zhong, H. A.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2002, 124, 1378–1399. doi:10.1021/ja011189x |
| 72. | Chen, G. S.; Labinger, J. A.; Bercaw, J. E. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6915–6920. doi:10.1073/pnas.0610981104 |
| 77. | Thomas, C. M.; Peters, J. C. Organometallics 2005, 24, 5858–5867. doi:10.1021/om050538j |
| 78. | Zhang, F.; Prokopchuk, E. M.; Broczkowski, M. E.; Jennings, M. C.; Puddepthatt, R. J. Organometallics 2006, 25, 1583–1591. doi:10.1021/om050982m |
| 79. | Karshtedt, D.; McBee, J. L.; Bell, A. T.; Tilley, T. D. Organometallics 2006, 25, 1801–1811. doi:10.1021/om0600902 |
| 80. | Britovsek, G. J. P.; Taylor, R. A.; Sunley, G. J.; Law, D. J.; White, A. J. P. Organometallics 2006, 25, 2074–2079. doi:10.1021/om060036l |
| 81. | Zhao, S.-B.; Wu, G.; Wang, S. Organometallics 2006, 25, 5979–5989. doi:10.1021/om0608861 |
| 103. |
Vedernikov, A. N.; Caulton, K. G. Angew. Chem. 2002, 114, 4276–4278. doi:10.1002/1521-3757(20021104)114:21<4276::AID-ANGE4276>3.0.CO;2-8
Angew. Chem. Int. Ed. 2002, 41, 4102–4104. doi:10.1002/1521-3773(20021104)41:21<4102::AID-ANIE4102>3.0.CO;2-# |
| 104. | Vedernikov, A. N.; Huffman, J. C.; Caulton, K. G. New J. Chem. 2003, 27, 665–667. doi:10.1039/b302055j |
| 105. | Jensen, M. P.; Wick, D. D.; Reinartz, S.; White, P. S.; Templeton, J. L.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 8614–8624. doi:10.1021/ja028477t |
| 99. | Fekl, U.; Goldberg, K. I. J. Am. Chem. Soc. 2002, 124, 6804–6805. doi:10.1021/ja0258677 |
| 100. | Fekl, U.; Kaminsky, W.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 15286–15287. doi:10.1021/ja037781z |
| 101. | Kloek, S. M.; Goldberg, K. I. J. Am. Chem. Soc. 2007, 129, 3460–3461. doi:10.1021/ja0669629 |
| 102. | Luedtke, A. T.; Goldberg, K. I. Inorg. Chem. 2007, 46, 8496–8498. doi:10.1021/ic701504z |
| 146. | Rashidi, M.; Nabavizadeh, S. M.; Zare, A.; Jamali, S.; Puddephatt, R. J. Inorg. Chem. 2010, 49, 8435–8443. doi:10.1021/ic1010067 |
| 114. | Omae, I. J. Organomet. Chem. 2011, 696, 1128–1145. doi:10.1016/j.jorganchem.2010.11.023 |
| 110. |
Romeo, R.; Carnabuci, S.; Fenech, L.; Plutino, M. R.; Albinati, A. Angew. Chem. 2006, 118, 4606–4610. doi:10.1002/ange.200600827
Angew. Chem., Int. Ed. 2006, 45, 4494–4498. doi:10.1002/anie.200600827 |
| 111. | Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j |
| 111. | Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j |
| 113. | Romeo, R. Comments Inorg. Chem. 1990, 11, 21–57. doi:10.1080/02603599008035817 |
| 112. | Foley, P.; DiCosimo, R.; Whitesides, G. M. J. Am. Chem. Soc. 1980, 102, 6713–6725. doi:10.1021/ja00542a009 |
| 110. |
Romeo, R.; Carnabuci, S.; Fenech, L.; Plutino, M. R.; Albinati, A. Angew. Chem. 2006, 118, 4606–4610. doi:10.1002/ange.200600827
Angew. Chem., Int. Ed. 2006, 45, 4494–4498. doi:10.1002/anie.200600827 |
| 107. | Romeo, R.; Fenech, L.; Scolaro, L. M.; Albinati, A.; Macchioni, A.; Zuccaccia, C. Inorg. Chem. 2001, 40, 3293–3302. doi:10.1021/ic0014080 |
| 108. | Romeo, R.; Fenech, L.; Carnabuci, S.; Plutino, M. R.; Romeo, A. Inorg. Chem. 2002, 41, 2839–2847. doi:10.1021/ic011206j |
| 109. | Romeo, R.; Carnabuci, S.; Plutino, M. R.; Romeo, A.; Rizzato, S.; Albinati, A. Inorg. Chem. 2005, 44, 1248–1262. doi:10.1021/ic0485920 |
| 110. |
Romeo, R.; Carnabuci, S.; Fenech, L.; Plutino, M. R.; Albinati, A. Angew. Chem. 2006, 118, 4606–4610. doi:10.1002/ange.200600827
Angew. Chem., Int. Ed. 2006, 45, 4494–4498. doi:10.1002/anie.200600827 |
| 111. | Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j |
| 107. | Romeo, R.; Fenech, L.; Scolaro, L. M.; Albinati, A.; Macchioni, A.; Zuccaccia, C. Inorg. Chem. 2001, 40, 3293–3302. doi:10.1021/ic0014080 |
| 108. | Romeo, R.; Fenech, L.; Carnabuci, S.; Plutino, M. R.; Romeo, A. Inorg. Chem. 2002, 41, 2839–2847. doi:10.1021/ic011206j |
| 109. | Romeo, R.; Carnabuci, S.; Plutino, M. R.; Romeo, A.; Rizzato, S.; Albinati, A. Inorg. Chem. 2005, 44, 1248–1262. doi:10.1021/ic0485920 |
| 110. |
Romeo, R.; Carnabuci, S.; Fenech, L.; Plutino, M. R.; Albinati, A. Angew. Chem. 2006, 118, 4606–4610. doi:10.1002/ange.200600827
Angew. Chem., Int. Ed. 2006, 45, 4494–4498. doi:10.1002/anie.200600827 |
| 111. | Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j |
| 94. | Ortega-Carrasco, E.; Cossío, F. P.; Lledós, A.; Maréchal, J.-D. J. Inorg. Biochem. 2012, 117, 230–236. doi:10.1016/j.jinorgbio.2012.09.020 |
| 21. | Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879 |
| 93. | Casini, A.; Mastrobuoni, G.; Temperini, C.; Gabbiani, C.; Francese, S.; Moneti, G.; Supuran, C. T.; Scozzafava, A.; Messori, L. Chem. Commun. 2007, 156–158. doi:10.1039/b611122j |
| 94. | Ortega-Carrasco, E.; Cossío, F. P.; Lledós, A.; Maréchal, J.-D. J. Inorg. Biochem. 2012, 117, 230–236. doi:10.1016/j.jinorgbio.2012.09.020 |
| 93. | Casini, A.; Mastrobuoni, G.; Temperini, C.; Gabbiani, C.; Francese, S.; Moneti, G.; Supuran, C. T.; Scozzafava, A.; Messori, L. Chem. Commun. 2007, 156–158. doi:10.1039/b611122j |
| 102. | Luedtke, A. T.; Goldberg, K. I. Inorg. Chem. 2007, 46, 8496–8498. doi:10.1021/ic701504z |
| 92. |
Calderone, V.; Casini, A.; Mangani, S.; Messori, L.; Orioli, P. L. Angew. Chem. 2006, 118, 1289–1291. doi:10.1002/ange.200502599
Angew. Chem., Int. Ed. 2006, 45, 1267–1269 doi:10.1002/anie.200502599 |
| 102. | Luedtke, A. T.; Goldberg, K. I. Inorg. Chem. 2007, 46, 8496–8498. doi:10.1021/ic701504z |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 92. |
Calderone, V.; Casini, A.; Mangani, S.; Messori, L.; Orioli, P. L. Angew. Chem. 2006, 118, 1289–1291. doi:10.1002/ange.200502599
Angew. Chem., Int. Ed. 2006, 45, 1267–1269 doi:10.1002/anie.200502599 |
| 111. | Romeo, R.; D’Amico, G.; Guido, E.; Albinati, A.; Rizzato, S. Inorg. Chem. 2007, 46, 10681–10692. doi:10.1021/ic701396j |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 118. | Romeo, R.; Plutino, M. R.; Romeo, A. Helv. Chim. Acta 2005, 88, 507–522. doi:10.1002/hlca.200590035 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 19. | Goel, R. G.; Srivastava, R. C. Can. J. Chem. 1983, 61, 1352–1359. doi:10.1139/v83-238 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 120. | Marrone, A.; Re, N.; Romeo, R. Organometallics 2008, 27, 2215–2222. doi:10.1021/om701070f |
| 118. | Romeo, R.; Plutino, M. R.; Romeo, A. Helv. Chim. Acta 2005, 88, 507–522. doi:10.1002/hlca.200590035 |
| 121. | Fantasia, S.; Pasini, A.; Nolan, S. P. Dalton Trans. 2009, 8107–8110. doi:10.1039/b911164f |
| 121. | Fantasia, S.; Pasini, A.; Nolan, S. P. Dalton Trans. 2009, 8107–8110. doi:10.1039/b911164f |
| 119. | Scott, J. D.; Puddephatt, R. J. Organometallics 1983, 2, 1643–1648. doi:10.1021/om50005a028 |
| 120. | Marrone, A.; Re, N.; Romeo, R. Organometallics 2008, 27, 2215–2222. doi:10.1021/om701070f |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 139. | Iron, M. A.; Lo, H. C.; Martin, J. M. L.; Keinan, E. J. Am. Chem. Soc. 2002, 124, 7041–7054. doi:10.1021/ja025667v |
| 139. | Iron, M. A.; Lo, H. C.; Martin, J. M. L.; Keinan, E. J. Am. Chem. Soc. 2002, 124, 7041–7054. doi:10.1021/ja025667v |
| 7. | Butschke, B.; Schröder, D.; Schwarz, H. Organometallics 2009, 28, 4340–43496. doi:10.1021/om900388k |
| 12. | DeMott, J. C.; Bhuvanesh, N.; Ozerov, O. V. Chem. Sci. 2013, 4, 642–649. doi:10.1039/c2sc21385k |
| 7. | Butschke, B.; Schröder, D.; Schwarz, H. Organometallics 2009, 28, 4340–43496. doi:10.1021/om900388k |
| 141. | Moret, M.-E.; Chen, P. Organometallics 2007, 26, 1523–1530. doi:10.1021/om0610039 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 12. | DeMott, J. C.; Bhuvanesh, N.; Ozerov, O. V. Chem. Sci. 2013, 4, 642–649. doi:10.1039/c2sc21385k |
| 50. | Harkins, S. B.; Peters, J. C. Organometallics 2002, 21, 1753–1755. doi:10.1021/om011044z |
| 140. | Liang, L.-C.; Lin, J.-M.; Lee, W.-Y. Chem. Commun. 2005, 2462–2464. doi:10.1039/b501520k |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 142. | Butschke, B.; Schwarz, H. Organometallics 2011, 30, 1588–1598. doi:10.1021/om101138d |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 54. | Butts, M. D.; Scott, B. L.; Kubas, G. K. J. Am. Chem. Soc. 1996, 118, 11831–11843. doi:10.1021/ja961836y |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 54. | Butts, M. D.; Scott, B. L.; Kubas, G. K. J. Am. Chem. Soc. 1996, 118, 11831–11843. doi:10.1021/ja961836y |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 54. | Butts, M. D.; Scott, B. L.; Kubas, G. K. J. Am. Chem. Soc. 1996, 118, 11831–11843. doi:10.1021/ja961836y |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 7. | Butschke, B.; Schröder, D.; Schwarz, H. Organometallics 2009, 28, 4340–43496. doi:10.1021/om900388k |
| 142. | Butschke, B.; Schwarz, H. Organometallics 2011, 30, 1588–1598. doi:10.1021/om101138d |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 95. | Wick, D. D.; Goldberg, K. I. J. Am. Chem. Soc. 1997, 119, 10235–10236. doi:10.1021/ja971952g |
| 61. | Romeo, R.; Alibrandi, G. Inorg. Chem. 1997, 36, 4822–4830. doi:10.1021/ic970210l |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 1. | Hartwig, J. F. Organotransition metal chemistry: from bonding to catalysis; University Science: Sausalito, CA, USA, 2010. |
| 22. | Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Jimenez-Halla, J. O. C.; Kupfer, T.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2013, 125, 3055–3058. doi:10.1002/anie.201209717 |
| 144. | Crosby, S. H.; Thomas, H. R.; Clarkson, G. J.; Rourke, J. P. Chem. Commun. 2012, 48, 5775–5777. doi:10.1039/c2cc31572f |
| 19. | Goel, R. G.; Srivastava, R. C. Can. J. Chem. 1983, 61, 1352–1359. doi:10.1139/v83-238 |
| 20. |
Braunschweig, H.; Radacki, K.; Rais, D.; Scheschkewitz, D. Angew. Chem. 2005, 117, 5796–5799. doi:10.1002/ange.200501588
Angew. Chem. Int. Ed. 2005, 44, 5651–5654. doi:10.1002/anie.200501588 |
| 21. | Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 144. | Crosby, S. H.; Thomas, H. R.; Clarkson, G. J.; Rourke, J. P. Chem. Commun. 2012, 48, 5775–5777. doi:10.1039/c2cc31572f |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 61. | Romeo, R.; Alibrandi, G. Inorg. Chem. 1997, 36, 4822–4830. doi:10.1021/ic970210l |
| 22. | Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Jimenez-Halla, J. O. C.; Kupfer, T.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2013, 125, 3055–3058. doi:10.1002/anie.201209717 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 61. | Romeo, R.; Alibrandi, G. Inorg. Chem. 1997, 36, 4822–4830. doi:10.1021/ic970210l |
| 6. | Gerdes, G.; Chen, P. Organometallics 2003, 22, 2217–2225. doi:10.1021/om0209982 |
| 7. | Butschke, B.; Schröder, D.; Schwarz, H. Organometallics 2009, 28, 4340–43496. doi:10.1021/om900388k |
| 143. | Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; García-Garrido, S. E.; Maya, C.; Lledós, A.; Conejero, S. J. Am. Chem. Soc. 2012, 134, 15261–15264. doi:10.1021/ja307139p |
| 5. | Moncho, S.; Ujaque, G.; Lledós, A.; Espinet, P. Chem.–Eur. J. 2008, 14, 8986–8994. doi:10.1002/chem.200800423 |
| 3. | Yamashita, M.; Hartwig, J. F. J. Am. Chem. Soc. 2004, 126, 5344–5345. doi:10.1021/ja0315107 |
| 4. | Walter, M. D.; White, P. S.; Brookhart, M. New J. Chem. 2013, 37, 1128–1133. doi:10.1039/c3nj41145a |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 143. | Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; García-Garrido, S. E.; Maya, C.; Lledós, A.; Conejero, S. J. Am. Chem. Soc. 2012, 134, 15261–15264. doi:10.1021/ja307139p |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 143. | Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; García-Garrido, S. E.; Maya, C.; Lledós, A.; Conejero, S. J. Am. Chem. Soc. 2012, 134, 15261–15264. doi:10.1021/ja307139p |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 10. | Albright, T. A.; Burdett, J. K.; Whango, M. H. Orbital interactions in chemistry; Wiley: New York, 1985. |
| 11. | Jean, Y. Molecular orbitals of transition metal complexes; Oxford University Press: New York, 2005. |
| 9. | Alvarez, S. Coord. Chem. Rev. 1999, 193–195, 13–41. doi:10.1016/S0010-8545(99)00085-5 |
| 8. | Ozerov, O. V.; Watson, L. A.; Pink, M.; Caulton, K. G. J. Am. Chem. Soc. 2007, 129, 6003–6016. doi:10.1021/ja062327r |
| 58. | Konze, W. V.; Scott, B. L.; Kubas, G. J. J. Am. Chem. Soc. 2002, 124, 12550–12556. doi:10.1021/ja020798h |
| 146. | Rashidi, M.; Nabavizadeh, S. M.; Zare, A.; Jamali, S.; Puddephatt, R. J. Inorg. Chem. 2010, 49, 8435–8443. doi:10.1021/ic1010067 |
| 43. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 74. | Heyduk, A. F.; Driver, T. G.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2004, 126, 15034–15035. doi:10.1021/ja045078k |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 36. | Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026 |
| 145. | Romeo, R.; Grassi, A.; Scolaro, L. M. Inorg. Chem. 1992, 31, 4383–4390. doi:10.1021/ic00047a028 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 119. | Scott, J. D.; Puddephatt, R. J. Organometallics 1983, 2, 1643–1648. doi:10.1021/om50005a028 |
| 43. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n |
| 117. | Plutino, M. R.; Scolaro, L. M.; Albinati, A.; Romeo, R. J. Am. Chem. Soc. 2004, 126, 6470–6484. doi:10.1021/ja030486u |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 36. | Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 62. | Guido, E.; D’Amico, G.; Russo, N.; Sicilia, E.; Rizzato, S.; Albinati, A.; Romeo, A.; Plutino, M. R.; Romeo, R. Inorg. Chem. 2011, 50, 2224–2239. doi:10.1021/ic101879s |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 62. | Guido, E.; D’Amico, G.; Russo, N.; Sicilia, E.; Rizzato, S.; Albinati, A.; Romeo, A.; Plutino, M. R.; Romeo, R. Inorg. Chem. 2011, 50, 2224–2239. doi:10.1021/ic101879s |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 29. | Brookhart, M.; Green, M. L. H. J. Organomet. Chem. 1983, 250, 395–408. doi:10.1016/0022-328X(83)85065-7 |
| 30. | Brookhart, M.; Green, M. L. H.; Wong, L. L. Carbon-Hydrogen-Transition Metal Bonds. In Progress in Inorganic Chemistry; Lippard, S. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; Vol. 36, pp 1–124. doi:10.1002/9780470166376.ch1 |
| 31. | Brookhart, M.; Green, M. L. H.; Parkin, G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6908–6914. doi:10.1073/pnas.0610747104 |
| 29. | Brookhart, M.; Green, M. L. H. J. Organomet. Chem. 1983, 250, 395–408. doi:10.1016/0022-328X(83)85065-7 |
| 30. | Brookhart, M.; Green, M. L. H.; Wong, L. L. Carbon-Hydrogen-Transition Metal Bonds. In Progress in Inorganic Chemistry; Lippard, S. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; Vol. 36, pp 1–124. doi:10.1002/9780470166376.ch1 |
| 40. |
Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. 2012, 124, 8380–8383. doi:10.1002/ange.201202655
Angew. Chem. Int. Ed. 2012, 51, 8255–8258. doi:10.1002/anie.201202655 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 36. | Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026 |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 36. | Mole, L.; Spencer, J. L.; Carr, N.; Orpen, A. G. Organometallics 1991, 10, 49–52. doi:10.1021/om00047a026 |
| 37. | Carr, N.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1992, 2653–2662. doi:10.1039/DT9920002653 |
| 38. | Spencer, J. L.; Mhinzi, G. S. J. Chem. Soc., Dalton Trans. 1995, 3819–3824. doi:10.1039/DT9950003819 |
| 34. | Carr, N.; Dunne, B. J.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Chem. Commun. 1988, 926–928. doi:10.1039/C39880000926 |
| 32. | Clot, E.; Eisenstein, O. Struct. Bond. 2004, 113, 1–36. doi:10.1007/b97940 |
| 33. | Lein, M. Coord. Chem. Rev. 2009, 253, 625–634. doi:10.1016/j.ccr.2008.07.007 |
| 34. | Carr, N.; Dunne, B. J.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Chem. Commun. 1988, 926–928. doi:10.1039/C39880000926 |
| 35. | Carr, N.; Dunne, B. J.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1991, 863–871. doi:10.1039/DT9910000863 |
| 21. | Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879 |
| 42. | Thakur, T. S.; Desiraju, G. R. J. Mol. Struct.: THEOCHEM 2007, 810, 143–154. doi:10.1016/j.theochem.2007.02.012 |
| 43. | Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. J. Am. Chem. Soc. 2009, 131, 14142–14143. doi:10.1021/ja905046n |
| 39. |
Baratta, W.; Stoccoro, S.; Doppiu, A.; Herdtweck, E.; Zucca, A.; Rigo, P. Angew. Chem. 2003, 115, 109–113. doi:10.1002/ange.200390003
Angew. Chem. Int. Ed. 2003, 42, 105–108. doi:10.1002/anie.200390035 |
| 41. | Ingleson, M.; Mahon, M. F.; Weller, A. S. Chem. Commun. 2004, 2398–2399. doi:10.1039/b410846a |
| 34. | Carr, N.; Dunne, B. J.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Chem. Commun. 1988, 926–928. doi:10.1039/C39880000926 |
| 37. | Carr, N.; Mole, L.; Orpen, A. G.; Spencer, J. L. J. Chem. Soc., Dalton Trans. 1992, 2653–2662. doi:10.1039/DT9920002653 |
| 23. | Lam, W. H.; Lam, K. C.; Lin, Z.; Shimada, S.; Perutz, R. N.; Marder, T. B. Dalton Trans. 2004, 1556–1562. doi:10.1039/b402632b |
| 24. | Toner, A.; Matthes, J.; Gründemann, S.; Limbach, H.-H.; Chaudret, B.; Clot, E.; Sabo-Etienne, S. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6945–6950. doi:10.1073/pnas.0608979104 |
| 45. | Zhu, J.; Lin, Z. Inorg. Chem. 2005, 44, 9384–9390. doi:10.1021/ic0513641 |
| 46. | Braunschweig, H.; Brenner, P.; Müller, A.; Radacki, K.; Rais, D.; Uttinger, K. Chem.–Eur. J. 2007, 13, 7171–7176. doi:10.1002/chem.200700539 |
| 44. | Rivada-Wheelaghan, O.; Donnadieu, B.; Maya, C.; Conejero, S. Chem.–Eur. J. 2010, 16, 10323–10326. doi:10.1002/chem.201000955 |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 16. | Takagi, N.; Sakaki, S. J. Am. Chem. Soc. 2012, 134, 11749–11759. doi:10.1021/ja304110h |
| 15. |
Berthon-Gelloz, G.; de Bruin, B.; Tinant, B.; Markó, I. E. Angew. Chem. 2009, 121, 3207–3210. doi:10.1002/ange.200900435
Angew. Chem. Int. Ed. 2009, 48, 3161–3164. doi:10.1002/anie.200900435 |
| 2. | Romeo, R.; D’Amico, G.; Sicilia, E.; Russo, N.; Rizzato, S. J. Am. Chem. Soc. 2007, 129, 5744–5755. doi:10.1021/ja0702162 |
| 15. |
Berthon-Gelloz, G.; de Bruin, B.; Tinant, B.; Markó, I. E. Angew. Chem. 2009, 121, 3207–3210. doi:10.1002/ange.200900435
Angew. Chem. Int. Ed. 2009, 48, 3161–3164. doi:10.1002/anie.200900435 |
| 13. | Su, M.-D.; Chu, S.-Y. J. Phys. Chem. A 1998, 102, 10159–10166. doi:10.1021/jp982861o |
| 10. | Albright, T. A.; Burdett, J. K.; Whango, M. H. Orbital interactions in chemistry; Wiley: New York, 1985. |
| 14. | Komiya, S.; Albright, T. A.; Hoffmann, R.; Kochi, J. K. J. Am. Chem. Soc. 1976, 98, 7255–7265. doi:10.1021/ja00439a024 |
| 12. | DeMott, J. C.; Bhuvanesh, N.; Ozerov, O. V. Chem. Sci. 2013, 4, 642–649. doi:10.1039/c2sc21385k |
| 19. | Goel, R. G.; Srivastava, R. C. Can. J. Chem. 1983, 61, 1352–1359. doi:10.1139/v83-238 |
| 18. | Sturmayr, D.; Schubert, U. Monatsh. Chem. 2003, 134, 791–795. doi:10.1007/s00706-002-0568-6 |
| 95. | Wick, D. D.; Goldberg, K. I. J. Am. Chem. Soc. 1997, 119, 10235–10236. doi:10.1021/ja971952g |
| 95. | Wick, D. D.; Goldberg, K. I. J. Am. Chem. Soc. 1997, 119, 10235–10236. doi:10.1021/ja971952g |
| 105. | Jensen, M. P.; Wick, D. D.; Reinartz, S.; White, P. S.; Templeton, J. L.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 8614–8624. doi:10.1021/ja028477t |
| 105. | Jensen, M. P.; Wick, D. D.; Reinartz, S.; White, P. S.; Templeton, J. L.; Goldberg, K. I. J. Am. Chem. Soc. 2003, 125, 8614–8624. doi:10.1021/ja028477t |
| 132. | Shilov, A. E.; Shul’pin, G. B. Chem. Rev. 1997, 97, 2879–2932. doi:10.1021/cr9411886 |
| 133. | Periana, R. A.; Taube, D. J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Science 1998, 280, 560–564. doi:10.1126/science.280.5363.560 |
| 134. | Labinger, J. A.; Bercaw, J. E. Nature 2002, 417, 507–514. doi:10.1038/417507a |
| 135. | Fekl, U.; Goldberg, K. I. Adv. Inorg. Chem. 2003, 54, 259–320. doi:10.1016/S0898-8838(03)54005-3 |
| 136. | Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471–2526. doi:10.1021/cr030710y |
| 137. | Balcells, D.; Clot, E.; Eisenstein, O. Chem. Rev. 2010, 110, 749–823. doi:10.1021/cr900315k |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 135. | Fekl, U.; Goldberg, K. I. Adv. Inorg. Chem. 2003, 54, 259–320. doi:10.1016/S0898-8838(03)54005-3 |
| 136. | Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471–2526. doi:10.1021/cr030710y |
| 73. | Johansson, L.; Ryan, O. B.; Rømming, C.; Tilset, M. J. Am. Chem. Soc. 2001, 123, 6579–6590. doi:10.1021/ja010277e |
| 138. | Johansson, L.; Tilset, M. J. Am. Chem. Soc. 2001, 123, 739–740. doi:10.1021/ja002505v |
| 25. | Dorta, R.; Stevens, E. D.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc. 2003, 125, 10490–10491. doi:10.1021/ja0362151 |
| 26. |
Lavallo, V.; Canac, Y.; De Hope, A.; Donnadieu, B.; Bertrand, G. Angew. Chem. 2005, 117, 7402–7405. doi:10.1002/ange.200502566
Angew. Chem., Int. Ed. 2005, 44, 7236–7239 doi:10.1002/anie.200502566 |
| 27. | Clavier, H.; Nolan, S. P. Chem. Commun. 2010, 46, 841–861. doi:10.1039/b922984a |
| 28. |
Rivada-Wheelaghan, O.; Ortuño, M. A.; Díez, J.; Lledós, A.; Conejero, S. Angew. Chem. 2012, 124, 4002–4005. doi:10.1002/ange.201200070
Angew. Chem., Int. Ed. 2012, 51, 3936–3939. doi:10.1002/anie.201200070 |
| 22. | Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Jimenez-Halla, J. O. C.; Kupfer, T.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2013, 125, 3055–3058. doi:10.1002/anie.201209717 |
| 23. | Lam, W. H.; Lam, K. C.; Lin, Z.; Shimada, S.; Perutz, R. N.; Marder, T. B. Dalton Trans. 2004, 1556–1562. doi:10.1039/b402632b |
| 24. | Toner, A.; Matthes, J.; Gründemann, S.; Limbach, H.-H.; Chaudret, B.; Clot, E.; Sabo-Etienne, S. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6945–6950. doi:10.1073/pnas.0608979104 |
| 22. | Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Jimenez-Halla, J. O. C.; Kupfer, T.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2013, 125, 3055–3058. doi:10.1002/anie.201209717 |
| 20. |
Braunschweig, H.; Radacki, K.; Rais, D.; Scheschkewitz, D. Angew. Chem. 2005, 117, 5796–5799. doi:10.1002/ange.200501588
Angew. Chem. Int. Ed. 2005, 44, 5651–5654. doi:10.1002/anie.200501588 |
| 20. |
Braunschweig, H.; Radacki, K.; Rais, D.; Scheschkewitz, D. Angew. Chem. 2005, 117, 5796–5799. doi:10.1002/ange.200501588
Angew. Chem. Int. Ed. 2005, 44, 5651–5654. doi:10.1002/anie.200501588 |
| 21. | Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.–Eur. J. 2008, 14, 7858–7866. doi:10.1002/chem.200800879 |
© 2013 Ortuño et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)