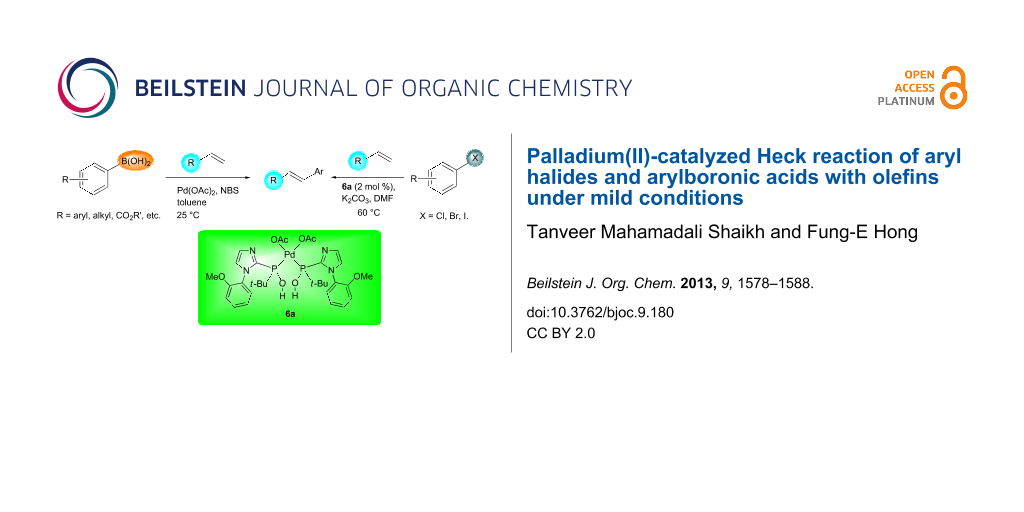
Abstract
A series of general and selective Pd(II)-catalyzed Heck reactions were investigated under mild reaction conditions. The first protocol has been developed employing an imidazole-based secondary phosphine oxide (SPO) ligated palladium complex (6) as a precatalyst. The catalytic coupling of aryl halides and olefins led to the formation of the corresponding coupled products in excellent yields. A variety of substrates, both electron-rich and electron-poor olefins, were converted smoothly to the targeted products in high yields. Compared with the existing approaches employing SPO–Pd complexes in a Heck reaction, the current strategy features mild reaction conditions and broad substrate scope. Furthermore, we described the coupling of arylboronic acids with olefins, which were catalyzed by Pd(OAc)2 and employed N-bromosuccinimide as an additive under ambient conditions. The resulted biaryls have been obtained in moderate to good yields.

Graphical Abstract
Introduction
Substituted olefins are important structural motifs in natural products, pharmaceuticals, bioactive compounds and organic materials [1,2]. Olefins such as stilbene derivatives normally show antitumor [3], antiinflammatory [4], neuroprotective [5], and cardioprotective [6] properties. Due to its importance in the synthesis of leading molecules, a variety of preparative methodologies have been developed. Particularly, the Heck reaction is one of the most chosen methods in the synthesis of aryl-substituted olefins [7-9]. Aryl halides or arylboronic acids are among the most commonly employed arylpalladium precursors in the Heck reaction.
In the early 1970s, Mizoroki [10] and Heck [11] developed a palladium(0)-catalyzed cross-coupling of olefins with organic halides. Later, several other catalytic protocols were used with variations in their coupling procedures by changing metal sources, ligands, additives or substrates [12-16]. The class of phosphine-ligated palladium complexes [17-21] represents the most frequently employed precatalysts to achieve high reactivities and selectivities for such reactions. However, such trisubstituted phosphines in the palladium complexes are often air-sensitive in nature and easily oxidized [22,23]. Therefore, a new class of secondary phosphine oxide ligands (SPO) has been explored for these ligand-assisted palladium-catalyzed cross-coupling reactions [24-27]. This type of SPO ligand is stable towards air and moisture and convenient to handle compared to the conventional trisubstituted phosphine ligands. Despite this advantage, the potential of these ligands has not been fully realized in Heck arylation reactions. Up to now, only a few examples of utilizing SPO-ligated palladium complexes in oxidative Heck reactions have been demonstrated [28-31]. Previously, we also reported the synthesis of cobalt-containing SPO ligands and their palladium complex. This was successfully applied as a catalytic precursor in oxidative Heck reactions [32]. However, these reactions were carried out at high temperatures with limited substrate scope. Therefore, the development of an alternative general and mild procedure employing a stable and inexpensive ligand is still in great demand.
Furthermore, the application of palladium complexes in the oxidative coupling of organo-boron compounds with olefins has attracted much attention in recent years [33-38]. Various catalytic systems have been developed by Jung [39] and Larhed et al. [40-43] by employing diverse variations in oxidants, ligands or palladium complexes [44-47]. Nowadays, several competent methods are also known to achieve this transformation with different reaction conditions employing base-free, ligand-free conditions or by using conventional oxidants such as oxygen, benzoquinone, Cu salts, etc. [48-53].
In this article we report two new protocols for Heck cross-coupling reactions: (i) a stable SPO ligated palladium complex 6 catalyzed cross-coupling of aryl halides 1 with olefins 2 at 60 °C; and (ii) Pd(OAc)2 catalyzed arylation of arylboronic acids 4 with olefins at 25 °C (Scheme 1).
![[1860-5397-9-180-i1]](/bjoc/content/inline/1860-5397-9-180-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Heck reaction of olefins with aryl halides and arylboronic acids.
Scheme 1: Heck reaction of olefins with aryl halides and arylboronic acids.
Results and Discussion
Heck reaction of aryl halides with olefins
In the presence of solvents, secondary phosphine oxide (RR'P(O)H) might undergo tautomerization, which generates a less stable phosphinous acid (RR'POH) species. Subsequently, its coordination to the metal center through the phosphorus atom forms a phosphinous acid–metal complex [54-56]. Thus, the resulting transition-metal complex might function as an active catalyst in various C–C-bond-forming reactions. Ackermann et al. reported the synthesis of stable N-aryl-substituted pyrrole and indole-derived SPO-preligands, which were utilized in Kumada–Corriu cross-coupling reactions [57]. Recently, we reported the synthesis and characterization of imidazole-based secondary phosphine oxide ligand 5 and its application in C–C-bond-forming reactions (Scheme 2) [58]. Furthermore, the application of complex 6 in cross-coupling reactions has been carefully studied. We found that complex 6 is an active catalyst for the Heck reaction of aryl halides with olefins under mild conditions.
![[1860-5397-9-180-i2]](/bjoc/content/inline/1860-5397-9-180-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of imidazole-based SPO–Pd complex 6.
Scheme 2: Synthesis of imidazole-based SPO–Pd complex 6.
To optimize the reaction conditions, a series of reactions under various combinations of bases, solvents and temperatures, employing complex 6 as precatalyst, was pursued. Bromobenzene (1a) and styrene (2a) were chosen as the model substrates in this coupling reaction and the results are presented in Table 1.
Table 1: Palladium complex (6) catalyzed Heck reaction of bromobenzene and styrene: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-180-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Complex 6 (mol %) | Base (equiv) | Solvent | Temp (°C) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | 1 | NaOH (1) | DMSO | rt | – |
| 2 | 2 | NaOAc (1) | DMSO | rt | – |
| 3 | 2 | Et3N (2) | DMSO | 40 | – |
| 4 | 1 | K2CO3 (1) | toluene | rt | – |
| 5 | 1 | K2CO3 (1) | CH3CN | rt | –c |
| 6 | 1 | K3PO4 (1) | THF | 40 | 17 |
| 7 | 1 | K3PO4 (1) | THF | 60 | 24 |
| 8 | 1 | K2CO3 (1) | THF | 60 | 46 |
| 9 | 1 | K2CO3 (1) | DMF | 100 | 65 |
| 10 | 2 | K2CO3 (2) | DMF | 100 | 84 |
| 11 | 2 | K2CO3 (2) | DMF | 80 | 92 |
| 12 | 2 | K2CO3 (2) | DMF | 50 | 73 |
| 13 | 2 | K2CO3 (1) | DMF | 60 | 89 |
| 14 | 2 | K2CO3 (2) | DMF | 60 | 96 |
| 15 | 2 | K2CO3 (2) | no solv. | 60 | 82 |
aReaction conditions: styrene (1.0 mmol), bromobenzene (1.0 mmol), base, solvent (1 mL), stirred for 12 h. bIsolated yield. cReaction mixture was stirred for 24 h.
Initially, the coupling was carried out by using 1 mol % loading of Pd-complex 6 as a precatalyst, with styrene (2a, 1 mmol), and bromobenzene (1a, 1 mmol) in DMSO (2 mL), and at ambient temperature in the presence of NaOH (1 equiv, Table 1, entry 1). The reaction did not give the coupled product 3a. Moreover, the use of other bases such as NaOAc, Et3N and K2CO3 in the presence of the solvents, DMSO, toluene or acetonitrile were not useful and no coupled product was observed. Interestingly, the reaction showed little progress in the presence of K3PO4 and tetrahydrofuran at 40 °C to obtain 3a in 17% yield (Table 1, entry 6). The yield was slightly improved when the reaction was heated at 60 °C (Table 1, entry 7). When K2CO3 (1.0 equiv) in THF was employed under similar reaction conditions, the yield of trans-stilbene was improved to 46% (Table 1, entry 8). Once K2CO3 had been selected as the most effective base, the next step involved the enhancement of the product yield. The combination of K2CO3 (2 equiv) and DMF (2 mL) resulted in the formation of 84% of 3a at 100 °C (Table 1, entry 10). A further increase in the reaction temperature would lead to decomposition of the palladium complex, which was formed in situ, thus lowered the yield of the product. Therefore, the loading of the precatalyst 6 was increased to 2 mol % and resulted in the formation of trans-stilbene in 92% yield at 80 °C (Table 1, entry 11). Synthetically, it is important to carry out reactions under mild reaction conditions. Nevertheless, low yield (73%) of the product was obtained by reducing the reaction temperature to 50 °C. Thus, a substrate survey was conducted at 60 °C. The optimized reaction conditions were found to be the use of styrene (2a, 1 mmol), bromobenzene (1a, 1 mmol), K2CO3 (2 mmol), and precatalyst 6 (2 mol %) with heating at 60 °C in DMF (1 mL, Table 1, entry 14). It is worthy of noting that the coupling reaction was also performed in the absence of solvent, which gave 82% yield (Table 1, entry 15) of the coupled product.
A wide range of olefins with different and diversely substituted aryl bromides were subjected to cross-coupling to produce the corresponding 1,2-disubstituted olefins. The results are summarized in Table 2.
Table 2: Heck reaction of olefins and aryl halides: Scope of substrate.a
| Entry | Olefin (2) | Aryl halide (1) | Product (3) | Yield (%)b,c |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-180-i4.svg?max-width=637&scale=1.0)
2a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-180-i5.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-180-i6.svg?max-width=637&scale=1.0)
3a |
96 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-180-i7.svg?max-width=637&scale=1.0)
2a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-180-i8.svg?max-width=637&scale=1.0)
1b |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-180-i9.svg?max-width=637&scale=1.0)
3a |
98 |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-180-i10.svg?max-width=637&scale=1.0)
2a |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-180-i11.svg?max-width=637&scale=1.0)
1c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-180-i12.svg?max-width=637&scale=1.0)
3a |
62 |
| 4 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-180-i13.svg?max-width=637&scale=1.0)
2b |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-180-i14.svg?max-width=637&scale=1.0)
1a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-180-i15.svg?max-width=637&scale=1.0)
3b |
90 |
| 5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-180-i16.svg?max-width=637&scale=1.0)
2c |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-180-i17.svg?max-width=637&scale=1.0)
1a |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-180-i18.svg?max-width=637&scale=1.0)
3c |
92 |
| 6 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-180-i19.svg?max-width=637&scale=1.0)
2a |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-180-i20.svg?max-width=637&scale=1.0)
1d |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-180-i21.svg?max-width=637&scale=1.0)
3d |
88 |
| 7 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-180-i22.svg?max-width=637&scale=1.0)
2d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-180-i23.svg?max-width=637&scale=1.0)
1a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-180-i24.svg?max-width=637&scale=1.0)
3e |
90 |
| 8 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-180-i25.svg?max-width=637&scale=1.0)
2e |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-180-i26.svg?max-width=637&scale=1.0)
1a |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-180-i27.svg?max-width=637&scale=1.0)
3f |
95 (35)d |
| 9 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-180-i28.svg?max-width=637&scale=1.0)
2f |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-180-i29.svg?max-width=637&scale=1.0)
1a |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-180-i30.svg?max-width=637&scale=1.0)
3d |
91 |
| 10 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-180-i31.svg?max-width=637&scale=1.0)
2g |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-180-i32.svg?max-width=637&scale=1.0)
1a |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-180-i33.svg?max-width=637&scale=1.0)
3g |
80 |
| 11 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-180-i34.svg?max-width=637&scale=1.0)
2h |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-180-i35.svg?max-width=637&scale=1.0)
1a |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-180-i36.svg?max-width=637&scale=1.0)
3h |
85 |
| 12 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-180-i37.svg?max-width=637&scale=1.0)
2i |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-180-i38.svg?max-width=637&scale=1.0)
1a |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-180-i39.svg?max-width=637&scale=1.0)
3i |
87 |
| 13 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-180-i40.svg?max-width=637&scale=1.0)
2j |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-180-i41.svg?max-width=637&scale=1.0)
1a |
![[Graphic 40]](/bjoc/content/inline/1860-5397-9-180-i42.svg?max-width=637&scale=1.0)
3j |
90 |
| 14 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-9-180-i43.svg?max-width=637&scale=1.0)
2k |
![[Graphic 42]](/bjoc/content/inline/1860-5397-9-180-i44.svg?max-width=637&scale=1.0)
1a |
![[Graphic 43]](/bjoc/content/inline/1860-5397-9-180-i45.svg?max-width=637&scale=1.0)
3k |
89 |
| 15 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-9-180-i46.svg?max-width=637&scale=1.0)
2l |
![[Graphic 45]](/bjoc/content/inline/1860-5397-9-180-i47.svg?max-width=637&scale=1.0)
1a |
![[Graphic 46]](/bjoc/content/inline/1860-5397-9-180-i48.svg?max-width=637&scale=1.0)
3l |
88 |
| 16 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-9-180-i49.svg?max-width=637&scale=1.0)
2a |
![[Graphic 48]](/bjoc/content/inline/1860-5397-9-180-i50.svg?max-width=637&scale=1.0)
1e |
![[Graphic 49]](/bjoc/content/inline/1860-5397-9-180-i51.svg?max-width=637&scale=1.0)
3m |
90 |
| 17 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-9-180-i52.svg?max-width=637&scale=1.0)
2m |
![[Graphic 51]](/bjoc/content/inline/1860-5397-9-180-i53.svg?max-width=637&scale=1.0)
1a |
![[Graphic 52]](/bjoc/content/inline/1860-5397-9-180-i54.svg?max-width=637&scale=1.0)
3n |
78 |
| 18 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-9-180-i55.svg?max-width=637&scale=1.0)
2n |
![[Graphic 54]](/bjoc/content/inline/1860-5397-9-180-i56.svg?max-width=637&scale=1.0)
1a |
![[Graphic 55]](/bjoc/content/inline/1860-5397-9-180-i57.svg?max-width=637&scale=1.0)
3o |
95 |
| 19 |
![[Graphic 56]](/bjoc/content/inline/1860-5397-9-180-i58.svg?max-width=637&scale=1.0)
2o |
![[Graphic 57]](/bjoc/content/inline/1860-5397-9-180-i59.svg?max-width=637&scale=1.0)
1a |
![[Graphic 58]](/bjoc/content/inline/1860-5397-9-180-i60.svg?max-width=637&scale=1.0)
3p |
90 |
| 20 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-9-180-i61.svg?max-width=637&scale=1.0)
2p |
![[Graphic 60]](/bjoc/content/inline/1860-5397-9-180-i62.svg?max-width=637&scale=1.0)
1a |
![[Graphic 61]](/bjoc/content/inline/1860-5397-9-180-i63.svg?max-width=637&scale=1.0)
3q |
92 |
aReaction conditions: olefin (1.0 mmol), aryl halide (1.0 mmol), Pd-complex 6 (2.0 mol %), K2CO3 (2.0 mmol), DMF (1 mL), 60 °C, 12 h. bIsolated yield. cProducts were characterized by 1H, 13C NMR and GC–MS. dThe yield corresponds to employing 4-chloro anisole as the aryl halide source.
Both aryl bromide and aryl iodide performed well (Table 2, entries 1 and 2) under these conditions. However, the aryl chloride was found to be less reactive giving the corresponding product 3a in 62% yield (Table 2, entry 3). The oxidative coupling was found to be selective in the case of 4-bromostyrene (2b), which gives 90% yield of 4-bromo trans-stilbene (3b) without the observation of any side product (Table 2, entry 4). The presence of either an electron-withdrawing or electron-donating group on the aromatic ring of olefin did not affect the reactivity and yield of product. The reactions led to the formation of excellent yields of the corresponding products 3e and 3f in 90% and 95% yields, respectively (Table 2, entries 7 and 8). As known, aromatic rings having substituents such as, -CH2OH, -CHO, -COCH3 -CN and -CF3 are rather useful in organic synthesis. However, in earlier reported oxidative coupling conditions these functional groups were not compatible and gave low yields of products. Therefore, these highly modifiable groups were screened under these catalytic conditions. Thus, 4-vinylbenzyl alcohol (2g), 4-vinyl benzaldehyde (2h), 4-vinylacetophenone (2i), 4-cyanostyrene (2j) and 4-trifluoromethylstyrene (2k) were smoothly converted to their corresponding coupled products 3g–3k in excellent yields (Table 2, entries 10–14). The selectivities and yields of the coupled products were excellent regardless of ortho-, meta-, or para-substitution patterns on either styrenes or aryl halides under these catalytic conditions. For example, the coupling of substituted methylstyrenes (Table 2, entry 15) or alkyl-substituted aryl halides (Table 2, entry 16) gave 88–90% isolated yields of 3l and 3m. To investigate whether the reaction was compatible with a heteroaryl olefin, 2-vinylpyridine (2m) was subjected to this reaction. It produced the corresponding coupled product 3n in 78% yield (Table 2, entry 17). Furthermore, using these optimized conditions, bromobenzene (1a) was examined with different vinyl esters to determine the scope of this procedure. The results are given in Table 2, entries 18–20. Notably, the performances were in agreement with the previous expectations and yields are excellent in the preparation of α,β–unsaturated esters. The corresponding α,β-unsaturated esters 3o–3q were obtained in 90–95% yields, respectively.
Heck reaction of arylboronic acids with olefins
The phosphine- and base-free coupling of arylboronic acids with olefins under mild reaction conditions were studied as well to broaden the scope of cross-coupling reactions. To search for the optimized reaction conditions, phenylboronic acid (4a) and styrene (2a) were chosen as the model substrates and Pd(OAc)2 was employed as the catalyst. Various reaction conditions were tested and the results are presented in Table 3. Initially, a Pd(OAc)2 catalyzed Heck reaction was performed employing polar sovents, dimethylacetamide (DMAc) and DMF, at 25 °C in the presence of 0.5 equiv of N-bromosuccinimide (NBS). This resulted in the formation of trans-stilbene (3a) in 52% and 40% yield, respectively (Table 3, entries 1 and 2). However, the same reaction under the control conditions (i.e., in the absence of NBS) resulted in production of a trace amount of the coupled product 3a (Table 3, entry 3). When the coupling reaction was carried out at 90 °C in DMAc solvent, the yield of 3a decreased, due to the formation of side product, such as bromobenzene, from the corresponding phenylboronic acid (Table 3, entry 4). Therefore, it is believed that NBS plays an important role in this catalytic reaction. Furthermore, we focused our attention to other solvents such as MeOH, CH2Cl2, CH3CN, Me2O, t-Bu2O, THF, DMSO and 1,4-dioxane, which resulted in low yields of arylated product. Subsequently, the reaction was subjected to the apolar solvent toluene. The expected product trans-stilbene (3a) was obtained in 68% yield at 25 °C for 18 h (Table 3, entry 5). The yield of the desired product did not improve even when the reaction was stirred for 24 h (Table 3, entry 6). On the other hand, lowering the additive (NBS) to 10 mol % did not show any improvement to the formation of trans-stilbene (3a) (Table 3, entries 7 and 8). A sharp decline in the formation of trans-stilbene (3a) (Table 3, entry 9) was observed on increasing the quantity of NBS to a stoichiometric amount (1.0 equiv). This was probably due to the formation of other competitive side product(s). Interestingly, the coupled product was obtained with improved yield of 76% by using 30 mol % NBS (Table 3, entry 10). Next, we turned our attention to the improvement of the yields of trans-stilbene by adjusting other reaction parameters. Thus, the addition of K2CO3 as base along with NBS under similarly performed reaction conditions led to no formation of the targeted product. The addition of molecular sieves was not a good choice either [59]. The other additives such as LiBr and CuBr were also examined. Still, no coupled product was obtained in the presence of LiBr (30 mol %, Table 3, entry 13). On the other hand, the employment of CuBr (30 mol %) with the presence of Pd(OAc)2 resulted in a 42% yield of trans-stilbene (3a) (Table 3, entry 14). Thus, the optimized reaction conditions for the Heck reaction here is the use of arylboronic acid (1 mmol), olefin (1 mmol), Pd(OAc)2 (5 mol %), NBS (30 mol %), toluene (1 mL) at 25 °C under stirring for 12 h.
Table 3: Heck reaction of phenylboronic acid and styrene: Optimization of the reaction conditions.a
![[Graphic 62]](/bjoc/content/inline/1860-5397-9-180-i64.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Additive (equiv) | Solvent | Time (h) | Temp. (°C) | Yield (%)b,c |
|---|---|---|---|---|---|
| 1 | NBS (0.5) | DMAc | 18 | 25 | 52 |
| 2 | NBS (0.5) | DMF | 18 | 25 | 40 |
| 3 | – | DMAc | 24 | 25 | trace |
| 4 | NBS (0.5) | DMAc | 18 | 90 | 34 |
| 5 | NBS (0.5) | toluene | 18 | 25 | 68 |
| 6 | NBS (0.5) | toluene | 24 | 25 | 69 |
| 7 | NBS (0.1) | toluene | 18 | 25 | 30 |
| 8 | NBS (0.1) | toluene | 18 | 80 | 47 |
| 9 | NBS (1) | toluene | 18 | 25 | 40 |
| 10 | NBS (0.3) | toluene | 12 | 25 | 76 |
| 11 | NBS/K2CO3 (0.3:1) | toluene | 18 | 25 | nrd |
| 12 | NBS/4 Å MS (0.3:2) | toluene | 12 | 25 | 15 |
| 13 | LiBr (0.3) | toluene | 12 | 25 | nr |
| 14 | CuBr | toluene | 12 | 25 | 42 |
aReaction conditions: styrene (0.5 mmol), phenylboronic acid (0.5 mmol), Pd(OAc)2 (5 mol %), additive and dry solvent (1 mL) for 12 h at 25 °C. bIsolated yield. cProduct was characterized by GC–MS, 1H and 13C NMR. dReaction was stirred under air.
The optimized Heck cross-coupling conditions were employed to examine the arylation of substituted olefins and phenylboronic acid. The results are presented in Table 4. As shown in Table 4, this coupling procedure tolerates various functional groups to afford the desired product (3). The compatibility of halo-substituted styrenes is synthetically useful since the products could be easily modified further to form synthetic building blocks. Thus, the coupling of 4-fluorostyrene (2q), 4-bromostyrene (2b) and 4-chlorostyrene (2r) through oxidative Heck reaction led to the corresponding products in 65–69% yields, respectively (Table 4, entries 2–4). Furthermore, the electron-withdrawing groups on styrene, such as 3-nitrostilbene (2d) and 4-trifluorostilbene (2k) resulted in the formation 3e and 3k in 76% and 70% yields, respectively (Table 4, entries 5 and 6). However, the electron-donating substituent on olefin lessened the reaction rate and thus led to poor yield of product 3f (Table 4, entry 7). The reaction with aliphatic alkenes, such as tert-butyl acrylate (2n) or ethyl acrylate (2o), allyl acetate (2s) and n-heptene (2t) afforded the corresponding coupled products in moderate yields, respectively (Table 4, entries 9–12).
Table 4: Substrate scope in the Heck arylation reaction of phenylboronic acids with olefins.a
![[Graphic 63]](/bjoc/content/inline/1860-5397-9-180-i65.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate (2) | Product (3)b | Yield (%)c |
|---|---|---|---|
| 1 |
![[Graphic 64]](/bjoc/content/inline/1860-5397-9-180-i66.svg?max-width=637&scale=1.0)
2a |
![[Graphic 65]](/bjoc/content/inline/1860-5397-9-180-i67.svg?max-width=637&scale=1.0)
3a |
76 |
| 2 |
![[Graphic 66]](/bjoc/content/inline/1860-5397-9-180-i68.svg?max-width=637&scale=1.0)
2q |
![[Graphic 67]](/bjoc/content/inline/1860-5397-9-180-i69.svg?max-width=637&scale=1.0)
3r |
65d |
| 3 |
![[Graphic 68]](/bjoc/content/inline/1860-5397-9-180-i70.svg?max-width=637&scale=1.0)
2b |
![[Graphic 69]](/bjoc/content/inline/1860-5397-9-180-i71.svg?max-width=637&scale=1.0)
3b |
69 |
| 4 |
![[Graphic 70]](/bjoc/content/inline/1860-5397-9-180-i72.svg?max-width=637&scale=1.0)
2r |
![[Graphic 71]](/bjoc/content/inline/1860-5397-9-180-i73.svg?max-width=637&scale=1.0)
3s |
66 |
| 5 |
![[Graphic 72]](/bjoc/content/inline/1860-5397-9-180-i74.svg?max-width=637&scale=1.0)
2d |
![[Graphic 73]](/bjoc/content/inline/1860-5397-9-180-i75.svg?max-width=637&scale=1.0)
3e |
76 |
| 6 |
![[Graphic 74]](/bjoc/content/inline/1860-5397-9-180-i76.svg?max-width=637&scale=1.0)
2k |
![[Graphic 75]](/bjoc/content/inline/1860-5397-9-180-i77.svg?max-width=637&scale=1.0)
3k |
70 |
| 7 |
![[Graphic 76]](/bjoc/content/inline/1860-5397-9-180-i78.svg?max-width=637&scale=1.0)
2e |
![[Graphic 77]](/bjoc/content/inline/1860-5397-9-180-i79.svg?max-width=637&scale=1.0)
3f |
30 |
| 8 |
![[Graphic 78]](/bjoc/content/inline/1860-5397-9-180-i80.svg?max-width=637&scale=1.0)
2c |
![[Graphic 79]](/bjoc/content/inline/1860-5397-9-180-i81.svg?max-width=637&scale=1.0)
3c |
70 |
| 9 |
![[Graphic 80]](/bjoc/content/inline/1860-5397-9-180-i82.svg?max-width=637&scale=1.0)
2n |
![[Graphic 81]](/bjoc/content/inline/1860-5397-9-180-i83.svg?max-width=637&scale=1.0)
3o |
50 |
| 10 |
![[Graphic 82]](/bjoc/content/inline/1860-5397-9-180-i84.svg?max-width=637&scale=1.0)
2o |
![[Graphic 83]](/bjoc/content/inline/1860-5397-9-180-i85.svg?max-width=637&scale=1.0)
3p |
42 |
| 11 |
![[Graphic 84]](/bjoc/content/inline/1860-5397-9-180-i86.svg?max-width=637&scale=1.0)
2s |
![[Graphic 85]](/bjoc/content/inline/1860-5397-9-180-i87.svg?max-width=637&scale=1.0)
3t |
38 |
| 12 |
![[Graphic 86]](/bjoc/content/inline/1860-5397-9-180-i88.svg?max-width=637&scale=1.0)
2t |
![[Graphic 87]](/bjoc/content/inline/1860-5397-9-180-i89.svg?max-width=637&scale=1.0)
3u |
44e |
aReaction conditions: styrene (1.0 mmol), phenylboronic acid (1.0 mmol), catalyst (5 mol %), N-bromosuccinimide (30 mol %), and toluene (2 mL) under nitrogen for 12 h. bProduct was characterized by GC–MS, 1H and 13C NMR. cIsolated yield. dDetermined by GC–MS. eE/Z ratio 20:1 by 1H NMR, terminal/internal 4/1.
To expand the scope of this cross-coupling, these conditions were then applied to a variety of boronic acids and styrene (Table 5). For a diverse set of boronic acids, cross-coupling proceeded smoothly with 2a in moderate to good yields. In this case, the procedure also tolerated a range of functional groups, such as bromo, chloro, nitro, methoxy, and alkyl groups. The arylboronic acids with electron-withdrawing substituents furnished good yields of coupled product as compared to the electron-donating substituents. For example, 4-nitro (4e) and 3-nitrophenylboronic acid (4f) were reacted smoothly with styrene to afford the corresponding products in 75% and 73% yields, respectively (Table 5, entries 5 and 6).
Table 5: Substrate scope in Heck arylation reaction of phenylboronic acids with olefins.a
![[Graphic 88]](/bjoc/content/inline/1860-5397-9-180-i90.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Productb | Yield (%)c |
|---|---|---|---|
| 1 |
![[Graphic 89]](/bjoc/content/inline/1860-5397-9-180-i91.svg?max-width=637&scale=1.0)
4a |
![[Graphic 90]](/bjoc/content/inline/1860-5397-9-180-i92.svg?max-width=637&scale=1.0)
3a |
76 |
| 2 |
![[Graphic 91]](/bjoc/content/inline/1860-5397-9-180-i93.svg?max-width=637&scale=1.0)
4b |
![[Graphic 92]](/bjoc/content/inline/1860-5397-9-180-i94.svg?max-width=637&scale=1.0)
3b |
69 |
| 3 |
![[Graphic 93]](/bjoc/content/inline/1860-5397-9-180-i95.svg?max-width=637&scale=1.0)
4c |
![[Graphic 94]](/bjoc/content/inline/1860-5397-9-180-i96.svg?max-width=637&scale=1.0)
3c |
67 |
| 4 |
![[Graphic 95]](/bjoc/content/inline/1860-5397-9-180-i97.svg?max-width=637&scale=1.0)
4d |
![[Graphic 96]](/bjoc/content/inline/1860-5397-9-180-i98.svg?max-width=637&scale=1.0)
3k |
72 |
| 5 |
![[Graphic 97]](/bjoc/content/inline/1860-5397-9-180-i99.svg?max-width=637&scale=1.0)
4e |
![[Graphic 98]](/bjoc/content/inline/1860-5397-9-180-i100.svg?max-width=637&scale=1.0)
3v |
75 |
| 6 |
![[Graphic 99]](/bjoc/content/inline/1860-5397-9-180-i101.svg?max-width=637&scale=1.0)
4f |
![[Graphic 100]](/bjoc/content/inline/1860-5397-9-180-i102.svg?max-width=637&scale=1.0)
3e |
73 |
| 7 |
![[Graphic 101]](/bjoc/content/inline/1860-5397-9-180-i103.svg?max-width=637&scale=1.0)
4g |
![[Graphic 102]](/bjoc/content/inline/1860-5397-9-180-i104.svg?max-width=637&scale=1.0)
3l |
60 |
| 8 |
![[Graphic 103]](/bjoc/content/inline/1860-5397-9-180-i105.svg?max-width=637&scale=1.0)
4h |
![[Graphic 104]](/bjoc/content/inline/1860-5397-9-180-i106.svg?max-width=637&scale=1.0)
3s |
62 |
| 9 |
![[Graphic 105]](/bjoc/content/inline/1860-5397-9-180-i107.svg?max-width=637&scale=1.0)
4i |
![[Graphic 106]](/bjoc/content/inline/1860-5397-9-180-i108.svg?max-width=637&scale=1.0)
3f |
40 |
aReaction conditions: similar to Table 4. bProduct was characterized by GC–MS, 1H and 13C NMR. cIsolated yield.
Conclusion
In summary, we have developed two new protocols for oxidative Heck reactions employing Pd(OAc)2 as a catalytic precursor. The first method is based on coupling between various olefins and aryl halides utilizing an imidazole-based secondary phosphine oxide ligated palladium complex (6) under mild conditions. The yields of products obtained were excellent and in high regioselectivity. Compared with the previously described procedures for the Heck reaction of aryl halides as substrates employing a SPO–Pd complex as a catalyst, the method reported here has the advantages of having a stable catalyst system, general substrate scope, and mild reaction conditions (60 °C). Secondly, we also developed the Heck reaction of arylboronic acids with various alkenes employing N-bromosuccinimide as an additive and catalyzed by Pd(OAc)2, under base- and ligand-free conditions at 25 °C. The yields of the coupled products are moderate to good.
Supporting Information
| Supporting Information File 1: General procedure for Heck reactions, preparation of complex 6 and characterization data. | ||
| Format: PDF | Size: 267.9 KB | Download |
References
-
Douney, A. M.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2963. doi:10.1021/cr020039h
Return to citation in text: [1] -
Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442–4489. doi:10.1002/anie.200500368
Return to citation in text: [1] -
Jang, M.; Cai, L.; Udeani, G. O.; Slowing, K. V.; Thomas, C. F.; Beecher, C. W. W.; Fong, H. H. S.; Farnsworth, N. R.; Kinghorn, A. D.; Mehta, R. G.; Moon, R. C.; Pezzuto, J. M. Science 1997, 275, 218–220. doi:10.1126/science.275.5297.218
Return to citation in text: [1] -
Elmali, N.; Baysal, O.; Harma, A.; Esenkaya, I.; Mizrak, B. Inflammation 2007, 30, 1–6. doi:10.1007/s10753-006-9012-0
Return to citation in text: [1] -
Karuppagounder, S. S.; Pinto, J. T.; Xu, H.; Chen, L.-H.; Beal, M. F.; Gibson, G. E. Neurochem. Int. 2009, 54, 111–118. doi:10.1016/j.neuint.2008.10.008
Return to citation in text: [1] -
Gurusamy, N.; Lekli, I.; Mukherjee, S.; Ray, D.; Ahsan, M. K.; Gherghiceanu, M.; Popescu, L. M.; Das, D. K. Cardiovasc. Res. 2010, 86, 103–112. doi:10.1093/cvr/cvp384
Return to citation in text: [1] -
Tsuji, J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis; Wiley: Chichester, U.K., 1995.
Return to citation in text: [1] -
Bräse, S.; de Meijere, A. Cross-Coupling of Organyl Halides with Alkenes: the Heck Reaction. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; Stang, P. J., Eds.; Wiley-VCH: Weinheim, Germany, 1998.
Return to citation in text: [1] -
Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824–889. doi:10.1021/cr9003242
Return to citation in text: [1] -
Mizoroki, T.; Mori, K.; Ozaki, A. Bull. Chem. Soc. Jpn. 1971, 44, 581. doi:10.1246/bcsj.44.581
Return to citation in text: [1] -
Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024
Return to citation in text: [1] -
Crisp, G. T. Chem. Soc. Rev. 1998, 27, 427–436. doi:10.1039/a827427z
Return to citation in text: [1] -
Link, J. T.; Overman, L. E. Intramolecular Heck Reactions in Natural Product Chemistry. In Metal-Catalyzed Cross-Coupling Reactions; Diedrich, F.; Stang, P. J., Eds.; Wiley-VCH: Weinheim, Germany, 1998.
Return to citation in text: [1] -
Beletskaya, I.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048
Return to citation in text: [1] -
Liu, L.-j.; Wang, F.; Wang, W.; Zhao, M.-x.; Shi, M. Beilstein J. Org. Chem. 2011, 7, 555–564. doi:10.3762/bjoc.7.64
Return to citation in text: [1] -
Grasa, G. A.; Singh, R.; Stevens, E. D.; Nolan, S. P. J. Organomet. Chem. 2003, 687, 269–279. doi:10.1016/S0022-328X(03)00375-9
Return to citation in text: [1] -
Ozawa, F.; Kubo, A.; Hayashi, T. Chem. Lett. 1992, 2177–2180. doi:10.1246/cl.1992.2177
Return to citation in text: [1] -
Littke, A. F.; Fu, G. C. J. Org. Chem. 1999, 64, 10–11. doi:10.1021/jo9820059
Return to citation in text: [1] -
Stambuli, J. P.; Stauffer, S. R.; Shaughnessy, K. H.; Hartwig, J. F. J. Am. Chem. Soc. 2001, 123, 2677–2678. doi:10.1021/ja0058435
Return to citation in text: [1] -
Hansen, A. L.; Ebran, J.-P.; Ahlquist, M.; Norrby, P.-O.; Skrydstrup, T. Angew. Chem., Int. Ed. 2006, 45, 3349–3353. doi:10.1002/anie.200600442
Return to citation in text: [1] -
Fleckenstein, C. A.; Plenio, H. Chem. Soc. Rev. 2010, 39, 694–711. doi:10.1039/b903646f
Return to citation in text: [1] -
Parshall, G. W.; Ittel, S. D. Homogeneous Catalysis; J. Wiley and Sons: New York, 1992.
Return to citation in text: [1] -
Albéniz, A. C.; Carrera, N. Eur. J. Inorg. Chem. 2011, 2347–2360. doi:10.1002/ejic.201100162
Return to citation in text: [1] -
Li, G. Y. Angew. Chem., Int. Ed. 2001, 40, 1513–1516. doi:10.1002/1521-3773(20010417)40:8<1513::AID-ANIE1513>3.0.CO;2-C
Return to citation in text: [1] -
Jiang, X.-b.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G. J. Org. Chem. 2004, 69, 2327–2331. doi:10.1021/jo035487j
Return to citation in text: [1] -
Ackermann, L.; Born, R. Angew. Chem., Int. Ed. 2005, 44, 2444–2447. doi:10.1002/anie.200462371
Return to citation in text: [1] -
Xu, H.; Ekoue-Kovi, K.; Wolf, C. J. Org. Chem. 2008, 73, 7638–7650. doi:10.1021/jo801445y
Return to citation in text: [1] -
Ackermann, L.; Potukuchi, H. K.; Kapdi, A. R.; Schulzke, C. Chem.–Eur. J. 2010, 16, 3300–3303. doi:10.1002/chem.201000032
Return to citation in text: [1] -
Li, G. Y.; Zheng, G.; Noonan, A. F. J. Org. Chem. 2001, 66, 8677–8681. doi:10.1021/jo010764c
Return to citation in text: [1] -
Wolf, C.; Lerebours, R. J. Org. Chem. 2003, 68, 7077–7084. doi:10.1021/jo034758n
Return to citation in text: [1] -
Punji, B.; Mague, J. T.; Balakrishna, M. S. Inorg. Chem. 2007, 46, 11316–11327. doi:10.1021/ic701674x
Return to citation in text: [1] -
Wei, C.-H.; Wu, C.-E.; Huang, Y.-L.; Kultyshev, R. G.; Hong, F.-E. Chem.–Eur. J. 2007, 13, 1583–1593. doi:10.1002/chem.200601051
Return to citation in text: [1] -
Dieck, H. A.; Heck, R. F. J. Org. Chem. 1975, 40, 1083–1090. doi:10.1021/jo00896a020
Return to citation in text: [1] -
Hayashi, T.; Yamasaki, K. Chem. Rev. 2003, 103, 2829–2844. doi:10.1021/cr020022z
Return to citation in text: [1] -
Itoh, T.; Mase, T.; Nishikata, T.; Iyama, T.; Tachikawa, H.; Kobayashi, Y.; Yamamoto, Y.; Miyaura, N. Tetrahedron 2006, 62, 9610–9621. doi:10.1016/j.tet.2006.07.075
Return to citation in text: [1] -
Vandyck, K.; Mattys, B.; Willen, M.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, J. Org. Lett. 2006, 8, 363–366. doi:10.1021/ol0522788
Return to citation in text: [1] -
Bazin, M.-A.; El Kihel, L.; Lancelot, J.-C.; Rault, S. Tetrahedron Lett. 2007, 48, 4347–4351. doi:10.1016/j.tetlet.2007.04.114
Return to citation in text: [1] -
Motokura, K.; Hashimoto, N.; Hara, T.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Green Chem. 2011, 13, 2416–2422. doi:10.1039/c1gc15146k
Return to citation in text: [1] -
Yoo, K. S.; Park, C. P.; Yoon, C. H.; Sakaguchi, S.; O’Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933–3935. doi:10.1021/ol701584f
Return to citation in text: [1] -
Andappan, M. M. S.; Nilsson, P.; Larhed, M. Chem. Commun. 2004, 218–219. doi:10.1039/b311492a
Return to citation in text: [1] -
Lindh, J.; Sävmarker, J.; Nilsson, P.; Sjöberg, P. J. R.; Larhed, M. Chem.–Eur. J. 2009, 15, 4630–4636. doi:10.1002/chem.200802744
Return to citation in text: [1] -
Odell, L. R.; Lindh, J.; Gustafsson, T.; Larhed, M. Eur. J. Org. Chem. 2010, 2270–2274. doi:10.1002/ejoc.201000063
Return to citation in text: [1] -
Nordqvist, A.; Björkelid, C.; Andaloussi, M.; Jansson, A. M.; Mowbray, S. L.; Karlén, A.; Larhed, M. J. Org. Chem. 2011, 76, 8986–8998. doi:10.1021/jo201715x
Return to citation in text: [1] -
Likhar, P. R.; Roy, M.; Roy, S.; Subhas, M. S.; Kantam, M. L.; Sreedhar, B. Adv. Synth. Catal. 2008, 350, 1968–1974. doi:10.1002/adsc.200800329
Return to citation in text: [1] -
Delcamp, J. H.; Brucks, A. P.; White, M. C. J. Am. Chem. Soc. 2008, 130, 11270–11271. doi:10.1021/ja804120r
Return to citation in text: [1] -
Leng, Y.; Yang, F.; Wei, K.; Wu, Y. Tetrahedron 2010, 66, 1244–1248. doi:10.1016/j.tet.2009.12.027
Return to citation in text: [1] -
Sakaguchi, S.; Yoo, K. S.; O’Neill, J.; Lee, J. H.; Stewart, T.; Jung, K. W. Angew. Chem., Int. Ed. 2008, 47, 9326–9329. doi:10.1002/anie.200803793
Return to citation in text: [1] -
Ruan, J.; Li, X.; Saidi, O.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 2424–2425. doi:10.1021/ja0782955
Return to citation in text: [1] -
Gottumukkala, A. L.; Teichert, J. F.; Heijnen, D.; Eisink, N.; Van Dijk, S.; Ferrer, C.; van den Hoogenband, A.; Minnaard, A. J. J. Org. Chem. 2011, 76, 3498–3501. doi:10.1021/jo101942f
Return to citation in text: [1] -
Li, T.; Qu, X.; Zhu, Y.; Sun, P.; Yang, H.; Shan, Y.; Zhang, H.; Liu, D.; Zhang, X.; Mao, J. Adv. Synth. Catal. 2011, 353, 2731–2738. doi:10.1002/adsc.201100238
Return to citation in text: [1] -
Werner, E. W.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 13981–13983. doi:10.1021/ja1060998
Return to citation in text: [1] -
Sun, P.; Zhu, Y.; Yang, H.; Yan, H.; Lu, L.; Zhang, X.; Mao, J. Org. Biomol. Chem. 2012, 10, 4512–4515. doi:10.1039/c2ob25462j
Return to citation in text: [1] -
Mino, T.; Koizumi, T.; Suzuki, S.; Hirai, K.; Kajiwara, K.; Sakamoto, M.; Fujita, T. Eur. J. Org. Chem. 2012, 678–680. doi:10.1002/ejoc.201101533
Return to citation in text: [1] -
Dubrovina, N. V.; Borner, A. Angew. Chem., Int. Ed. 2004, 43, 5883–5886. doi:10.1002/anie.200460848
Return to citation in text: [1] -
Ackermann, L. Isr. J. Chem. 2010, 50, 652–663. doi:10.1002/ijch.201000043
Return to citation in text: [1] -
Shaikh, T. M.; Weng, C.-M.; Hong, F.-E. Coord. Chem. Rev. 2012, 256, 771–803. doi:10.1016/j.ccr.2011.11.007
Return to citation in text: [1] -
Ackermann, L.; Kapdi, A. R.; Schulzke, C. Org. Lett. 2010, 12, 2298–2301. doi:10.1021/ol100658y
Return to citation in text: [1] -
Hu, D.-F.; Weng, C.-M.; Hong, F.-E. Organometallics 2011, 30, 1139–1147. doi:10.1021/om101132t
Return to citation in text: [1] -
Zhang, Y.; Xing, H.; Xie, W.; Wan, X.; Lai, Y.; Ma, D. Adv. Synth. Catal. 2013, 355, 68–72. doi:10.1002/adsc.201200782
Return to citation in text: [1]
| 48. | Ruan, J.; Li, X.; Saidi, O.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 2424–2425. doi:10.1021/ja0782955 |
| 49. | Gottumukkala, A. L.; Teichert, J. F.; Heijnen, D.; Eisink, N.; Van Dijk, S.; Ferrer, C.; van den Hoogenband, A.; Minnaard, A. J. J. Org. Chem. 2011, 76, 3498–3501. doi:10.1021/jo101942f |
| 50. | Li, T.; Qu, X.; Zhu, Y.; Sun, P.; Yang, H.; Shan, Y.; Zhang, H.; Liu, D.; Zhang, X.; Mao, J. Adv. Synth. Catal. 2011, 353, 2731–2738. doi:10.1002/adsc.201100238 |
| 51. | Werner, E. W.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 13981–13983. doi:10.1021/ja1060998 |
| 52. | Sun, P.; Zhu, Y.; Yang, H.; Yan, H.; Lu, L.; Zhang, X.; Mao, J. Org. Biomol. Chem. 2012, 10, 4512–4515. doi:10.1039/c2ob25462j |
| 53. | Mino, T.; Koizumi, T.; Suzuki, S.; Hirai, K.; Kajiwara, K.; Sakamoto, M.; Fujita, T. Eur. J. Org. Chem. 2012, 678–680. doi:10.1002/ejoc.201101533 |
| 40. | Andappan, M. M. S.; Nilsson, P.; Larhed, M. Chem. Commun. 2004, 218–219. doi:10.1039/b311492a |
| 41. | Lindh, J.; Sävmarker, J.; Nilsson, P.; Sjöberg, P. J. R.; Larhed, M. Chem.–Eur. J. 2009, 15, 4630–4636. doi:10.1002/chem.200802744 |
| 42. | Odell, L. R.; Lindh, J.; Gustafsson, T.; Larhed, M. Eur. J. Org. Chem. 2010, 2270–2274. doi:10.1002/ejoc.201000063 |
| 43. | Nordqvist, A.; Björkelid, C.; Andaloussi, M.; Jansson, A. M.; Mowbray, S. L.; Karlén, A.; Larhed, M. J. Org. Chem. 2011, 76, 8986–8998. doi:10.1021/jo201715x |
| 44. | Likhar, P. R.; Roy, M.; Roy, S.; Subhas, M. S.; Kantam, M. L.; Sreedhar, B. Adv. Synth. Catal. 2008, 350, 1968–1974. doi:10.1002/adsc.200800329 |
| 45. | Delcamp, J. H.; Brucks, A. P.; White, M. C. J. Am. Chem. Soc. 2008, 130, 11270–11271. doi:10.1021/ja804120r |
| 46. | Leng, Y.; Yang, F.; Wei, K.; Wu, Y. Tetrahedron 2010, 66, 1244–1248. doi:10.1016/j.tet.2009.12.027 |
| 47. | Sakaguchi, S.; Yoo, K. S.; O’Neill, J.; Lee, J. H.; Stewart, T.; Jung, K. W. Angew. Chem., Int. Ed. 2008, 47, 9326–9329. doi:10.1002/anie.200803793 |
| 1. | Douney, A. M.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2963. doi:10.1021/cr020039h |
| 2. | Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442–4489. doi:10.1002/anie.200500368 |
| 6. | Gurusamy, N.; Lekli, I.; Mukherjee, S.; Ray, D.; Ahsan, M. K.; Gherghiceanu, M.; Popescu, L. M.; Das, D. K. Cardiovasc. Res. 2010, 86, 103–112. doi:10.1093/cvr/cvp384 |
| 33. | Dieck, H. A.; Heck, R. F. J. Org. Chem. 1975, 40, 1083–1090. doi:10.1021/jo00896a020 |
| 34. | Hayashi, T.; Yamasaki, K. Chem. Rev. 2003, 103, 2829–2844. doi:10.1021/cr020022z |
| 35. | Itoh, T.; Mase, T.; Nishikata, T.; Iyama, T.; Tachikawa, H.; Kobayashi, Y.; Yamamoto, Y.; Miyaura, N. Tetrahedron 2006, 62, 9610–9621. doi:10.1016/j.tet.2006.07.075 |
| 36. | Vandyck, K.; Mattys, B.; Willen, M.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, J. Org. Lett. 2006, 8, 363–366. doi:10.1021/ol0522788 |
| 37. | Bazin, M.-A.; El Kihel, L.; Lancelot, J.-C.; Rault, S. Tetrahedron Lett. 2007, 48, 4347–4351. doi:10.1016/j.tetlet.2007.04.114 |
| 38. | Motokura, K.; Hashimoto, N.; Hara, T.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Green Chem. 2011, 13, 2416–2422. doi:10.1039/c1gc15146k |
| 5. | Karuppagounder, S. S.; Pinto, J. T.; Xu, H.; Chen, L.-H.; Beal, M. F.; Gibson, G. E. Neurochem. Int. 2009, 54, 111–118. doi:10.1016/j.neuint.2008.10.008 |
| 39. | Yoo, K. S.; Park, C. P.; Yoon, C. H.; Sakaguchi, S.; O’Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933–3935. doi:10.1021/ol701584f |
| 4. | Elmali, N.; Baysal, O.; Harma, A.; Esenkaya, I.; Mizrak, B. Inflammation 2007, 30, 1–6. doi:10.1007/s10753-006-9012-0 |
| 28. | Ackermann, L.; Potukuchi, H. K.; Kapdi, A. R.; Schulzke, C. Chem.–Eur. J. 2010, 16, 3300–3303. doi:10.1002/chem.201000032 |
| 29. | Li, G. Y.; Zheng, G.; Noonan, A. F. J. Org. Chem. 2001, 66, 8677–8681. doi:10.1021/jo010764c |
| 30. | Wolf, C.; Lerebours, R. J. Org. Chem. 2003, 68, 7077–7084. doi:10.1021/jo034758n |
| 31. | Punji, B.; Mague, J. T.; Balakrishna, M. S. Inorg. Chem. 2007, 46, 11316–11327. doi:10.1021/ic701674x |
| 3. | Jang, M.; Cai, L.; Udeani, G. O.; Slowing, K. V.; Thomas, C. F.; Beecher, C. W. W.; Fong, H. H. S.; Farnsworth, N. R.; Kinghorn, A. D.; Mehta, R. G.; Moon, R. C.; Pezzuto, J. M. Science 1997, 275, 218–220. doi:10.1126/science.275.5297.218 |
| 32. | Wei, C.-H.; Wu, C.-E.; Huang, Y.-L.; Kultyshev, R. G.; Hong, F.-E. Chem.–Eur. J. 2007, 13, 1583–1593. doi:10.1002/chem.200601051 |
| 12. | Crisp, G. T. Chem. Soc. Rev. 1998, 27, 427–436. doi:10.1039/a827427z |
| 13. | Link, J. T.; Overman, L. E. Intramolecular Heck Reactions in Natural Product Chemistry. In Metal-Catalyzed Cross-Coupling Reactions; Diedrich, F.; Stang, P. J., Eds.; Wiley-VCH: Weinheim, Germany, 1998. |
| 14. | Beletskaya, I.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048 |
| 15. | Liu, L.-j.; Wang, F.; Wang, W.; Zhao, M.-x.; Shi, M. Beilstein J. Org. Chem. 2011, 7, 555–564. doi:10.3762/bjoc.7.64 |
| 16. | Grasa, G. A.; Singh, R.; Stevens, E. D.; Nolan, S. P. J. Organomet. Chem. 2003, 687, 269–279. doi:10.1016/S0022-328X(03)00375-9 |
| 22. | Parshall, G. W.; Ittel, S. D. Homogeneous Catalysis; J. Wiley and Sons: New York, 1992. |
| 23. | Albéniz, A. C.; Carrera, N. Eur. J. Inorg. Chem. 2011, 2347–2360. doi:10.1002/ejic.201100162 |
| 58. | Hu, D.-F.; Weng, C.-M.; Hong, F.-E. Organometallics 2011, 30, 1139–1147. doi:10.1021/om101132t |
| 11. | Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024 |
| 24. | Li, G. Y. Angew. Chem., Int. Ed. 2001, 40, 1513–1516. doi:10.1002/1521-3773(20010417)40:8<1513::AID-ANIE1513>3.0.CO;2-C |
| 25. | Jiang, X.-b.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G. J. Org. Chem. 2004, 69, 2327–2331. doi:10.1021/jo035487j |
| 26. | Ackermann, L.; Born, R. Angew. Chem., Int. Ed. 2005, 44, 2444–2447. doi:10.1002/anie.200462371 |
| 27. | Xu, H.; Ekoue-Kovi, K.; Wolf, C. J. Org. Chem. 2008, 73, 7638–7650. doi:10.1021/jo801445y |
| 59. | Zhang, Y.; Xing, H.; Xie, W.; Wan, X.; Lai, Y.; Ma, D. Adv. Synth. Catal. 2013, 355, 68–72. doi:10.1002/adsc.201200782 |
| 10. | Mizoroki, T.; Mori, K.; Ozaki, A. Bull. Chem. Soc. Jpn. 1971, 44, 581. doi:10.1246/bcsj.44.581 |
| 54. | Dubrovina, N. V.; Borner, A. Angew. Chem., Int. Ed. 2004, 43, 5883–5886. doi:10.1002/anie.200460848 |
| 55. | Ackermann, L. Isr. J. Chem. 2010, 50, 652–663. doi:10.1002/ijch.201000043 |
| 56. | Shaikh, T. M.; Weng, C.-M.; Hong, F.-E. Coord. Chem. Rev. 2012, 256, 771–803. doi:10.1016/j.ccr.2011.11.007 |
| 7. | Tsuji, J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis; Wiley: Chichester, U.K., 1995. |
| 8. | Bräse, S.; de Meijere, A. Cross-Coupling of Organyl Halides with Alkenes: the Heck Reaction. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; Stang, P. J., Eds.; Wiley-VCH: Weinheim, Germany, 1998. |
| 9. | Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824–889. doi:10.1021/cr9003242 |
| 17. | Ozawa, F.; Kubo, A.; Hayashi, T. Chem. Lett. 1992, 2177–2180. doi:10.1246/cl.1992.2177 |
| 18. | Littke, A. F.; Fu, G. C. J. Org. Chem. 1999, 64, 10–11. doi:10.1021/jo9820059 |
| 19. | Stambuli, J. P.; Stauffer, S. R.; Shaughnessy, K. H.; Hartwig, J. F. J. Am. Chem. Soc. 2001, 123, 2677–2678. doi:10.1021/ja0058435 |
| 20. | Hansen, A. L.; Ebran, J.-P.; Ahlquist, M.; Norrby, P.-O.; Skrydstrup, T. Angew. Chem., Int. Ed. 2006, 45, 3349–3353. doi:10.1002/anie.200600442 |
| 21. | Fleckenstein, C. A.; Plenio, H. Chem. Soc. Rev. 2010, 39, 694–711. doi:10.1039/b903646f |
| 57. | Ackermann, L.; Kapdi, A. R.; Schulzke, C. Org. Lett. 2010, 12, 2298–2301. doi:10.1021/ol100658y |
© 2013 Shaikh and Hong; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)