Abstract
A silyl glyoxylate three-component-coupling methodology has been exploited to achieve a formal synthesis, an analogue to an intermediate in a distinct formal synthetic route, and a third (unique) approach to the natural product alternaric acid. Highlighted in this study is the versatility of silyl glyoxylates to engage a variety of nucleophile and electrophile pairs to provide wide latitude in the approach to complex molecule synthesis.

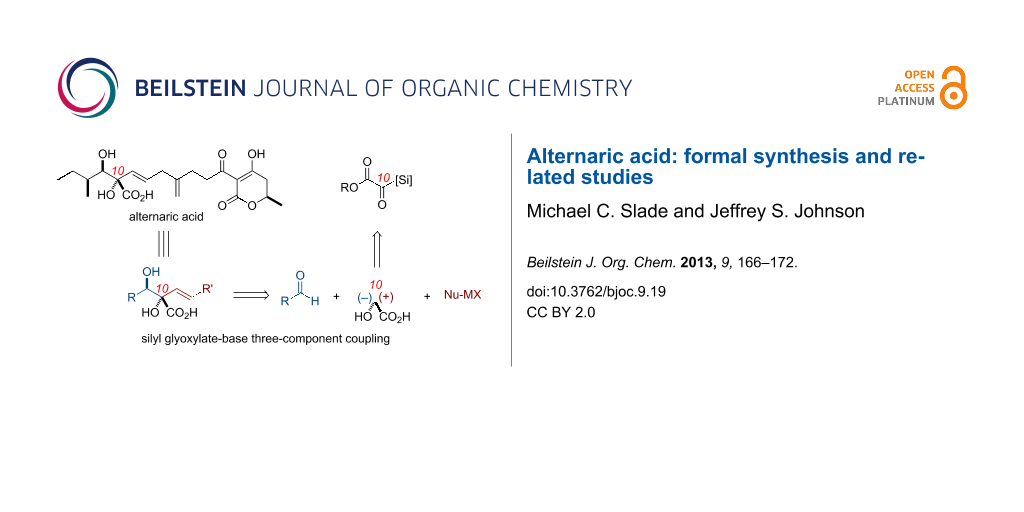
Graphical Abstract
Introduction
The rapid development of molecular complexity from simple starting materials is an important goal in modern synthetic organic chemistry. In this context, streamlined one-pot transformations, cascade reactions, and multicomponent couplings have emerged as enabling tools for the synthesis of complex molecules [1,2]. Our laboratory [3-16] and others have developed [17] and employed [18,19] silyl glyoxylates 1 in a variety of synthetic endeavors, both in natural-product synthesis and synthetic methodologies [20]. Key to their use in a variety of contexts is the ability of silyl glyoxylates to function as linchpin synthons for geminal coupling of nucleophile/electrophile pairs at a glycolic acid subunit (Scheme 1A), which allows the rapid build-up of molecular complexity. Alternaric acid (2) [21-23] is an antifungal and phytotoxic natural product, which bears a substituted glycolic acid in the functionally and stereochemically dense core of the molecule; the potential application of silyl glyoxylate technology emerged as an attractive starting point for synthetic planning (Scheme 1B). This paper summarizes our synthetic work in this arena, which culminated in a formal synthesis, an analogue of another formal synthesis, and a unique approach to the target; each of the routes was enabled by distinct coupling partners.
![[1860-5397-9-19-i1]](/bjoc/content/inline/1860-5397-9-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (A) Silyl glyoxylates as versatile reagents for three-component coupling reactions: representative nucleophiles and electrophiles. (B) Alternaric acid as a potential application of a silyl glyoxylate-enabled three-component coupling reaction.
Scheme 1: (A) Silyl glyoxylates as versatile reagents for three-component coupling reactions: representative ...
Alternaric acid is a particularly interesting target to demonstrate the utility of silyl glyoxylates, as it has been the subject of one total synthesis [24], one formal synthesis [25], and a potential application of an asymmetric glycolate aldol methodology [26]. These precedents serve as fruitful comparison points for application of a silyl glyoxylate three-component coupling methodology. Scheme 2 highlights three potential avenues toward the natural product and their precedents, by using the readily available (S)-2-methylbutanal (3) [27] and silyl glyoxylates 1 as two of the three key components for a coupling reaction.
![[1860-5397-9-19-i2]](/bjoc/content/inline/1860-5397-9-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Potential applications of silyl glyoxylate couplings and precedent synthetic intermediates toward the synthesis of alternaric acid.
Scheme 2: Potential applications of silyl glyoxylate couplings and precedent synthetic intermediates toward t...
Results and Discussion
Synthetic studies were initiated to explore paths a and b in Scheme 2, given the perceived rapidity with which the known intermediates could be intercepted after the proposed three-component coupling reactions. These initial studies revealed a limitation to this approach, inherent in the use of aldehyde 3 as a coupling partner: inherently poor Felkin–Anh facial selectivity with respect to the aldehyde electrophile due to minor differentiation between the Et/Me groups [28-30]. In all cases, the facial selectivity was rather poor, i.e., approximately 1.7:1, regardless of nucleophile, counterion, solvent, and temperature. Brief optimization efforts for each nucleophile thus focused on maximizing the coupling efficiency and syn-/anti-aldol selectivity (see Supporting Information File 1).
The optimal conditions for use of a vinyl nucleophile involved addition of a solution of vinylmagnesium bromide (4) and (−)-sparteine [31] in toluene to a solution of the tert-butyl silyl glyoxylate 1a and (S)-2-methylbutanal (3) in toluene at −78 °C followed by warming to room temperature, which provided the three-component-coupling product 5 with excellent (>20:1) syn-/anti-aldol selectivity in 65% yield (Scheme 3). Ichihara’s aldehyde intermediate could be intercepted in three additional steps, in high overall yield. Simultaneous cleavage of the silyl ether and transesterification from the tert-butyl to the methyl ester in 6 was effected by warming in acidic methanol. Subsequent acetonide formation provided 7, and ozonolysis afforded Ichihara’s aldehyde 8 (Scheme 3).
![[1860-5397-9-19-i3]](/bjoc/content/inline/1860-5397-9-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Three-component coupling with a vinyl nucleophile and elaboration to Ichihara’s aldehyde.
Scheme 3: Three-component coupling with a vinyl nucleophile and elaboration to Ichihara’s aldehyde.
Interception of this intermediate thus constituted a formal synthesis; the precedent for the C8–C9 olefination involved a classical, three-step Julia olefination sequence [24]. To demonstrate proof-of-concept for a more step-efficient endgame, test substrates were prepared for exploration of a modified Julia olefination [32]. As shown in Scheme 4, the phenyltetrazole heteroaromatic core in sulfones 9a and 9b provided excellent E-/Z- selectivity for formation of the C8–C9 olefin under typical modified Julia conditions with no optimization necessary. In particular, the vinyl bromide functional handle in 10b provides a potential avenue for elaboration to the natural product.
![[1860-5397-9-19-i4]](/bjoc/content/inline/1860-5397-9-19-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Modified Julia olefination as a step-efficient alternative endgame strategy.
Scheme 4: Modified Julia olefination as a step-efficient alternative endgame strategy.
With the promise of the approach thus demonstrated involving the use of the vinyl nucleophile, attention shifted toward exploration of the allyl nucleophile. The best conditions for the use of an allyl nucleophile involved the addition of allylzinc bromide (11) in THF to a THF solution of benzyl silyl glyoxylate 1b and (S)-2-methylbutanal (3) at 0 °C followed by warming to room temperature (Scheme 5). In the event, the three-component coupling product 12 was in 50% combined yield of all four possible diastereomers: 3.6:1 syn-/anti-selectivity and ~1.7:1 facial selectivity were observed. Thus, under these conditions both the control of enolate geometry as well as facial selectivity with respect to the aldehyde were incomplete. The four diastereomers could only be separated into syn/anti sets; within each set, the Felkin/anti-Felkin diastereomers could not be separated.
![[1860-5397-9-19-i5]](/bjoc/content/inline/1860-5397-9-19-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Three-component coupling with an allyl nucleophile and demonstration of successful ruthenium-catalyzed Alder–ene approach.
Scheme 5: Three-component coupling with an allyl nucleophile and demonstration of successful ruthenium-cataly...
As with three-component coupling product 5, advancement of intermediate 12 proved straightforward (Scheme 5). Deprotection of the silyl ether with TBAF afforded diol 13, which is a benzyl ester analogue of one of Trost’s substrates employed in the ruthenium-catalyzed Alder–ene reaction [25]. It too proved to be a successful substrate for the reaction with alkyne 14, affording the 1,4-diene product 15 in 52% yield. This sequence thus demonstrated a second avenue for successful exploitation of a silyl glyoxylate coupling methodology to achieve a step-efficient approach toward the assembly of the carbon skeleton of alternaric acid.
The two approaches described above both highlighted an important limitation to the use of (S)-2-methylbutanal (3) as the third component in the silyl glyoxylate-based three-component coupling reaction: while this aldehyde directly affords the substructure of the natural product target, it is unable to adequately control the facial selectivity of the approach of the glycolate enolate revealed after nucleophile addition/[1,2]-Brook [33] rearrangement. Moreover, attempts to achieve separation of the resultant diastereomers at all synthetic intermediates in these two routes were unsuccessful. Thus, attention shifted to address the stereochemical issue.
Various approaches were considered to achieve a higher level of stereoselection in the three-component coupling reaction, which are summarized in Scheme 6 [34]. In light of the elegant precedent for overriding the moderate substrate bias from (S)-2-methylbutanal (3) [26], auxiliary modification of the silyl glyoxylate structure to generalized type 1c could be envisioned. As hydrolysis of an ester would be required as a late-stage deprotection in any silyl glyoxylate-based approach, this modification would represent a relatively minor departure from ideality in the form of additional concession steps [35]. Alternatively, modification of the aldehyde partner, as in generalized type 16, was also considered. For this purpose, any stereocontrolling element (Ω or Ψ in aldehyde types 16a and 16b, respectively) employed should meet the additional requirement that it be easily converted to a simple ethyl group to minimize the number of concession steps.
![[1860-5397-9-19-i6]](/bjoc/content/inline/1860-5397-9-19-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Approaches considered to address the stereochemical issue.
Scheme 6: Approaches considered to address the stereochemical issue.
The auxiliary approach using silyl glyoxylates 1c ([Si] = TES or TBS, Scheme 6) proved to be suboptimal: despite the successful formation of the desired three-component coupling product, yields were low and poor stereochemical control was observed. Likewise, even the extreme steric demand of the tris(trimethylsilyl) group in aldehyde 16aa was insufficient for adequate stereocontrol in the three-component coupling reaction [34]. An additional branch point in the carbon backbone, such as in 16b, was deemed necessary. The 1,3-dithiane group in aldehyde 16ba was conceived as a promising candidate for a stereocontrolling element due to its large size and the wealth of precedents for single-step desulfurization to alkanes [36-40]. The racemic synthesis of the requisite aldehyde proved straightforward (see Supporting Information File 1). Most importantly, in initial three-component coupling reactions with vinyl nucleophile 4 and silyl glyoxylate 1a under previously optimized conditions, high efficiency was achieved along with excellent (>20:1) stereochemical control for the formation of three-component-coupling product 17 (Scheme 7) [41-44]. To verify that the dithiane was acting in the desired fashion, and to rule out chelation from one of the Lewis basic sulfur atoms, derivatization to a lactone was carried out. The dithiane was cleaved to the ketone 18, which underwent a 1,3-syn-selective reduction [45]. The resultant diol 19 was subjected to acidic conditions to effect cleavage of the tert-butyl ester and lactonization to afford 20. The NOESY and coupling-constant data of 20 was consistent with the role of the dithiane in 16ba as a nonchelating RL group that led to Felkin selectivity in the three-component coupling reaction.
![[1860-5397-9-19-i7]](/bjoc/content/inline/1860-5397-9-19-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Use of a dithiane moiety to excert stereochemical control in the three-component coupling reaction and supporting evidence for its nature as a nonchelating RL.
Scheme 7: Use of a dithiane moiety to excert stereochemical control in the three-component coupling reaction ...
The complete diastereochemical control exerted by the dithiane moiety of the aldehyde 16ba provided the impetus for exploring the use of a functionalized vinyl nucleophile in the three-component coupling reaction. Use of a more complex nucleophile would maintain the convergence of the overall synthesis, which was deemed important because (1) the route to the aldehyde component was becoming more involved; and (2) one or more additional steps for the removal of the directing group would be required. Thus, we developed a synthesis of a nucleophile that would allow the vast majority of the alternaric acid carbon skeleton to be installed through the three-component coupling reaction (Scheme 8). It began from the known allylic alcohol 21 [46], which was acetylated to afford ester 22 as prelude for reaction as a π-allyl electrophile with the Reformatsky reagent 23 derived from tert-butyl bromoacetate. The TMS-alkyne in 24 was deprotected with buffered TBAF to afford free alkyne 25, and the vinyl iodide 26 was generated by hydrozirconation/iodination of the free alkyne with Schwartz’s reagent [47]. The vinyl nucleophile 27 could be generated by Knochel’s Mg/I exchange [48] and employed successfully in the three-component-coupling reaction with silyl glyoxylate 1a and aldehyde 16ba to assemble 28, which contains the bulk of the carbon skeleton of alternaric acid. Remarkably, this highly convergent coupling allows the majority of the carbon backbone of the natural product to be assembled in a single complexity-building step.
![[1860-5397-9-19-i8]](/bjoc/content/inline/1860-5397-9-19-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of a vinyl iodide for nucleophile generation and its use in a three-component coupling reaction.
Scheme 8: Synthesis of a vinyl iodide for nucleophile generation and its use in a three-component coupling re...
With this gratifying result, a third distinct route to alternaric acid was enabled. Most importantly, this provides the first example of such a highly functionalized nucleophile being used in a silyl glyoxylate based three-component coupling reaction. Remaining tasks for the complete formation of the natural product include desulfurization [49], deprotection [50], and appendage of the pyrone moiety [24].
Conclusion
In conclusion, we have described the application of silyl glyoxylate three-component-coupling reactions as the central feature of three distinct approaches to the total synthesis of alternaric acid. By judicious choice of coupling partner and reaction conditions, it has been possible to achieve a formal synthesis, an analogous formal synthesis via an alternative route, and significant progress toward a third distinct route reliant on a highly functionalized nucleophile/electrophile combination for the construction of the majority of the natural product in a single step. In particular, this underscores the unique utility of silyl glyoxylates to serve as crucial linchpins for the coupling of a variety of nucleophile/electrophile pairs at a glycolic acid junction for the rapid development of molecular complexity.
Supporting Information
Contains additional tables and schemes for the three-component coupling reactions and approaches to address the stereochemical problem. Also contains experimental procedures, characterization, and spectral data.
| Supporting Information File 1: Additional data. | ||
| Format: PDF | Size: 7.6 MB | Download |
References
-
Nicolaou, K. C.; Hale, C. R. H.; Nilewski, C.; Ioannidou, H. A. Chem. Soc. Rev. 2012, 41, 5185–5238. doi:10.1039/c2cs35116a
Return to citation in text: [1] -
Rapid complexity generation in natural product total synthesis. Chem. Soc. Rev. 2009, 38, 2969–3276. doi:10.1039/b920767h
See the thematic issue for a recent compilation of reviews on this topic in the context of natural-products synthesis.
Return to citation in text: [1] -
Nicewicz, D. A.; Johnson, J. S. J. Am. Chem. Soc. 2005, 127, 6170–6171. doi:10.1021/ja043884l
Return to citation in text: [1] -
Linghu, X.; Satterfield, A. D.; Johnson, J. S. J. Am. Chem. Soc. 2006, 128, 9302–9303. doi:10.1021/ja062637+
Return to citation in text: [1] -
Nicewicz, D. A.; Brétéché, G.; Johnson, J. S.; Bryan, C.; Lautens, M. Org. Synth. 2008, 85, 278–286.
Return to citation in text: [1] -
Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Johnson, J. S. J. Am. Chem. Soc. 2008, 130, 17281–17283. doi:10.1021/ja808347q
Return to citation in text: [1] -
Greszler, S. N.; Johnson, J. S. Org. Lett. 2009, 11, 827–830. doi:10.1021/ol802828d
Return to citation in text: [1] -
Greszler, S. N.; Johnson, J. S. Angew. Chem., Int. Ed. 2009, 48, 3689–3691. doi:10.1002/anie.200900215
Return to citation in text: [1] -
Schmitt, D. C.; Johnson, J. S. Org. Lett. 2010, 12, 944–947. doi:10.1021/ol9029353
Return to citation in text: [1] -
Steward, K. M.; Johnson, J. S. Org. Lett. 2010, 12, 2864–2867. doi:10.1021/ol100996w
Return to citation in text: [1] -
Boyce, G. R.; Johnson, J. S. Angew. Chem., Int. Ed. 2010, 49, 8930–8933. doi:10.1002/anie.201003470
Return to citation in text: [1] -
Greszler, S. N.; Malinowski, J. T.; Johnson, J. S. J. Am. Chem. Soc. 2010, 132, 17393–17395. doi:10.1021/ja108848d
Return to citation in text: [1] -
Greszler, S. N.; Malinowski, J. T.; Johnson, J. S. Org. Lett. 2011, 13, 3206–3209. doi:10.1021/ol2011192
Return to citation in text: [1] -
Schmitt, D. C.; Lam, L.; Johnson, J. S. Org. Lett. 2011, 13, 5136–5139. doi:10.1021/ol202002r
Return to citation in text: [1] -
Boyce, G. R.; Liu, S.; Johnson, J. S. Org. Lett. 2012, 14, 652–655. doi:10.1021/ol2033527
Return to citation in text: [1] -
Schmitt, D. C.; Malow, E. J.; Johnson, J. S. J. Org. Chem. 2012, 77, 3246–3251. doi:10.1021/jo202679u
Return to citation in text: [1] -
Bolm, C.; Kasyan, A.; Heider, P.; Saladin, S.; Drauz, K.; Günther, K.; Wagner, C. Org. Lett. 2002, 4, 2265–2267. doi:10.1021/ol025911n
Return to citation in text: [1] -
Lettan, R. B.; Galliford, C. V.; Woodward, C. C.; Scheidt, K. A. J. Am. Chem. Soc. 2009, 131, 8805–8814. doi:10.1021/ja808811u
Return to citation in text: [1] -
Yao, M.; Lu, C. D. Org. Lett. 2011, 13, 2782–2785. doi:10.1021/ol201029b
Return to citation in text: [1] -
Boyce, G. R.; Greszler, S. N.; Johnson, J. S.; Linghu, X.; Malinowski, J. T.; Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Steward, K. M. J. Org. Chem. 2012, 77, 4503–4515. doi:10.1021/jo300184h
Perspective article.
Return to citation in text: [1] -
Brian, P. W.; Curtis, P. J.; Hemming, H. G.; Unwin, C. H.; Wright, J. M. Nature 1949, 164, 534. doi:10.1038/164534a0
Return to citation in text: [1] -
Brian, P. W.; Curtis, P. J.; Hemming, H. G.; Jefferys, E. G.; Unwin, C. H.; Wright, J. M. J. Gen. Microbiol. 1951, 5, 619–632. doi:10.1099/00221287-5-4-619
Return to citation in text: [1] -
Bartels-Keith, J. R. J. Chem. Soc. 1960, 1662–1665. doi:10.1039/JR9600001662
Return to citation in text: [1] -
Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. J. Org. Chem. 1994, 59, 4749–4759. doi:10.1021/jo00096a016
Return to citation in text: [1] [2] [3] -
Trost, B. M.; Probst, G. D.; Schoop, A. J. Am. Chem. Soc. 1998, 120, 9228–9236. doi:10.1021/ja981540n
Return to citation in text: [1] [2] -
Giampietro, N. C.; Kampf, J. W.; Wolfe, J. P. J. Am. Chem. Soc. 2009, 131, 12556–12557. doi:10.1021/ja905930s
Return to citation in text: [1] [2] -
Anelli, P. L.; Montanari, F.; Quici, S.; Nonoshita, K.; Yamamot, H. Org. Synth. 1990, 69, 212.
Return to citation in text: [1] -
Our results are consistent with the additions of nucleophiles to 3 (cf. [24-26]). For rare, successful examples of Me/Et differentiation, see [29,30].
Return to citation in text: [1] -
Evans, D. A.; Kozlowski, M. C.; Burgey, C. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 1997, 119, 7893–7894. doi:10.1021/ja971521y
Return to citation in text: [1] [2] -
Copeland, G. T.; Miller, S. J. J. Am. Chem. Soc. 2001, 123, 6496–6502. doi:10.1021/ja0108584
Return to citation in text: [1] [2] -
The precise role of the (−)-sparteine is uncertain at this time. It is possible that the Lewis basic nitrogens chelate the magnesium, facilitating the reversibility of the aldolization step leading to high syn-selectivity. See [20], footnote 42.
Return to citation in text: [1] -
Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H
Return to citation in text: [1] -
Brook, A. G. Acc. Chem. Res. 1974, 7, 77–84. doi:10.1021/ar50075a003
Return to citation in text: [1] -
The results of efforts with silylglyoxylates 1c, and other specific examples for the identities of Ω and Ψ, were also pursued. See Supporting Information File 1 for additional details.
Return to citation in text: [1] [2] -
Gaich, T.; Baran, P. S. J. Org. Chem. 2010, 75, 4657–4673. doi:10.1021/jo1006812
Return to citation in text: [1] -
Yang, T.-K.; Lee, D.-S.; Haas, J. Raney Nickel. e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: New York, N. Y., 2006. doi:10.1002/047084289X.rr001.pub2
Return to citation in text: [1] -
Back, T. G.; Baron, D. L.; Yang, K. J. Org. Chem. 1993, 58, 2407–2413. doi:10.1021/jo00061a011
Return to citation in text: [1] -
Chan, M. C.; Cheng, K. M.; Ho, K. M.; Ng, C. T.; Yam, T. M.; Wang, B. S. L.; Luh, T. Y. J. Org. Chem. 1988, 53, 4466–4471. doi:10.1021/jo00254a009
Return to citation in text: [1] -
Becker, S.; Fort, Y.; Vanderesse, R.; Caubere, P. Tetrahedron Lett. 1988, 29, 2963–2966. doi:10.1016/0040-4039(88)85058-5
Return to citation in text: [1] -
Becker, S.; Fort, Y.; Caubere, P. J. Org. Chem. 1990, 55, 6194–6198. doi:10.1021/jo00312a029
Return to citation in text: [1] -
Aldehyde 16ba has previously been used in aldol reactions; no comment on the diastereoselectivity was made, likely because it was immaterial in those applications (see [42,43]).
Return to citation in text: [1] -
Kim, H.; Baker, J. B.; Lee, S.-U.; Park, Y.; Bolduc, K. L.; Park, H.-B.; Dickens, M. G.; Lee, D.-S.; Kim, Y.; Kim, S. H.; Hong, J. J. Am. Chem. Soc. 2009, 131, 3192–3194. doi:10.1021/ja8101192
Return to citation in text: [1] [2] -
Kim, H.; Baker, J. B.; Park, Y.; Park, H. B.; DeArmond, P. D.; Kim, S. H.; Fitzgerald, M. C.; Lee, D.-S.; Hong, J. Chem.–Asian J. 2010, 5, 1902–1910. doi:10.1002/asia.201000147
Return to citation in text: [1] [2] -
Ward, D. E.; Kazemeini, A. J. Org. Chem. 2012, 77, 10789–10803. doi:10.1021/jo302142v
While this manuscript was in review, a thorough study of the stereochemical outcomes using structurally related aldehydes was reported.
Return to citation in text: [1] -
Chen, K.-M.; Gunderson, K. G.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Chem. Lett. 1987, 16, 1923–1926. doi:10.1246/cl.1987.1923
Return to citation in text: [1] -
Renaud, J.-L.; Aubert, C.; Malacria, M. Tetrahedron Lett. 1999, 40, 5015–5018. doi:10.1016/S0040-4039(99)00981-8
Return to citation in text: [1] -
Reichard, H. A.; Rieger, J. C.; Micalizio, G. C. Angew. Chem., Int. Ed. 2008, 47, 7837–7840. doi:10.1002/anie.200803031
Return to citation in text: [1] -
Ren, H.; Krasovskiy, A.; Knochel, P. Org. Lett. 2004, 6, 4215–4217. doi:10.1021/ol048363h
Return to citation in text: [1] -
Initial attempts to effect this transformation in a model system have demonstrated that this may not be as straightforward as anticipated. See Supporting Information File 1 for details.
Return to citation in text: [1] -
Likewise, this may prove more difficult than anticipated; acid-promoted hydrolysis of the tert-butyl esters and silyl ether was planned, due to the success of such a transformation with the simpler substrate 5, as in Scheme 3. However, substrate 28 tends to decompose when treated under a variety of acidic conditions.
Return to citation in text: [1]
| 50. | Likewise, this may prove more difficult than anticipated; acid-promoted hydrolysis of the tert-butyl esters and silyl ether was planned, due to the success of such a transformation with the simpler substrate 5, as in Scheme 3. However, substrate 28 tends to decompose when treated under a variety of acidic conditions. |
| 24. | Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. J. Org. Chem. 1994, 59, 4749–4759. doi:10.1021/jo00096a016 |
| 24. | Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. J. Org. Chem. 1994, 59, 4749–4759. doi:10.1021/jo00096a016 |
| 25. | Trost, B. M.; Probst, G. D.; Schoop, A. J. Am. Chem. Soc. 1998, 120, 9228–9236. doi:10.1021/ja981540n |
| 26. | Giampietro, N. C.; Kampf, J. W.; Wolfe, J. P. J. Am. Chem. Soc. 2009, 131, 12556–12557. doi:10.1021/ja905930s |
| 1. | Nicolaou, K. C.; Hale, C. R. H.; Nilewski, C.; Ioannidou, H. A. Chem. Soc. Rev. 2012, 41, 5185–5238. doi:10.1039/c2cs35116a |
| 2. |
Rapid complexity generation in natural product total synthesis. Chem. Soc. Rev. 2009, 38, 2969–3276. doi:10.1039/b920767h
See the thematic issue for a recent compilation of reviews on this topic in the context of natural-products synthesis. |
| 20. |
Boyce, G. R.; Greszler, S. N.; Johnson, J. S.; Linghu, X.; Malinowski, J. T.; Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Steward, K. M. J. Org. Chem. 2012, 77, 4503–4515. doi:10.1021/jo300184h
Perspective article. |
| 25. | Trost, B. M.; Probst, G. D.; Schoop, A. J. Am. Chem. Soc. 1998, 120, 9228–9236. doi:10.1021/ja981540n |
| 18. | Lettan, R. B.; Galliford, C. V.; Woodward, C. C.; Scheidt, K. A. J. Am. Chem. Soc. 2009, 131, 8805–8814. doi:10.1021/ja808811u |
| 19. | Yao, M.; Lu, C. D. Org. Lett. 2011, 13, 2782–2785. doi:10.1021/ol201029b |
| 17. | Bolm, C.; Kasyan, A.; Heider, P.; Saladin, S.; Drauz, K.; Günther, K.; Wagner, C. Org. Lett. 2002, 4, 2265–2267. doi:10.1021/ol025911n |
| 24. | Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. J. Org. Chem. 1994, 59, 4749–4759. doi:10.1021/jo00096a016 |
| 3. | Nicewicz, D. A.; Johnson, J. S. J. Am. Chem. Soc. 2005, 127, 6170–6171. doi:10.1021/ja043884l |
| 4. | Linghu, X.; Satterfield, A. D.; Johnson, J. S. J. Am. Chem. Soc. 2006, 128, 9302–9303. doi:10.1021/ja062637+ |
| 5. | Nicewicz, D. A.; Brétéché, G.; Johnson, J. S.; Bryan, C.; Lautens, M. Org. Synth. 2008, 85, 278–286. |
| 6. | Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Johnson, J. S. J. Am. Chem. Soc. 2008, 130, 17281–17283. doi:10.1021/ja808347q |
| 7. | Greszler, S. N.; Johnson, J. S. Org. Lett. 2009, 11, 827–830. doi:10.1021/ol802828d |
| 8. | Greszler, S. N.; Johnson, J. S. Angew. Chem., Int. Ed. 2009, 48, 3689–3691. doi:10.1002/anie.200900215 |
| 9. | Schmitt, D. C.; Johnson, J. S. Org. Lett. 2010, 12, 944–947. doi:10.1021/ol9029353 |
| 10. | Steward, K. M.; Johnson, J. S. Org. Lett. 2010, 12, 2864–2867. doi:10.1021/ol100996w |
| 11. | Boyce, G. R.; Johnson, J. S. Angew. Chem., Int. Ed. 2010, 49, 8930–8933. doi:10.1002/anie.201003470 |
| 12. | Greszler, S. N.; Malinowski, J. T.; Johnson, J. S. J. Am. Chem. Soc. 2010, 132, 17393–17395. doi:10.1021/ja108848d |
| 13. | Greszler, S. N.; Malinowski, J. T.; Johnson, J. S. Org. Lett. 2011, 13, 3206–3209. doi:10.1021/ol2011192 |
| 14. | Schmitt, D. C.; Lam, L.; Johnson, J. S. Org. Lett. 2011, 13, 5136–5139. doi:10.1021/ol202002r |
| 15. | Boyce, G. R.; Liu, S.; Johnson, J. S. Org. Lett. 2012, 14, 652–655. doi:10.1021/ol2033527 |
| 16. | Schmitt, D. C.; Malow, E. J.; Johnson, J. S. J. Org. Chem. 2012, 77, 3246–3251. doi:10.1021/jo202679u |
| 32. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H |
| 26. | Giampietro, N. C.; Kampf, J. W.; Wolfe, J. P. J. Am. Chem. Soc. 2009, 131, 12556–12557. doi:10.1021/ja905930s |
| 28. | Our results are consistent with the additions of nucleophiles to 3 (cf. [24-26]). For rare, successful examples of Me/Et differentiation, see [29,30]. |
| 29. | Evans, D. A.; Kozlowski, M. C.; Burgey, C. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 1997, 119, 7893–7894. doi:10.1021/ja971521y |
| 30. | Copeland, G. T.; Miller, S. J. J. Am. Chem. Soc. 2001, 123, 6496–6502. doi:10.1021/ja0108584 |
| 42. | Kim, H.; Baker, J. B.; Lee, S.-U.; Park, Y.; Bolduc, K. L.; Park, H.-B.; Dickens, M. G.; Lee, D.-S.; Kim, Y.; Kim, S. H.; Hong, J. J. Am. Chem. Soc. 2009, 131, 3192–3194. doi:10.1021/ja8101192 |
| 43. | Kim, H.; Baker, J. B.; Park, Y.; Park, H. B.; DeArmond, P. D.; Kim, S. H.; Fitzgerald, M. C.; Lee, D.-S.; Hong, J. Chem.–Asian J. 2010, 5, 1902–1910. doi:10.1002/asia.201000147 |
| 25. | Trost, B. M.; Probst, G. D.; Schoop, A. J. Am. Chem. Soc. 1998, 120, 9228–9236. doi:10.1021/ja981540n |
| 31. | The precise role of the (−)-sparteine is uncertain at this time. It is possible that the Lewis basic nitrogens chelate the magnesium, facilitating the reversibility of the aldolization step leading to high syn-selectivity. See [20], footnote 42. |
| 24. | Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. J. Org. Chem. 1994, 59, 4749–4759. doi:10.1021/jo00096a016 |
| 29. | Evans, D. A.; Kozlowski, M. C.; Burgey, C. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 1997, 119, 7893–7894. doi:10.1021/ja971521y |
| 30. | Copeland, G. T.; Miller, S. J. J. Am. Chem. Soc. 2001, 123, 6496–6502. doi:10.1021/ja0108584 |
| 21. | Brian, P. W.; Curtis, P. J.; Hemming, H. G.; Unwin, C. H.; Wright, J. M. Nature 1949, 164, 534. doi:10.1038/164534a0 |
| 22. | Brian, P. W.; Curtis, P. J.; Hemming, H. G.; Jefferys, E. G.; Unwin, C. H.; Wright, J. M. J. Gen. Microbiol. 1951, 5, 619–632. doi:10.1099/00221287-5-4-619 |
| 23. | Bartels-Keith, J. R. J. Chem. Soc. 1960, 1662–1665. doi:10.1039/JR9600001662 |
| 27. | Anelli, P. L.; Montanari, F.; Quici, S.; Nonoshita, K.; Yamamot, H. Org. Synth. 1990, 69, 212. |
| 20. |
Boyce, G. R.; Greszler, S. N.; Johnson, J. S.; Linghu, X.; Malinowski, J. T.; Nicewicz, D. A.; Satterfield, A. D.; Schmitt, D. C.; Steward, K. M. J. Org. Chem. 2012, 77, 4503–4515. doi:10.1021/jo300184h
Perspective article. |
| 35. | Gaich, T.; Baran, P. S. J. Org. Chem. 2010, 75, 4657–4673. doi:10.1021/jo1006812 |
| 34. | The results of efforts with silylglyoxylates 1c, and other specific examples for the identities of Ω and Ψ, were also pursued. See Supporting Information File 1 for additional details. |
| 26. | Giampietro, N. C.; Kampf, J. W.; Wolfe, J. P. J. Am. Chem. Soc. 2009, 131, 12556–12557. doi:10.1021/ja905930s |
| 48. | Ren, H.; Krasovskiy, A.; Knochel, P. Org. Lett. 2004, 6, 4215–4217. doi:10.1021/ol048363h |
| 49. | Initial attempts to effect this transformation in a model system have demonstrated that this may not be as straightforward as anticipated. See Supporting Information File 1 for details. |
| 46. | Renaud, J.-L.; Aubert, C.; Malacria, M. Tetrahedron Lett. 1999, 40, 5015–5018. doi:10.1016/S0040-4039(99)00981-8 |
| 47. | Reichard, H. A.; Rieger, J. C.; Micalizio, G. C. Angew. Chem., Int. Ed. 2008, 47, 7837–7840. doi:10.1002/anie.200803031 |
| 41. | Aldehyde 16ba has previously been used in aldol reactions; no comment on the diastereoselectivity was made, likely because it was immaterial in those applications (see [42,43]). |
| 42. | Kim, H.; Baker, J. B.; Lee, S.-U.; Park, Y.; Bolduc, K. L.; Park, H.-B.; Dickens, M. G.; Lee, D.-S.; Kim, Y.; Kim, S. H.; Hong, J. J. Am. Chem. Soc. 2009, 131, 3192–3194. doi:10.1021/ja8101192 |
| 43. | Kim, H.; Baker, J. B.; Park, Y.; Park, H. B.; DeArmond, P. D.; Kim, S. H.; Fitzgerald, M. C.; Lee, D.-S.; Hong, J. Chem.–Asian J. 2010, 5, 1902–1910. doi:10.1002/asia.201000147 |
| 44. |
Ward, D. E.; Kazemeini, A. J. Org. Chem. 2012, 77, 10789–10803. doi:10.1021/jo302142v
While this manuscript was in review, a thorough study of the stereochemical outcomes using structurally related aldehydes was reported. |
| 45. | Chen, K.-M.; Gunderson, K. G.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Chem. Lett. 1987, 16, 1923–1926. doi:10.1246/cl.1987.1923 |
| 34. | The results of efforts with silylglyoxylates 1c, and other specific examples for the identities of Ω and Ψ, were also pursued. See Supporting Information File 1 for additional details. |
| 36. | Yang, T.-K.; Lee, D.-S.; Haas, J. Raney Nickel. e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: New York, N. Y., 2006. doi:10.1002/047084289X.rr001.pub2 |
| 37. | Back, T. G.; Baron, D. L.; Yang, K. J. Org. Chem. 1993, 58, 2407–2413. doi:10.1021/jo00061a011 |
| 38. | Chan, M. C.; Cheng, K. M.; Ho, K. M.; Ng, C. T.; Yam, T. M.; Wang, B. S. L.; Luh, T. Y. J. Org. Chem. 1988, 53, 4466–4471. doi:10.1021/jo00254a009 |
| 39. | Becker, S.; Fort, Y.; Vanderesse, R.; Caubere, P. Tetrahedron Lett. 1988, 29, 2963–2966. doi:10.1016/0040-4039(88)85058-5 |
| 40. | Becker, S.; Fort, Y.; Caubere, P. J. Org. Chem. 1990, 55, 6194–6198. doi:10.1021/jo00312a029 |
© 2013 Slade and Johnson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)