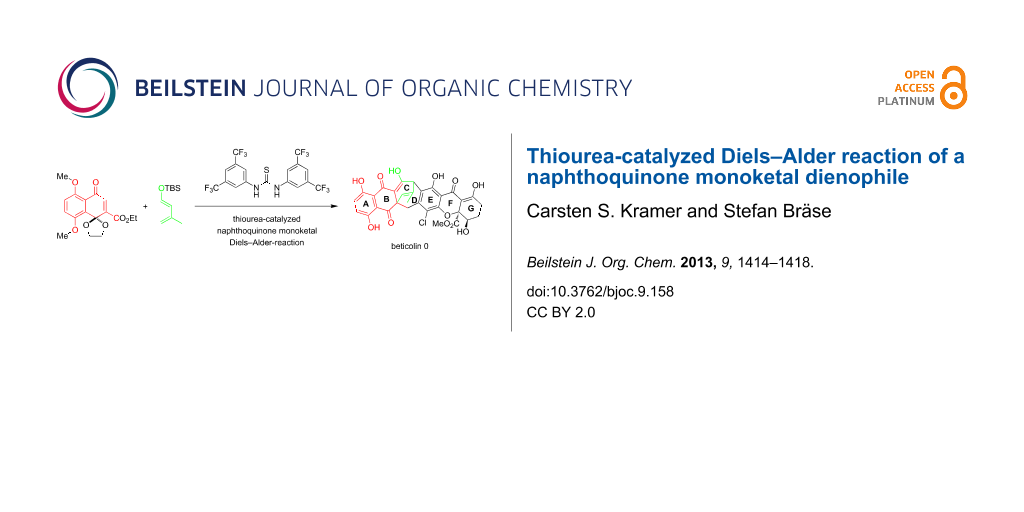
Abstract
A variety of organocatalysts were screened for the catalysis of the naphthoquinone monoketal Diels–Alder reaction. In this study we found that Schreiner's thiourea catalyst 10 and Jacobson's thiourea catalyst 12 facilitate the cycloaddition of the sterically hindered naphthoquinone monoketal dienophile 3 with diene 4. The use of thiourea catalysis allowed for the first time the highly selective synthesis of the exo-product 2a in up to 63% yield. In this reaction a new quaternary center was built. The so formed cycloaddition product 2a represents the ABC tricycle of beticolin 0 (1) and is also a valuable model substrate for the total synthesis of related natural products.

Graphical Abstract
Introduction
In the past few years, many different types of organocatalysts were found to accelerate a variety of reactions. Due to the value of the Diels–Alder reaction for the synthetic community, different organocatalysts have been developed to catalyze this atom-economical cycloaddition in a highly enantioselective fashion. Thereby, chiral amines, heterocyclic carbenes, guanidines, thioureas, amidinium ions, diols, and Brønsted acids showed their value to give densely functionalized Diels–Alder products in high selectivities [1-7]. In addition, some organocatalysts enabled even the formation of quaternary centers in Diels–Alder cycloadditions [3,8].
In our recent studies towards a synthetic access to the natural product beticolin 0 (1) [9] (Scheme 1), we have found that naphthoquinone monoketals are suitable building blocks for our designed synthesis [9]. In our retrosynthetic approach, the naphthoquinone monoketal dienophile 3 was found to undergo cycloaddition with diene 4 to tricycle 2, which represents the ABC-ring system of beticolin 0 (1) and other beticolins [10].
![[1860-5397-9-158-i1]](/bjoc/content/inline/1860-5397-9-158-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic dissection of the ABC-ring system of beticolin 0 (1).
Scheme 1: Retrosynthetic dissection of the ABC-ring system of beticolin 0 (1).
We found that dienophile 3 and diene 4 can undergo a cycloaddition by applying either thermal conditions or microwave irradiation to these both compounds (Table 1) [9]. Thereby, a mixture of regioisomers was always isolated. Regioisomer 5 was obtained diastereoselectively pure, the other isomer was received in a 3:1 (Table 1, entry 1) or 4:1 (Table 1, entry 2) mixture of 2a and 2b.
Table 1: Preliminary studies towards the ABC tricycle of beticolin 0 (1) [9].
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-158-i2.svg?max-width=637&scale=1.0)
|
|
| Entry | Product/Yield/d.r. |
|---|---|
| 1a | 2a/b (69%, d.r. 3:1), 5 (16%) |
| 2b | 2a/b (81%, d.r. 4:1), 5 (16%) |
aConditions: µW, 300 W, 0.5 h, 170 °C. bConditions: 170 °C, 1.5 h.
The rather harsh conditions and the lack of selectivity in the cycloaddition of dienophile 3 and diene 4 motivated us to find an appropriate catalyst system. Since the 1,3-dioxolane ring of 3 is prone to Lewis acid mediated ketal opening [11], we had to exclude metal catalysts and thus screened a variety of different organocatalysts.
Results
For the screening of an appropriate organocatalyst, dienophile 3 and diene 4 were stirred together at room temperature with an equimolar amount of the prospective organocatalyst in the absence of any solvent. The catalytic performance was classified by the acceleration of the reaction time compared to the uncatalyzed reaction (Table 2, entry 1).
Table 2: Catalyst screening for the naphthoquinone monoketal Diels–Alder reaction.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-158-i3.svg?max-width=637&scale=1.0)
|
||
| Entry | Catalysta | Catalyst performanceb |
|---|---|---|
| 1 | – | 0 |
| 2 | 6 | 0 |
| 3 | L-proline | + |
| 4 | 8 | 0 |
| 5 | 9 | ++ |
| 6 | 10 | +++ |
| 7 | 11 | ++ |
| 8 | 12 | +++ |
aDienophile 3 was stirred at rt with diene 4. In the case of entries 2–8, one equiv catalyst was added. bPerformance assessment: 0 = no acceleration, trace amounts of product appear after several weeks; + = slight acceleration, no full conversion even after 2 months; ++ = full conversion between 1–2 months; +++ = full conversion in less than one month.
At first, the use of MacMillan's imidazolidinone organocatalyst 6 [12] was examined, but no catalytic effect was observed (Table 2, entry 2). The usage of L-proline as a bifunctional catalyst only gave a slight improvement compared to the uncatalyzed reaction (Table 2, entry 3). Whereas the addition of one equivalent of Brønsted acid 8 had no effect (Table 2, entry 4), the addition of TADDOL (9) improved the reaction speed (Table 2, entry 5). By the use of Schreiner's catalyst 10 [7], the highest conversion speed was observed (Table 2, entry 6). After this finding, Jacobsen's thioureas 11 [13] (Table 2, entry 7) and 12 [14] (Table 2, entry 8) were also tested whereby the 3,5-bis(trifluoromethyl)phenyl-substituted thiourea 12 was superior to catalyst 11. The fact that thioureas as well as diol 9 improved the reaction speed was coherent, since both catalysts are well known to catalyze the Diels–Alder reaction by hydrogen-bond catalysis [1].
Subsequently, we examined the Diels–Alder reaction of dienophile 3 with diene 4 with 10 mol % of the superior catalysts 10 and 12 (Table 3). In this way, the desired tricycle 2a was obtained by usage of Schreiner's catalyst 10 in 63% yield (Table 3, entry 1). To our delight and in contrast to the uncatalyzed reaction conditions (Table 1), just one regioisomer was isolated. In addition, the cycloaddition with thiourea catalyst 10 afforded exclusively the exo-Diels–Alder product 2a, whereby thermal conditions afforded an inseparable mixture of 2a and 2b besides the separable product 5. The usage of Jacobsen's catalyst 12 also gave the neat exo-Diels–Alder product 2a without any accompanying isomers, but with lower yield (29%) and no remarkable ee value (6% ee, determined by HPLC) (Table 3, entry 2). The reaction of an electronically deactivated and sterically demanding dienophile, in which the ethyl ester was replaced with a CH2OTBS group, failed on using catalyst 10 and diene 4.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-158-i4.svg?max-width=637&scale=1.0)
Discussion
It is well known that thiourea 10 facilitates the Diels–Alder reaction by activation of the dienophile with hydrogen-bond catalysis [7]. The ester group in 3 is apparently the most basic carbonyl group in the molecule and the ester group should undergo hydrogen-bonding with 10. However, the resulting product 5 was not observed upon use of 10 or 12. To explain the observed regioselectivity, we can assume that the thiourea catalyst is more attracted to the carbonyl function within the monoketal naphthoquinone system due to steric reasons. Whereas the 1,3-dioxalane ring hampers bonding of the catalyst on the ester group, the aromatic methoxy group could facilitate catalyst attraction by supporting hydrogen bonds [6,15] (Figure 1). In addition, the 3,5-bis(trifluoromethyl)phenyl group could be involved in the substrate bonding [16]. That could explain the lower reaction acceleration of catalyst 11 compared to the 3,5-bis(trifluoromethyl)phenyl-substituted thiourea catalysts 10 and 12. Since the bulky OTBS-group from 4 points out of the favored transition state I, the high exo-selectivity for the Diels–Alder reaction can be explained. In the disfavored transition state II the bulky OTBS-group conflicts with the catalyst–substrate complex.
![[1860-5397-9-158-1]](/bjoc/content/figures/1860-5397-9-158-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proposed favored and disfavored transition states during the thiourea catalyzed Diels–Alder reaction of dienophile 2 and diene 3. R = 3,5-bis(trifluoromethyl)phenyl.
Figure 1: Proposed favored and disfavored transition states during the thiourea catalyzed Diels–Alder reactio...
Many organocatalytic reactions are known for their long reaction times because of the relatively weak interactions between catalyst and substrate. Also the naphthoquinone monoketal Diels–Alder reactions were quenched after 28 d. After that time no further conversion was observed and some starting material could be recovered. Nevertheless it has to be considered that the catalyst facilitates the construction of a very congested quarternary center in alpha-position to the obstructed spiroketal center, rendering the long reaction times valid.
Conclusion
In summary, we screened a variety of different organocatalysts to promote the naphthoquinone monoketal Diels–Alder reaction. Thereby we found that thioureas 10 and 12 facilitate the cycloaddition of the sterically hindered dienophile 3 with diene 4 in up to 63% yield in high selectivity. Although long reaction times were a drawback, this represents the first time and the sole method for the generation of neat exo-product 5a, bearing a newly formed quaternary center. Additionally, tricycle 5a is seen as a valuable intermediate for the total synthesis of beticolin 0 (1) and related natural products.
Experimental
IR: spectra were measured with a Bruker IFS 88 spectrometer, and wave numbers are given in cm−1. MS (EI): mass spectra were measured with a Finnigan MAT 95. NMR: Spectra were recorded on a Bruker Avance 400 or Avance DRX 500 spectrometer in the solvents indicated; 1H and 13C chemical shifts (δ) are given in parts per million relative to TMS, coupling constants (J) in hertz. The solvent signals were used as references and the chemical shifts were converted to the TMS scale. Unless stated otherwise, all commercially available compounds (Acros, Fluka, Aldrich) were used as received.
Ethyl 4-((tert-butyldimethylsilyl)oxy)-5,8-dimethoxy-2-methyl-10-oxo-4,4a,9a,10-tetrahydro-1H-spiro[anthracene-9,2'-[1,3]dioxolane]-9a-carboxylate (2a): In a small flask dienophile 3 (23.1 mg, 69.2 µmol) was covered with diene 4 (250 μL). The mixture was stirred together with Schreiner's catalyst 10 (3.50 mg, 6.92 µmol) at room temperature for 28 d. Purification by column chromatography (silica gel, ethyl acetate/cyclohexane 2:8 to 3:7) afforded product 2a (23.3 mg, 43.7 µmol, 63% yield) as a colorless oil as well as starting material 3 (6.73 mg, 20.1 µmol, 29% yield). If necessary, also diene 4 could be recovered by Kugelrohr distillation before column chromatography was done. Rf 0.48 (ethyl acetate/cyclohexane 1:1); 1H NMR (400 MHz, CDCl3) δ 6.99 (d, J = 9.1 Hz, 1H), 6.87 (d, J = 9.1 Hz, 1H), 5.44 (s, 1H), 4.60 (s, 1H), 4.27–4.08 (m, 5H), 3.98–3.88 (m, 1H), 3.77 (s, 3H), 3.73 (s, 3H), 3.68 (d, J = 5.1 Hz, 1H), 2.76 (d, J = 17.5 Hz, 1H), 2.14 (d, J = 17.8 Hz, 1H), 1.58 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H), 0.78 (s, 9H), 0.02 (s, 3H), 0.00 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.5, 172.0, 151.7, 151.3, 133.4, 129.9, 126.0, 125.3, 117.7, 115.0, 109.8, 67.9, 67.6, 67.2, 61.7, 58.3, 57.3, 57.1, 52.8, 32.6, 26.0, 25.8, 23.4, 18.3, 14.3, −4.3, −4.61; IR (thin film) νmax: 1710, 1477, 1269, 1208, 1002, 959, 920, 836, 805, 776, 723 cm−1; EIMS m/z: 532.2, 487.2, 476.2, 475.2, 413.1, 403.1, 402.1 358.1, 357.1; HRMS–EI (m/z): [M]+ calcd for C28H40O8Si, 532.2491; found, 532.2493.
References
-
Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1–26. doi:10.1055/s-0029-1217130
Return to citation in text: [1] [2] -
Duarte, F. J. S.; Santos, A. G. J. Org. Chem. 2012, 77, 3252–3261. doi:10.1021/jo300005e
Return to citation in text: [1] -
Jiang, H.; Rodríguez-Escrich, C.; Johansen, T. K.; Davis, R. L.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2012, 51, 10271–10274. doi:10.1002/anie.201205836
Angew. Chem. 2012, 124, 10417–10420. doi:10.1002/ange.201205836
Return to citation in text: [1] [2] -
Dieckmann, A.; Breugst, M.; Houk, K. N. J. Am. Chem. Soc. 2013, 135, 3237–3242. doi:10.1021/ja312043g
Return to citation in text: [1] -
Jiang, H.; Cruz, D. C.; Li, Y.; Lauridsen, V. H.; Jørgensen, K. A. J. Am. Chem. Soc. 2013, 135, 5200–5207. doi:10.1021/ja4007244
Return to citation in text: [1] -
Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217–220. doi:10.1021/ol017117s
Return to citation in text: [1] [2] -
Wittkopp, A.; Schreiner, P. R. Chem.–Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042
Return to citation in text: [1] [2] [3] -
Bella, M.; Gasperi, T. Synthesis 2009, 1583–1614. doi:10.1055/s-0029-1216796
Return to citation in text: [1] -
Kramer, C. S.; Nieger, M.; Bräse, S. unpublished results.
Return to citation in text: [1] [2] [3] [4] -
Masters, K.-S.; Bräse, S. Chem. Rev. 2012, 112, 3717–3776. doi:10.1021/cr100446h
Return to citation in text: [1] -
Balme, G.; Gore, J. J. Org. Chem. 1983, 48, 3336–3338. doi:10.1021/jo00167a038
Return to citation in text: [1] -
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s
Return to citation in text: [1] -
Zuend, S. J.; Jacobsen, E. N. J. Am. Chem. Soc. 2007, 129, 15872–15883. doi:10.1021/ja0735352
Return to citation in text: [1] -
Reisman, S. E.; Doyle, A. G.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 7198–7199. doi:10.1021/ja801514m
Return to citation in text: [1] -
Connon, S. J. Chem.–Eur. J. 2006, 12, 5418–5427. doi:10.1002/chem.200501076
Return to citation in text: [1] -
Lippert, K. M.; Hof, K.; Gerbig, D.; Ley, D.; Hausmann, H.; Guenther, S.; Schreiner, P. R. Eur. J. Org. Chem. 2012, 5919–5927. doi:10.1002/ejoc.201200739
Return to citation in text: [1]
| 1. | Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1–26. doi:10.1055/s-0029-1217130 |
| 2. | Duarte, F. J. S.; Santos, A. G. J. Org. Chem. 2012, 77, 3252–3261. doi:10.1021/jo300005e |
| 3. |
Jiang, H.; Rodríguez-Escrich, C.; Johansen, T. K.; Davis, R. L.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2012, 51, 10271–10274. doi:10.1002/anie.201205836
Angew. Chem. 2012, 124, 10417–10420. doi:10.1002/ange.201205836 |
| 4. | Dieckmann, A.; Breugst, M.; Houk, K. N. J. Am. Chem. Soc. 2013, 135, 3237–3242. doi:10.1021/ja312043g |
| 5. | Jiang, H.; Cruz, D. C.; Li, Y.; Lauridsen, V. H.; Jørgensen, K. A. J. Am. Chem. Soc. 2013, 135, 5200–5207. doi:10.1021/ja4007244 |
| 6. | Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217–220. doi:10.1021/ol017117s |
| 7. | Wittkopp, A.; Schreiner, P. R. Chem.–Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
| 10. | Masters, K.-S.; Bräse, S. Chem. Rev. 2012, 112, 3717–3776. doi:10.1021/cr100446h |
| 6. | Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217–220. doi:10.1021/ol017117s |
| 15. | Connon, S. J. Chem.–Eur. J. 2006, 12, 5418–5427. doi:10.1002/chem.200501076 |
| 16. | Lippert, K. M.; Hof, K.; Gerbig, D.; Ley, D.; Hausmann, H.; Guenther, S.; Schreiner, P. R. Eur. J. Org. Chem. 2012, 5919–5927. doi:10.1002/ejoc.201200739 |
| 1. | Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1–26. doi:10.1055/s-0029-1217130 |
| 3. |
Jiang, H.; Rodríguez-Escrich, C.; Johansen, T. K.; Davis, R. L.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2012, 51, 10271–10274. doi:10.1002/anie.201205836
Angew. Chem. 2012, 124, 10417–10420. doi:10.1002/ange.201205836 |
| 8. | Bella, M.; Gasperi, T. Synthesis 2009, 1583–1614. doi:10.1055/s-0029-1216796 |
| 7. | Wittkopp, A.; Schreiner, P. R. Chem.–Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
| 12. | Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s |
| 13. | Zuend, S. J.; Jacobsen, E. N. J. Am. Chem. Soc. 2007, 129, 15872–15883. doi:10.1021/ja0735352 |
| 11. | Balme, G.; Gore, J. J. Org. Chem. 1983, 48, 3336–3338. doi:10.1021/jo00167a038 |
| 14. | Reisman, S. E.; Doyle, A. G.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 7198–7199. doi:10.1021/ja801514m |
| 7. | Wittkopp, A.; Schreiner, P. R. Chem.–Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
© 2013 Kramer and Bräse; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)