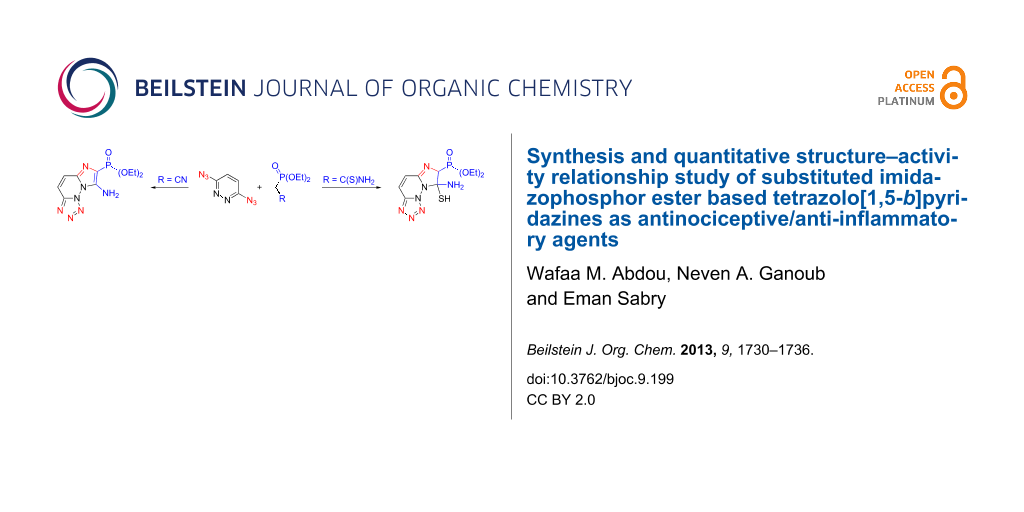
Abstract
A high-yielding general synthesis of imidazophosphor ester based tetrazolo[1,5-b]pyridazines is described. A conjugated reaction between 3,6-diazidopyridazine and different types of phosphonyl carbanion reagents followed by intramolecular cyclization afforded the target products, by using sodium ethanolate solution as a reaction medium. Among the products, five compounds, at a dose of 50 mg per kilogram body weight, showed a notable antinociceptive and anti-inflammatory activity without toxic side-effects.

Graphical Abstract
Introduction
Inflammation is a characteristic feature of disease pathology and progression in several neuro-degenerative disorders and physical functioning [1,2]. Recently, nonsteroidal anti-inflammatory drugs (NSAIDs) are well established for the treatment of inflammatory disorders [3-5]. The anti-inflammatory effect of NSAIDs is mainly based on the inhibition of the cyclooxygenase (COX) enzymes. Later on, it was reported that the second isoform of cyclooxygenase (COX-2) has a better effect on the inflammation with fewer side-effects [6-9]. Despite their widespread use, none of the presently available agents is ideal; each has its own shortcomings [3]. Subsequently, to improve the efficacy/safety profile of new NSAIDs, the structural–activity relationship (SAR) has been extensively studied, taking into account up-to-date knowledge about the mechanism of inflammation that balanced the inhibition of COX-1, COX-2, and lipoxygenase (LOX) [10-12].
As a part of our continued interest in the development of convenient synthetic approaches to β-enamino- and α-aminophosphonates with anti-inflammatory properties [13-20], we recently successfully synthesized a series of mono- and bisphosphonate-based tetrazolo[1,5-a]quinolines with marked anti-inflammatory properties [21,22]. Following this, synthesis of the target compounds, substituted tetrazolo[1,5-b]pyridazinphosphor esters, is described herein. In this context, we applied different types of phosphonyl carbanion reagents to 3,6-diazidopyridazine (1) as an adopted substrate. The anti-inflammatory and the antinociceptive properties of the prepared compounds were screened and the structure–activity relationships were studied. The anti-inflammatory properties of many tetrazole [21-23] and pyridazine derivatives have also led to their clinical application as NSAIDs (e.g. Bucolome) [24]. Several phosphonate derivatives also exhibit marked potency as inhibitors of COX-1 and COX-2 and are therefore believed to be useful as anti-inflammatory drugs [25,26]. Thus, we considered that it is of interest to gather these three motifs in one molecule.
Results and Discussion
It has been reported that 3,6-diazidopyridazine presents azido (1a) ![[Graphic 1]](/bjoc/content/inline/1860-5397-9-199-i8.svg?max-width=637&scale=1.18182) tetrazolo tautomerism (1b) whereby it is mainly in the tetrazole form 1b [27-29]. Accordingly, and as indicated from the spectral data of our products, we considered that the substrate 3,6-diazidopyridazine (1a) is exclusively, throughout our investigation, reacted in the tetrazolo-form 1b [28,29] (Scheme 1). The required [3,6-diazidopyridazine (1a) 6-azidotetrazolo[1,5-b]pyridazine (1b)] was prepared by treating the readily available 3,6-dichloropyridazine with sodium azide [30]. The reactions studied and the products obtained are depicted in Schemes 1–7 (see below). Reaction of 1b with 1.3 equivalents of methyl diethyl phosphonoacetate (2a) or the ethyl analogue 2b in sodium ethanolate solution resulted in the same product assigned as diethyl 8-oxo-7,8-dihydroimidazo[1,2-f]tetrazolo[1,5-b]pyridazin-7-ylphosphonate (4, 78%) as indicated from the analytical and the spectroscopic data.
tetrazolo tautomerism (1b) whereby it is mainly in the tetrazole form 1b [27-29]. Accordingly, and as indicated from the spectral data of our products, we considered that the substrate 3,6-diazidopyridazine (1a) is exclusively, throughout our investigation, reacted in the tetrazolo-form 1b [28,29] (Scheme 1). The required [3,6-diazidopyridazine (1a) 6-azidotetrazolo[1,5-b]pyridazine (1b)] was prepared by treating the readily available 3,6-dichloropyridazine with sodium azide [30]. The reactions studied and the products obtained are depicted in Schemes 1–7 (see below). Reaction of 1b with 1.3 equivalents of methyl diethyl phosphonoacetate (2a) or the ethyl analogue 2b in sodium ethanolate solution resulted in the same product assigned as diethyl 8-oxo-7,8-dihydroimidazo[1,2-f]tetrazolo[1,5-b]pyridazin-7-ylphosphonate (4, 78%) as indicated from the analytical and the spectroscopic data.
In the 1H NMR spectrum (CDCl3) of 4 (δP = 28.4 ppm) there is a doublet peak (2JP–H = 22.3 Hz) at δ 5.36 ppm corresponding to H(4) of the imidazole ring while its C(4)–P appeared at δC = 59.4 (d, 1JP–C = 148.6 Hz). A plausible mechanism for the formation of tetrazolopyridazinophosphonate 4 is presented in Scheme 1. Upon heating, the equilibrium between the azido-1a and its isomeric tetrazolo-1b form [1a 1b] lies exclusively at the tetrazole isomer 1b [29]. Compound 1b is then intercepted by the nucleophilic attack of the phosphonyl carbanion 2a or 2b on the azido-function in 1b yielding the phosphonate intermediate 3 along with the evolution of a nitrogen molecule. Subsequent intramolecular ring closure, the fused imidazolo-phosphonate 4 would be obtained under elimination of an appropriate alcohol moiety.
![[1860-5397-9-199-i1]](/bjoc/content/inline/1860-5397-9-199-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of substituted diethyl oxophosphonate 4.
Scheme 1: Synthesis of substituted diethyl oxophosphonate 4.
In the same fashion, the substrate 1b reacted with the phosphonyl carbanion, diethyl cyanomethylphosphonate 5, in ethanolate solution to yield diethyl 8-aminoimidazo[1,2-f]tetrazolo[1,5-b]pyridazin-7-ylphosphonate (7) in 74% yield. The IR spectrum of the phosphonate 7 (δP = 27.8 ppm) showed the NH2, P=O, and P–O–C motifs at ν 3377–3330, 1226, and 1123 cm−1, respectively. Its 1H NMR (CDCl3) spectrum showed the NH2-protons at δ 6.44 (HA, br) and 8.88 ppm (HB, br), which are attributed to the P=O bonding with one proton of the amino-group. Furthermore, the 13C NMR spectrum of 7 revealed, among others, three doublets at: δ 153.6 [d, 3JP–C = 11.4 Hz, C(2)], 141.2 (d, 1JP–C = 188.4 Hz, C(4)–P), and at δ 126.4 (d, 2JP–C = 14.6 Hz, C(5)–NH2) ppm. As displayed in Scheme 2, the formation of the fused imidazophosphonate 7 was formed via the initial condensation intermediate 6. Further alkaline hydrolysis of the cyano group and the intramolecular cyclization led to the product 7 (Scheme 2).
![[1860-5397-9-199-i2]](/bjoc/content/inline/1860-5397-9-199-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of substituted diethyl aminophosphonate 7.
Scheme 2: Synthesis of substituted diethyl aminophosphonate 7.
Conversely, similar treatment of 1b with diethyl (methylthioalkyl)phosphonates 8a and 8b under the same reaction conditions yielded the fused diazaphospholo-substituted compounds 10a and 10b in 72 and 74% yield, respectively. The 31P NMR spectrum (CDCl3) of the diazaphospholes 10a and10b showed a sharp singlet around 14.5 ppm, which is within the range expected for the assigned structure [31].
A mechanism for the formation of the heterophosphole structure 10 can be rationalized as in Scheme 3 through the condensation of 1b with 8a or 8b to elaborate the intermediates 9a or 9b accompanied by the elimination of a N2 molecule. Further intramolecular cyclization of 9 afforded the diazaphospholes 10a and 10b, respectively, through the loss of an ethanol molecule [31] (Scheme 3).
![[1860-5397-9-199-i3]](/bjoc/content/inline/1860-5397-9-199-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of fused diazaphospholo-substituted compounds 10a, 10b.
Scheme 3: Synthesis of fused diazaphospholo-substituted compounds 10a, 10b.
Next, the fused imidazophosphono-substituted compound 13 (68% yield) was obtained from the reaction of 1b with diethyl (2-amino-2-thioxoethyl)phosphonate (11) in ethanolate solution. Obviously, 13 resulted in the same manner from the intermediate 12 initially formed, as outlined in Scheme 4.
![[1860-5397-9-199-i4]](/bjoc/content/inline/1860-5397-9-199-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of fused imidazophosphono-substituted compound 13.
Scheme 4: Synthesis of fused imidazophosphono-substituted compound 13.
Further, the azidotetrazole 1b was allowed to react with the Horner–Emmons (HE) reactant, tetraethyl methylenebis(phosphonate) (14) under the same reaction conditions to give the respective β-enaminobisphosphonate 15 (≈73% yield). The gem-diphosphonate structure 15 was delineated from IR, NMR and MS spectra. The IR absorptions for 15 showed the 2 P=O stretching frequencies as two bands at 1262 (P=O, free) and 1226 (P=O, bonded) cm−1, which could be explained by a preferred conformation of intramolecular hydrogen bonding between the NH proton and one of the P=O moieties. The 31P NMR spectrum (CDCl3) of 15 showed the presence of two separate doublets with equal 2JP–P coupling constants 28.4 Hz at δ 25.6 and 24.8 ppm. The 1H and 13C NMR data were also in accordance with the assigned structure (see Supporting Information File 1). The formation of 15 can be rationalized as occurring in Scheme 5 through the addition of 14 to the azido group with concomitant evolution of a molecule of N2 (Scheme 5). Bisphosphonates belong to an important class of BP-drugs used for the treatment of bone diseases such as osteoporosis, hypocalcemia, inflammation and rheumatoid arthritis [32,33].
![[1860-5397-9-199-i5]](/bjoc/content/inline/1860-5397-9-199-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of β-enaminobisphosphonate 15.
Scheme 5: Synthesis of β-enaminobisphosphonate 15.
Finally, we studied the behavior of the azidotetrazole 1b toward the unsaturated HE reactant, diethyl vinylphosphonate (16), and diethyl 2-methylallylphosphonate (18) in EtONa solution to give the fused imidazophosphono-substituted compounds 17 (76%) and 19 (74%). Obviously, according to Scheme 6, the coupling cyclization reaction of 1b with 16 and auto-oxidation resulted in formation of the final product 17 in one step. This behavior is familiar in similar reactions. On the other hand, we presume that the allyl reagent 18, which could be present in the isomeric forms 18a ![[Graphic 2]](/bjoc/content/inline/1860-5397-9-199-i9.svg?max-width=637&scale=1.18182) 18b (Scheme 7), had been conjugated in 18b form with 1b to give the phosphonate 19.
18b (Scheme 7), had been conjugated in 18b form with 1b to give the phosphonate 19.
![[1860-5397-9-199-i6]](/bjoc/content/inline/1860-5397-9-199-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of fused imidazophosphono-substituted compounds 17 and 19.
Scheme 6: Synthesis of fused imidazophosphono-substituted compounds 17 and 19.
![[1860-5397-9-199-i7]](/bjoc/content/inline/1860-5397-9-199-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Isomeric forms of diethyl 2-methylallylphosphonate (18).
Scheme 7: Isomeric forms of diethyl 2-methylallylphosphonate (18).
In summary, it has been found that the substrate 3,6-diazidopyridazine reacts with nucleophilic phosphorus reagents, HE reactants, mainly in the tetrazole-form leading to the formation of tetrazolopyridazino-imidazophosphor esters or β-enaminophosphor esters.
Biological assays
Based on previous reports [24-26] that recognized the pyridazine nucleus is being suitable for anti-inflammatory and antinociceptive agents, and by the fact that ring-fused heterocycles containing more than one nitrogen atom (e.g., tetrazole nuclei [21-23]) are key structures in a large variety of biochemical processes, bioscreening of the synthesized substituted tetrazolo[1,5-b]pyridazine-phosphor derivatives was carried out. Thus, keeping the tetrazolopyridazine core structure intact, we studied the effect of different phosphorus-containing moieties on their antinociceptive and anti-inflammatory effects. Substrate 1 was also tested to reflect the effect of its transformations to our products.
Antinociceptive evaluation para-Benzoquinone (p-BQ)-induced writhing test
The evaluation of antinociceptive activity of the synthesized compounds was assessed in vivo in mice by using the p-benzoquinone-induced writhing test [26]. Ibuprofen was used as a positive control in our experiments; the antinociceptive capacity was expressed as the percentage change compared to writhing controls. The results shown in Table 1 indicated that β-enaminobisphosphonate 15 is the most potent antinociceptive structure (86.4%), which was followed by α-aminophosphonates 13 (84%) and 7 (81%). Indeed, while the azido substrate 1 showed only a weak antinociceptive effect (14%), the eight phosphor compounds demonstrated higher capacity than the reference ibuprofen drug (69.9%) at the same dose of 50 mg per kilogram body weight.
Table 1: Antinociceptive/anti-inflammatory effects of the tested compounds on the p-BQ-induced abdominal constriction test and carrageenan (CG)-induced hind paw edema model in mice.a
|
tested
compound |
no. of writhings ± SEM
(antinociceptive effect, %b) |
Swelling in thickness (× 10−2 mm) ± SEM
(inhibition of edema, %)c |
anti-inflammatory activity after 360 min, % | |||
|---|---|---|---|---|---|---|
| 90 min | 180 min | 270 min | 360 min | |||
| control | 28.6 ± 2.4 | 41.4 ± 3.3 | 55.2 ± 2.7 | 74.8 ± 3.6 | 78.2 ± 4.1 | — |
| 4d |
6.8 ± 2.2
(76.2)* |
30.2 ± 3.8
(27.1)* |
28.9 ± 4.6
(47.6)** |
37.4 ± 5.7
(50.0)** |
31.3 ± 5.6
(60)*** |
(96.8)** |
| 7d |
5.4 ± 4.3
(81.1)** |
31.8 ± 5.3
(23.2)* |
25.1 ± 5.6
(54.5)* |
33.8 ± 3.4
(54.8)*** |
27.4 ± 6.4
(65.0)* |
104.8 |
| 10ad |
5.8 ± 2.9
(79.7)*** |
30.6 ± 6.3
(26.1)** |
26.3 ± 5.6
(52.4)** |
34.6 ± 3.6
(53.7)*** |
27.9 ± 4.3
(64.3)*** |
103.7 |
| 10bd |
6.3 ± 2.9
(77.9)* |
30.8 ± 3.8
(25.6)* |
27.1 ± 4.7
(50.9)* |
35.6 ± 6.0
(52.4)*** |
28.8 ± 4.8
(63.2)*** |
102 |
| 13d |
4.4 ± 2.2
(84.6)* |
30.7 ± 3.3
(25.8)** |
23.4 ± 5.6
(57.6)** |
33.2 ± 3.6
(55.6)*** |
25.8 ± 4.3
(67.0)*** |
108 |
| 15d |
3.9 ± 2.1
(86.4)** |
28.9 ± 5.2
(30.2)*** |
21.6 ± 7.4
(60.9)*** |
28.2 ± 5.0
(62.3)*** |
22.8 ± 6.7
(70.8)*** |
114 |
| 17d |
7.6 ± 1.7
(73.4)*** |
30.4 ± 7.2
(26.5)*** |
29.3 ± 4.1
(46.9)** |
39.1 ± 4.2
(47.7)*** |
30.7 ± 4.5
(60.7)*** |
97.9 |
| 19d |
7.2 ± 2.1
(74.8)*** |
29.7 ± 4.6
(28.0)* |
28.7 ± 5.6
(48.0)** |
38.5 ± 5.2
(48.6)*** |
31.9 ± 3.8
(59.2)*** |
95.5 |
| 1 |
24.6 ± 2.3
(14.0)** |
36.8 ± 6.9
(11.1)** |
44.6 ± 5.4
(19.2)** |
56.8 ± 5.5
(24.0)** |
62.4 ± 3.8
(20.2)*** |
32.6 |
| ibuprofen |
8.7 ± 3.1
(69.6)*** |
|||||
| indomethacin |
34.6 ± 4.6
(16.4)** |
32.4 ± 4.4
(41.3)* |
31.4 ± 1.6
(58.02)*** |
29.8 ± 3.4
(62.0)*** |
100 | |
aData obtained from animal experiments are expressed as means ± SEM (dose = 50 mg per kilogram body weight, administered subcutaneously to mice (n = 6–8). bp-BQ-induced writhing; cCG-induced paw edema tests, respectively. dStatistical significance was evaluated from the control by one-way ANOVA post hoc Dunnett’s test (*p < 0.05, **p < 0.01, ***p < 0.001).
Anti-inflammatory screening
Carrageenan-induced hind paw edema test
Anti-inflammation properties of the new tetrazolo[1,5-b]pyridazines-bearing mono- (4, 7, 10a, 10b, 13, 17, 19) and diphosphonate nuclei (15) were evaluated in an animal model, by the carrageenan–induced paw edema (CPE) method. Following the standard procedures [34,35], these compounds were administered subcutaneously by using 50 mg per kilogram body weight, and the anti-inflammatory effect was measured at successive time intervals (90, 180, 270, and 360 min, after carrageenan injection). The results are profiled in Table 1 and are compared to the substrate 1 and to indomethacin (A). β-Enaminobisphosphonate 15 showed the most potent inflammatory properties (e.g., 114% after 360 min) relative to indomethacin. Nevertheless, other compounds displayed good to excellent effects (95 to 108%) in inflammation inhibition after 360 min comparing to A without toxic effects. Percentage inhibition of granuloma for the tested compounds at a dose of 50 mg per kilogram body weight at successive intervals is displayed in Figure 1.
![[1860-5397-9-199-1]](/bjoc/content/figures/1860-5397-9-199-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Percentage inhibition of granuloma for the tested compounds at a dose of 50 mg per kilogram body weight after the given time intervals. Error bar: 5%.
Figure 1: Percentage inhibition of granuloma for the tested compounds at a dose of 50 mg per kilogram body we...
According to the results of the biological assay in Table 1, we could deduce the structure–activity relationship (SAR) as follows: (1) among the tested compounds, β-enaminobisphosphonate 15 has the most antinociceptive/anti-inflammatory activity, even higher than the references ibuprofen and indomethacin; (2) there is a parallel correlation between the anti-inflammatory activities and the antinociceptive activity results (Table 1); (3) the amino group substituent has a positive effect (see 7 and 13); (4) like indomethacin, the tested phosphonates showed gradual increase in the second phase (after 270 min).
LD50 of the most promising products
In an acute-toxicity experiment, the most promising anti-inflammatory compounds 15, 7, and 13 were tested using the LD50 standard method in mice at doses of 500, 750 and 1000 mg per kilogram body weight, which is 10–20 times the used anti-inflammatory effective dose (50 mg per kilogram body weight). The assay did not show toxic symptoms or mortality rates throughout the following 24 h post-administration, indicating the safety of the used doses.
Conclusion
In summary, we have offered a practical and efficient procedure for the synthesis of imidazophosphor esters based tetrazolo[1,5-b]pyridazine in high yields by application of different types of Horner–Emmons (HE) reagents on 3,6-diazidopyridazine. Among the products, the β-enaminobisphosphonate compound demonstrated the highest antinociceptive and the anti-inflammatory activities.
Supporting Information
The experimental section, the general procedures, the experimental data, the results of the analyses, and the bioassay procedures are included in Supporting Information File 1.
| Supporting Information File 1: Full experimental details. | ||
| Format: PDF | Size: 276.0 KB | Download |
References
-
Hamor, G. H. Non-steroidal anti-inflammatory drugs. In Principles of Medicinal Chemistry, 3rd ed.; Foye, W. O., Ed.; LEA & Febiger: Philadelphia, London, 1989; pp 503–530.
Return to citation in text: [1] -
Harrak, K.; Rossel, G.; Daidone, G.; Plescia, S.; Schillaci, D.; Pujol, M. D. Bioorg. Med. Chem. 2007, 15, 4876–4890. doi:10.1016/j.bmc.2007.04.050
Return to citation in text: [1] -
Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; Kakudo, S.; Koizumi, K.; Suzuki, R.; Kato, M.; Kawai, S.; Matsumoto, S. J. Med. Chem. 2000, 43, 2040–2048. doi:10.1021/jm9906015
Return to citation in text: [1] [2] -
Flower, R. J. Nat. Rev. Drug Discovery 2003, 2, 179–191. doi:10.1038/nrd1034
Return to citation in text: [1] -
Dannhardt, G.; Kiefer, W. Eur. J. Med. Chem. 2001, 36, 109–126. doi:10.1016/S0223-5234(01)01197-7
Return to citation in text: [1] -
Penrose, J. F.; Austen, K. F.; Lam, B. K. Leukotrienes: Biosynthetic Pathways, Release and Receptor-Mediated Actions with Relevance to Disease States. In Inflammation Basic Principles and Clinical Correlates; Gallin, J. L.; Snyderman, R., Eds.; Lippicort Williams & Wilkins: Philadelphia, 1999; pp 361–372.
Return to citation in text: [1] -
Vane, J. R.; Bakhle, Y. S.; Botting, R. M. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. doi:10.1146/annurev.pharmtox.38.1.97
Return to citation in text: [1] -
Dirig, D. M.; Isakson, P. C.; Yaksh, T. L. J. Pharmacol. Exp. Ther. 1998, 285, 1031–1038.
Return to citation in text: [1] -
Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.-P. Ann. Rheum. Dis. 2003, 62, 501–509. doi:10.1136/ard.62.6.501
Return to citation in text: [1] -
Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Eur. J. Med. Chem. 2011, 46, 191–200. doi:10.1016/j.ejmech.2010.10.035
Return to citation in text: [1] -
Roussaki, M.; Kontogiorgis, C. A.; Hadjipavlou-Litina, D.; Hamilakis, S.; Detsi, A. Bioorg. Med. Chem. Lett. 2010, 20, 3889–3892. doi:10.1016/j.bmcl.2010.05.022
Return to citation in text: [1] -
Rajić, Z.; Hadjipavlou-Litina, D.; Pontiki, E.; Kralj, M.; Šuman, L.; Zorc, B. Chem. Biol. Drug Des. 2010, 75, 641–652. doi:10.1111/j.1747-0285.2010.00963.x
Return to citation in text: [1] -
Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Arch. Pharm. 2012, 345, 884–895. doi:10.1002/ardp.201200142
Return to citation in text: [1] -
Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Monatsh. Chem. 2011, 142, 649–656. doi:10.1007/s00706-011-0492-8
Return to citation in text: [1] -
Abdou, W. M.; Khidre, R. E.; Barghash, R. F. Synth. Commun. 2012, 42, 1967–1978. doi:10.1080/00397911.2010.551170
Return to citation in text: [1] -
Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. RSC Adv. 2013, 3, 1528–1540. doi:10.1039/c2ra22769j
Return to citation in text: [1] -
Sediek, A. A.; Shaddy, A. A.; Abdou, W. M. J. Heterocycl. Chem. 2011, 48, 1258–1263. doi:10.1002/jhet.743
Return to citation in text: [1] -
Abdou, W. M.; Khidre, M. D.; Khidre, R. E. Eur. J. Med. Chem. 2009, 44, 526–532. doi:10.1016/j.ejmech.2008.03.032
Return to citation in text: [1] -
Abdou, W. M.; Shaddy, A. A.; Sediek, A. A. J. Chem. Res. 2009, 8–13. doi:10.3184/030823409X393646
Return to citation in text: [1] -
Abdou, W. M.; Sediek, A. A.; Khidre, M. D. Monatsh. Chem. 2008, 139, 617–623. doi:10.1007/s00706-007-0806-z
Return to citation in text: [1] -
Abdou, W. M.; Khidre, R. E.; Shaddy, A. A. J. Heterocycl. Chem. 2013, 50, 33–41. doi:10.1002/jhet.968
Return to citation in text: [1] [2] [3] -
Abdou, W. M.; Khidre, R. E.; Kamel, A. A. Arch. Pharm. 2012, 345, 123–136. doi:10.1002/ardp.201100080
Return to citation in text: [1] [2] [3] -
Bhaskar, V. H.; Mohite, P. B. J. Optoelectron. Biomed. Mater. 2011, 3, 7–16.
Return to citation in text: [1] [2] -
Tewari, A. K.; Mishra, A. Bioorg. Med. Chem. 2001, 9, 715–718. doi:10.1016/S0968-0896(00)00285-6
Return to citation in text: [1] [2] -
Salari, H.; Bittman, R. Phosphonates as anti-inflammation agents. U.S. Patent US5219845, June 15, 1993.
Return to citation in text: [1] [2] -
Al Quntar, A. A. A.; Gallily, R.; Katzavian, G.; Srebnik, M. Eur. J. Pharmacol. 2007, 556, 9–13. doi:10.1016/j.ejphar.2006.10.041
Return to citation in text: [1] [2] [3] -
Deep, A.; Sterk, H.; Kappe, T. Liebigs Ann. Chem. 1991, 1225–1227. doi:10.1002/jlac.1991199101210
Return to citation in text: [1] -
Tisler, M. Synthesis 1973, 123–136. doi:10.1055/s-1973-22145
Return to citation in text: [1] [2] -
Kategaonkar, A. H.; Sapkal, S. B.; Madje, B. R.; Shingate, B. B.; Shingare, M. S. Chem. Heterocycl. Compd. 2010, 46, 754–758. doi:10.1007/s10593-010-0579-x
Return to citation in text: [1] [2] [3] -
Shin, M.-S.; Kang, Y.-J.; Chung, H.-A.; Park, J.-W.; Kweon, D.-H.; Lee, W. S.; Yoon, Y.-J.; Kim, S.-K. J. Heterocycl. Chem. 1999, 36, 1135–1142. doi:10.1002/jhet.5570360504
Return to citation in text: [1] -
He, L.-N.; Cai, F.; Chen, R.-Y.; Zhou, J. Phosphorus, Sulfur Silicon Relat. Elem. 1997, 130, 65–71. doi:10.1080/10426509708033698
Return to citation in text: [1] [2] -
Ghosh, S.; Chan, J. M. W.; Lea, C. R.; Meints, G. A.; Lewis, J. C.; Tovian, Z. S.; Flessner, R. M.; Loftus, T. C.; Bruchhaus, I.; Kendrick, H.; Croft, S. L.; Kemp, R. G.; Kobayashi, S.; Nozaki, T.; Oldfield, E. J. Med. Chem. 2004, 47, 175–187. doi:10.1021/jm030084x
Return to citation in text: [1] -
Russell, R. G. G. Ann. N. Y. Acad. Sci. 2006, 1068, 367–401. doi:10.1196/annals.1346.041
Return to citation in text: [1] -
Okun, R.; Liddon, S. C.; Lasagna, L. J. Pharmacol. Exp. Ther. 1963, 139, 107–109.
Return to citation in text: [1] -
Winter, C. A.; Risley, E. A.; Nuss, G. W. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547.
Return to citation in text: [1]
| 21. | Abdou, W. M.; Khidre, R. E.; Shaddy, A. A. J. Heterocycl. Chem. 2013, 50, 33–41. doi:10.1002/jhet.968 |
| 22. | Abdou, W. M.; Khidre, R. E.; Kamel, A. A. Arch. Pharm. 2012, 345, 123–136. doi:10.1002/ardp.201100080 |
| 23. | Bhaskar, V. H.; Mohite, P. B. J. Optoelectron. Biomed. Mater. 2011, 3, 7–16. |
| 32. | Ghosh, S.; Chan, J. M. W.; Lea, C. R.; Meints, G. A.; Lewis, J. C.; Tovian, Z. S.; Flessner, R. M.; Loftus, T. C.; Bruchhaus, I.; Kendrick, H.; Croft, S. L.; Kemp, R. G.; Kobayashi, S.; Nozaki, T.; Oldfield, E. J. Med. Chem. 2004, 47, 175–187. doi:10.1021/jm030084x |
| 33. | Russell, R. G. G. Ann. N. Y. Acad. Sci. 2006, 1068, 367–401. doi:10.1196/annals.1346.041 |
| 24. | Tewari, A. K.; Mishra, A. Bioorg. Med. Chem. 2001, 9, 715–718. doi:10.1016/S0968-0896(00)00285-6 |
| 25. | Salari, H.; Bittman, R. Phosphonates as anti-inflammation agents. U.S. Patent US5219845, June 15, 1993. |
| 26. | Al Quntar, A. A. A.; Gallily, R.; Katzavian, G.; Srebnik, M. Eur. J. Pharmacol. 2007, 556, 9–13. doi:10.1016/j.ejphar.2006.10.041 |
| 1. | Hamor, G. H. Non-steroidal anti-inflammatory drugs. In Principles of Medicinal Chemistry, 3rd ed.; Foye, W. O., Ed.; LEA & Febiger: Philadelphia, London, 1989; pp 503–530. |
| 2. | Harrak, K.; Rossel, G.; Daidone, G.; Plescia, S.; Schillaci, D.; Pujol, M. D. Bioorg. Med. Chem. 2007, 15, 4876–4890. doi:10.1016/j.bmc.2007.04.050 |
| 10. | Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Eur. J. Med. Chem. 2011, 46, 191–200. doi:10.1016/j.ejmech.2010.10.035 |
| 11. | Roussaki, M.; Kontogiorgis, C. A.; Hadjipavlou-Litina, D.; Hamilakis, S.; Detsi, A. Bioorg. Med. Chem. Lett. 2010, 20, 3889–3892. doi:10.1016/j.bmcl.2010.05.022 |
| 12. | Rajić, Z.; Hadjipavlou-Litina, D.; Pontiki, E.; Kralj, M.; Šuman, L.; Zorc, B. Chem. Biol. Drug Des. 2010, 75, 641–652. doi:10.1111/j.1747-0285.2010.00963.x |
| 31. | He, L.-N.; Cai, F.; Chen, R.-Y.; Zhou, J. Phosphorus, Sulfur Silicon Relat. Elem. 1997, 130, 65–71. doi:10.1080/10426509708033698 |
| 3. | Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; Kakudo, S.; Koizumi, K.; Suzuki, R.; Kato, M.; Kawai, S.; Matsumoto, S. J. Med. Chem. 2000, 43, 2040–2048. doi:10.1021/jm9906015 |
| 31. | He, L.-N.; Cai, F.; Chen, R.-Y.; Zhou, J. Phosphorus, Sulfur Silicon Relat. Elem. 1997, 130, 65–71. doi:10.1080/10426509708033698 |
| 6. | Penrose, J. F.; Austen, K. F.; Lam, B. K. Leukotrienes: Biosynthetic Pathways, Release and Receptor-Mediated Actions with Relevance to Disease States. In Inflammation Basic Principles and Clinical Correlates; Gallin, J. L.; Snyderman, R., Eds.; Lippicort Williams & Wilkins: Philadelphia, 1999; pp 361–372. |
| 7. | Vane, J. R.; Bakhle, Y. S.; Botting, R. M. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. doi:10.1146/annurev.pharmtox.38.1.97 |
| 8. | Dirig, D. M.; Isakson, P. C.; Yaksh, T. L. J. Pharmacol. Exp. Ther. 1998, 285, 1031–1038. |
| 9. | Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.-P. Ann. Rheum. Dis. 2003, 62, 501–509. doi:10.1136/ard.62.6.501 |
| 30. | Shin, M.-S.; Kang, Y.-J.; Chung, H.-A.; Park, J.-W.; Kweon, D.-H.; Lee, W. S.; Yoon, Y.-J.; Kim, S.-K. J. Heterocycl. Chem. 1999, 36, 1135–1142. doi:10.1002/jhet.5570360504 |
| 3. | Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; Kakudo, S.; Koizumi, K.; Suzuki, R.; Kato, M.; Kawai, S.; Matsumoto, S. J. Med. Chem. 2000, 43, 2040–2048. doi:10.1021/jm9906015 |
| 4. | Flower, R. J. Nat. Rev. Drug Discovery 2003, 2, 179–191. doi:10.1038/nrd1034 |
| 5. | Dannhardt, G.; Kiefer, W. Eur. J. Med. Chem. 2001, 36, 109–126. doi:10.1016/S0223-5234(01)01197-7 |
| 29. | Kategaonkar, A. H.; Sapkal, S. B.; Madje, B. R.; Shingate, B. B.; Shingare, M. S. Chem. Heterocycl. Compd. 2010, 46, 754–758. doi:10.1007/s10593-010-0579-x |
| 24. | Tewari, A. K.; Mishra, A. Bioorg. Med. Chem. 2001, 9, 715–718. doi:10.1016/S0968-0896(00)00285-6 |
| 27. | Deep, A.; Sterk, H.; Kappe, T. Liebigs Ann. Chem. 1991, 1225–1227. doi:10.1002/jlac.1991199101210 |
| 28. | Tisler, M. Synthesis 1973, 123–136. doi:10.1055/s-1973-22145 |
| 29. | Kategaonkar, A. H.; Sapkal, S. B.; Madje, B. R.; Shingate, B. B.; Shingare, M. S. Chem. Heterocycl. Compd. 2010, 46, 754–758. doi:10.1007/s10593-010-0579-x |
| 21. | Abdou, W. M.; Khidre, R. E.; Shaddy, A. A. J. Heterocycl. Chem. 2013, 50, 33–41. doi:10.1002/jhet.968 |
| 22. | Abdou, W. M.; Khidre, R. E.; Kamel, A. A. Arch. Pharm. 2012, 345, 123–136. doi:10.1002/ardp.201100080 |
| 23. | Bhaskar, V. H.; Mohite, P. B. J. Optoelectron. Biomed. Mater. 2011, 3, 7–16. |
| 28. | Tisler, M. Synthesis 1973, 123–136. doi:10.1055/s-1973-22145 |
| 29. | Kategaonkar, A. H.; Sapkal, S. B.; Madje, B. R.; Shingate, B. B.; Shingare, M. S. Chem. Heterocycl. Compd. 2010, 46, 754–758. doi:10.1007/s10593-010-0579-x |
| 21. | Abdou, W. M.; Khidre, R. E.; Shaddy, A. A. J. Heterocycl. Chem. 2013, 50, 33–41. doi:10.1002/jhet.968 |
| 22. | Abdou, W. M.; Khidre, R. E.; Kamel, A. A. Arch. Pharm. 2012, 345, 123–136. doi:10.1002/ardp.201100080 |
| 26. | Al Quntar, A. A. A.; Gallily, R.; Katzavian, G.; Srebnik, M. Eur. J. Pharmacol. 2007, 556, 9–13. doi:10.1016/j.ejphar.2006.10.041 |
| 13. | Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Arch. Pharm. 2012, 345, 884–895. doi:10.1002/ardp.201200142 |
| 14. | Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Monatsh. Chem. 2011, 142, 649–656. doi:10.1007/s00706-011-0492-8 |
| 15. | Abdou, W. M.; Khidre, R. E.; Barghash, R. F. Synth. Commun. 2012, 42, 1967–1978. doi:10.1080/00397911.2010.551170 |
| 16. | Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. RSC Adv. 2013, 3, 1528–1540. doi:10.1039/c2ra22769j |
| 17. | Sediek, A. A.; Shaddy, A. A.; Abdou, W. M. J. Heterocycl. Chem. 2011, 48, 1258–1263. doi:10.1002/jhet.743 |
| 18. | Abdou, W. M.; Khidre, M. D.; Khidre, R. E. Eur. J. Med. Chem. 2009, 44, 526–532. doi:10.1016/j.ejmech.2008.03.032 |
| 19. | Abdou, W. M.; Shaddy, A. A.; Sediek, A. A. J. Chem. Res. 2009, 8–13. doi:10.3184/030823409X393646 |
| 20. | Abdou, W. M.; Sediek, A. A.; Khidre, M. D. Monatsh. Chem. 2008, 139, 617–623. doi:10.1007/s00706-007-0806-z |
| 25. | Salari, H.; Bittman, R. Phosphonates as anti-inflammation agents. U.S. Patent US5219845, June 15, 1993. |
| 26. | Al Quntar, A. A. A.; Gallily, R.; Katzavian, G.; Srebnik, M. Eur. J. Pharmacol. 2007, 556, 9–13. doi:10.1016/j.ejphar.2006.10.041 |
| 34. | Okun, R.; Liddon, S. C.; Lasagna, L. J. Pharmacol. Exp. Ther. 1963, 139, 107–109. |
| 35. | Winter, C. A.; Risley, E. A.; Nuss, G. W. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. |
© 2013 Abdou et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)