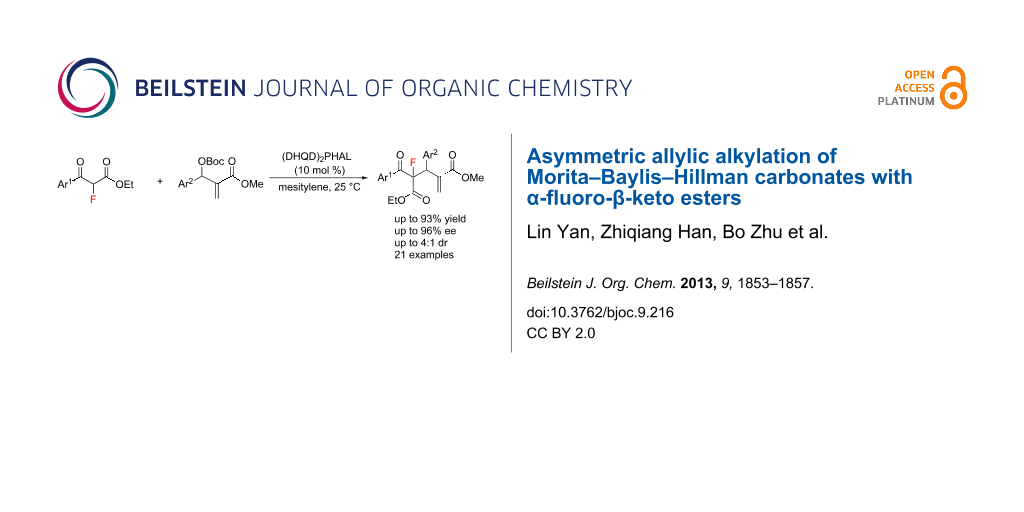
Abstract
In the presence of a commercially available Cinchona alkaloid as catalyst, the asymmetric allylic alkylation of Morita–Baylis–Hillman carbonates, with α-fluoro-β-keto esters as nucleophiles, have been successfully developed. A series of important fluorinated adducts, with chiral quaternary carbon centres containing a fluorine atom, was achieved in good yields (up to 93%), with good to excellent enantioselectivities (up to 96% ee) and moderate diastereoselectivities (up to 4:1 dr).

Graphical Abstract
Introduction
Fluorine is the most electronegative element in the periodic table, resulting in a highly polar C–F bond. This gives fluoro-organic compounds unique properties, compared with their parent compounds [1]. Due to the rareness of organofluorine compounds in nature, synthetic fluorinated compounds have been widely applied in numerous areas, including materials, agrochemicals, pharmaceuticals and fine chemicals [2-4]. In this context, the stereoselective introduction of fluorine atoms in molecules has become one of the most exciting and intense research areas in the recent years.
Lewis base-catalyzed asymmetric allylic alkylations (AAA) of Morita–Baylis–Hillman (MBH) adducts [5,6], such as acetates and carbonates, have become an attractive option to access various chiral C- [7-19], N- [20-25], O- [26-30], P- [31-33] and S-allylic [34] and spirocyclic compounds [35-37]. Several protocols have been established to introduce fluorine atoms in AAA of MBH adducts. For example, to introduce a CF3 group, Shibata and co-workers [13] and Jiang and co-workers [14] successively reported the asymmetric allylic trifluoromethylation of MBH adducts with Ruppert’s reagent [(trifluoromethyl)trimethylsilane, Me3SiCF3] in the presence of (DHQD)2PHAL as catalyst. In 2011, our research group [15], Shibata and co-workers [16] and Rios and co-workers [17] reported the addition of fluoromethyl(bisphenylsulfones) to MBH carbonates to access chiral monofluoromethyl derivatives. Furthermore, Rios and co-workers presented an asymmetric substitution of MBH carbonates with 2-fluoromalonates in good enantioselectivities [38]. Notably, the reaction between an achiral fluorocarbon nucleophile with MBH carbonates, to afford compounds with chiral quaternary carbon centres bearing a fluorine atom, remains a formidable task. Since 2009, we developed a highly enantioselective and diastereoselective guanidine-catalyzed conjugate addition and Mannich reaction of α-fluoro-β-ketoesters with excellent results [38-40]. Herein, we wish to report the first allylic alkylation of MBH carbonates with α-fluoro-β-ketoesters in excellent enantioselectivities and moderate diastereoselectivities, furnishing enantiopure fluorinated compounds with chiral quaternary carbon centres containing a fluorine atom.
Results and Discussion
In the preliminary experiments, we investigated the reaction of α-fluoro-β-ketoester 1a with MBH carbonate 2a as the model substrate, in the presence of several commercially available Cinchona alkaloids as Lewis base catalysts (Table 1). First, the reaction was conducted in the presence of quinine at 50 °C in dichloroethane (DCE) as the solvent (Table 1, entry 1). The desired adduct 3aa was obtained in 53% yield with poor enantio- and diastereoselectivity. Cinchonine provided similarly poor results (Table 1, entry 2). Next, we screened a series of C2-symmetric bis-Cinchona alkaloids as catalysts under the same conditions (Table 1, entries 3–7). (DHQD)2PHAL showed moderate catalytic activity; 3aa was obtained in 67% yield with 71% ee and 60:40 dr (entry 3). The effects of solvent were then investigated (Table 1, entries 8–18). The best-performing solvent was mesitylene with respect to enantio- and diastereoselectivity; providing 3aa in 78% yield with 89% ee and 71:29 dr (entry 18). The reaction temperature can be decreased to 25 °C and 67% yield of 3aa with 92% ee and 74:26 dr was obtained (entry 19). A slight increase in enantio- and diastereoselectivity could be obtained when the reaction temperature was decreased to 10 °C, but the reaction rate became too sluggish to be useful (Table 1, entry 20).
Table 1: Catalyst screeninga.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-216-i1.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Solvent | Yield (%)b | ee (%)c | drc |
|---|---|---|---|---|---|
| 1 | quinine | DCE | 53 | 32 (27) | 55:45 |
| 2 | cinchonine | DCE | 59 | 21 (10) | 55:45 |
| 3 | (DHQD)2PHAL | DCE | 67 | 71 (57) | 60:40 |
| 4 | (DHQD)2AQN | DCE | 53 | –5 (–5) | 56:44 |
| 5 | (DHQ)2PHAL | DCE | 64 | –35 (–1) | 52:48 |
| 6 | (DHQ)2PYR | DCE | 60 | –25 (–1) | 59:41 |
| 7 | (DHQ)2AQN | DCE | 47 | –11 (–10) | 55:45 |
| 8 | (DHQD)2PHAL | DCM | 56 | 69 (55) | 58:42 |
| 9 | (DHQD)2PHAL | toluene | 78 | 85 (65) | 67:33 |
| 10 | (DHQD)2PHAL | Et2O | 59 | 45 (30) | 55:45 |
| 11 | (DHQD)2PHAL | EA | 58 | 55 (30) | 55:45 |
| 12 | (DHQD)2PHAL | THF | 61 | 31 (49) | 63:37 |
| 13 | (DHQD)2PHAL | MeCN | 57 | 49 (19) | 65:35 |
| 14 | (DHQD)2PHAL | MeOH | 63 | 35 (20) | 60:40 |
| 15 | (DHQD)2PHAL | o-xylene | 74 | 85 (65) | 72:28 |
| 16 | (DHQD)2PHAL | m-xylene | 65 | 87 (74) | 70:30 |
| 17 | (DHQD)2PHAL | p-xylene | 72 | 85 (55) | 68:32 |
| 18 | (DHQD)2PHAL | mesitylene | 78 | 89 (72) | 71:29 |
| 19d | (DHQD)2PHAL | mesitylene | 67 | 92 (69) | 74:26 |
| 20e | (DHQD)2PHAL | mesitylene | 45 | 94 (55) | 75:25 |
aUnless otherwise noted, reactions were performed with 0.05 mmol of 1a, 0.15 of 2a, and 0.005 mmol of catalyst in 0.5 mL solvent. bYield of isolated product. cDetermined by HPLC methods. The data in parenthesis is the ee value of the minor diastereoisomer dThe reaction was conducted at 25 °C, 1.0 mmol scale in 1.0 mL of mesitylene. eThe reaction was conducted at 10 °C, 1.0 mmol scale in 1.0 mL of mesitylene.
Using the established conditions, allylic alkylations of α-fluoro-β-ketoesters 1b–g with MBH carbonate 2a were found to afford the products 3ba–ga in 67–79% yield with 88–96% ee and 3:1 to 4:1 dr (Table 2, entries 1–6). The results showed that the introduction of various aryl substituents in α-fluoro-β-ketoesters did not affect the reactivity and stereoselectivity. Subsequently, the scope of the allylic alkylation with respect to various MBH carbonates 2 and α-fluoro-β-ketoester 1a was investigated (Table 2, entries 7–20). The desired allylic alkylation adducts 3ab–o were achieved in moderate to good yields with good to excellent enantioselectivities and moderate diastereoselectivities. MBH carbonates (Table 2, 2b–k) with electron-withdrawing groups appended on the aromatic rings were more active than those (Table 2, 2l–m) with electron-neutral and donating groups. Excellent ee values with moderate dr values were obtained when the phenyl groups of MBH carbonates were replaced with hetereoaromatic groups, such as thiophene and furan (Table 2, 2n–o).
Table 2: Allylic alkylation of α-fluoro-β-ketoesters 1 with MBH carbonates 2a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-216-i2.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Ar1, 1 | Ar2, 2 | Time (h) | 3 | Yield (%)b | ee (%)c | drd |
|---|---|---|---|---|---|---|---|
| 1 | p-FPh, 1b | Ph, 2a | 40 | 3ba | 71 | 88 | 3:1 |
| 2 | p-ClPh, 1c | Ph, 2a | 70 | 3ca | 79 | 93 | 3:1 |
| 3 | p-BrPh, 1d | Ph, 2a | 70 | 3da | 75 | 96 | 3:1 |
| 4 | m-BrPh, 1e | Ph, 2a | 70 | 3ea | 72 | 90 | 3:1 |
| 5 | 3,5-Cl2Ph, 1f | Ph, 2a | 70 | 3fa | 69 | 88 | 3:1 |
| 6 | p-MePh, 1g | Ph, 2a | 50 | 3ga | 67 | 94 | 4:1 |
| 7 | Ph, 1a | p-NO2Ph, 2b | 70 | 3ab | 91 | 95 | 3:1 |
| 8 | Ph, 1a | p-CF3Ph, 2c | 70 | 3ac | 65 | 87 | 4:1 |
| 9 | Ph, 1a | p-FPh, 2d | 70 | 3ad | 71 | 90 | 3:1 |
| 10 | Ph, 1a | p-ClPh, 2e | 70 | 3ae | 73 | 93 | 4:1 |
| 11 | Ph, 1a | p-BrPh, 2f | 96 | 3af | 64 | 91 | 4:1 |
| 12 | Ph, 1a | m-NO2Ph, 2g | 96 | 3ag | 93 | 95 | 3:1 |
| 13 | Ph, 1a | m-ClPh, 2h | 70 | 3ah | 81 | 91 | 3:1 |
| 14 | Ph, 1a | m-BrPh, 2i | 70 | 3ai | 78 | 90 | 4:1 |
| 15 | Ph, 1a | o-FPh, 2j | 96 | 3aj | 73 | 86 | 4:1 |
| 16 | Ph, 1a | o-ClPh, 2k | 70 | 3ak | 84 | 86 | 4:1 |
| 17 | Ph, 1a | p-MePh, 2l | 70 | 3al | 53 | 91 | 4:1 |
| 18 | Ph, 1a | p-MeOPh, 2m | 70 | 3am | 50 | 91 | 3:1 |
| 19 | Ph, 1a | 2-thienyl, 2n | 90 | 3an | 78 | 92 | 4:1 |
| 20 | Ph, 1a | 2-furyl, 2o | 96 | 3ao | 73 | 84 | 3:1 |
aReactions were performed with 0.1 mmol of 1, 0.3 mmol of 2, and 0.005 mmol of (DHQD)2PHAL in 1.0 mL mesitylene. bYield of isolated product. cDetermined by chiral HPLC on the major diastereoisomer. dDetermined by 1H NMR analysis.
Conclusion
We have developed an asymmetric allylic alkylation of MBH carbonates with α-fluoro-β-ketoesters, catalyzed by a commercially available Cinchona alkaloid. Several fluorinated adducts, with chiral quaternary carbon centres containing a fluorine atom, were successfully prepared in 50–93% yields with 84–96% ee and a dr of 3:1 to 4:1. The absolute configurations of adducts still have to be determined and will be reported in due course.
Experimental
Representative procedure for the synthesis of 3aa: α-Fluoro-β-ketoester 1a (21.0 mg, 1.0 equiv, 0.1 mmol) and (DHQD)2PHAL (7.8 mg, 0.1 equiv, 0.01 mmol) were dissolved in mesitylene (1.0 mL) at 25 °C. After the addition of MBH carbonate 2a (3.0 equiv, 0.3 mmol) the reaction mixture was stirred at 25 °C. The reaction was monitored by TLC. After 96 hours, flash chromatography affords product 3aa (25.7 mg, 67% yield) as colorless oil.
Supporting Information
| Supporting Information File 1: Experimental details and spectroscopic data. | ||
| Format: PDF | Size: 2.2 MB | Download |
Acknowledgements
This work was supported by NSFC (nos. 21072044, 21202034), the Program for New Century Excellent Talents in University of the Ministry of Education (NCET-11-0938) and Excellent Youth Foundation of Henan Scientific Committee (114100510003). Z.J. also appreciates the generous financial support from Nanyang Technological University for the senior research fellow funding.
References
-
O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer: Berlin, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Uneyama, K. Organo-fluorine Chemisry; Blackwell: Oxford, 2006.
Return to citation in text: [1] -
Filler, R.; Kobayashi, Y.; Yagupolskii, L. M., Eds. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications; Elservier, 1993.
Return to citation in text: [1] -
Rios, R. Catal. Sci. Technol. 2012, 2, 267–278. doi:10.1039/c1cy00387a
Return to citation in text: [1] -
Liu, T.-Y.; Xie, M.; Chen, Y.-C. Chem. Soc. Rev. 2012, 41, 4101–4112. doi:10.1039/c2cs35017c
Return to citation in text: [1] -
Cho, C.-W.; Krische, M. J. Angew. Chem., Int. Ed. 2004, 43, 6689–6691. doi:10.1002/anie.200461381
Return to citation in text: [1] -
van Steenis, D. J. V. C.; Marcelli, T.; Lutz, M.; Spek, A. L.; van Maarseveen, J. H.; Hiemstra, H. Adv. Synth. Catal. 2007, 349, 281–286. doi:10.1002/adsc.200600467
Return to citation in text: [1] -
Jiang, Y.-Q.; Shi, Y.-L.; Shi, M. J. Am. Chem. Soc. 2008, 130, 7202–7203. doi:10.1021/ja802422d
Return to citation in text: [1] -
Cui, H.-L.; Peng, J.; Feng, X.; Du, W.; Jiang, K.; Chen, Y.-C. Chem.–Eur. J. 2009, 15, 1574–1577. doi:10.1002/chem.200802534
Return to citation in text: [1] -
Jiang, K.; Peng, J.; Cui, H.-L.; Chen, Y.-C. Chem. Commun. 2009, 45, 3955–3957. doi:10.1039/b905177e
Return to citation in text: [1] -
Cui, H.-L.; Huang, J.-R.; Lei, J.; Wang, Z.-F.; Chen, S.; Wu, L.; Chen, Y.-C. Org. Lett. 2010, 12, 720–723. doi:10.1021/ol100014m
Return to citation in text: [1] -
Furukawa, T.; Nishimine, T.; Tokunaga, E.; Hasegawa, K.; Shiro, M.; Shibata, N. Org. Lett. 2011, 13, 3972–3975. doi:10.1021/ol201490e
Return to citation in text: [1] [2] -
Li, Y.; Liang, F.; Li, Q.; Xu, Y.-C.; Wang, Q.-R.; Jiang, L. Org. Lett. 2011, 13, 6082–56085. doi:10.1021/ol202572u
Return to citation in text: [1] [2] -
Yang, W.; Wei, X.; Pan, Y.; Lee, R.; Zhu, B.; Liu, H.; Yan, L.; Huang, K.-W.; Jiang, Z.; Tan, C.-H. Chem.–Eur. J. 2011, 17, 8066–8070. doi:10.1002/chem.201100929
Return to citation in text: [1] [2] -
Furukawa, T.; Kawazoe, J.; Zhang, W.; Nishimine, T.; Tojunaga, E.; Matsumoto, T.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 9684–9688. doi:10.1002/anie.201103748
Return to citation in text: [1] [2] -
Company, X.; Valero, G.; Ceban, V.; Calvet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Org. Biomol. Chem. 2011, 9, 7986–7989. doi:10.1039/C1OB06308A
Return to citation in text: [1] [2] -
Yang, W.; Tan, D.; Li, L.; Han, Z.; Yan, L.; Huang, K.-W.; Tan, C.-H.; Jiang, Z. J. Org. Chem. 2012, 77, 6600–6607. doi:10.1021/jo3012539
Return to citation in text: [1] -
Tong, G.; Zhu, B.; Lee, R.; Yang, W.; Tan, D.; Yang, C.; Han, Z.; Yan, L.; Huang, K.-W.; Jiang, Z. J. Org. Chem. 2013, 78, 5067–5072. doi:10.1021/jo400496z
Return to citation in text: [1] -
Cho, C.-W.; Kong, J.-R.; Krische, M. J. Org. Lett. 2004, 6, 1337–1339. doi:10.1021/ol049600j
Return to citation in text: [1] -
Park, H.; Cho, C.-W.; Krische, M. J. J. Org. Chem. 2006, 71, 7892–7894. doi:10.1021/jo061218s
Return to citation in text: [1] -
Cui, H.-L.; Feng, X.; Peng, J.; Lei, J.; Jiang, K.; Chen, Y.-C. Angew. Chem., Int. Ed. 2009, 48, 5737–5740. doi:10.1002/anie.200902093
Return to citation in text: [1] -
Huang, J.-R.; Cui, H.-L.; Lei, J.; Sun, X.-H.; Chen, Y.-C. Chem. Commun. 2011, 47, 4784–4786. doi:10.1039/c0cc05616b
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 1956–1960. doi:10.1002/ejoc.201001660
Return to citation in text: [1] -
Lin, A.; Mao, H.; Zhu, X.; Ge, H.; Tan, R.; Zhu, C.; Cheng, Y. Chem.–Eur. J. 2011, 17, 13676–13679. doi:10.1002/chem.201102522
Return to citation in text: [1] -
Trost, B. M.; Tsui, H.-C.; Toste, F. D. J. Am. Chem. Soc. 2000, 122, 3534–3535. doi:10.1021/ja994326n
Return to citation in text: [1] -
Kim, J. N.; Lee, H. J.; Gong, J. H. Tetrahedron Lett. 2002, 43, 9141–9146. doi:10.1016/S0040-4039(02)02274-8
Return to citation in text: [1] -
Feng, X.; Yuan, Y.-Q.; Jiang, K.; Chen, Y.-C. Org. Biomol. Chem. 2009, 7, 3660–3662. doi:10.1039/b912110b
Return to citation in text: [1] -
Hu, Z.; Cui, H.; Jiang, K.; Chen, Y. Sci. China, Ser. B: Chem. 2009, 52, 1309–1313. doi:10.1007/s11426-009-0187-8
Return to citation in text: [1] -
Zhu, B.; Yan, L.; Pan, Y.; Lee, R.; Liu, H.; Han, Z.; Huang, K.-W.; Tan, C.-H.; Jiang, Z. J. Org. Chem. 2011, 76, 6894–6900. doi:10.1021/jo201096e
Return to citation in text: [1] -
Hong, L.; Sun, W.; Liu, C.; Zhao, D.; Wang, R. Chem. Commun. 2010, 46, 2856–2858. doi:10.1039/b926037d
Return to citation in text: [1] -
Sun, W.; Hong, L.; Liu, C.; Wang, R. Org. Lett. 2010, 12, 3914–3917. doi:10.1021/ol101601d
Return to citation in text: [1] -
Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 2012, 183–187. doi:10.1002/ejoc.201101365
Return to citation in text: [1] -
Lin, A.; Mao, H.; Zhu, X.; Ge, H.; Tan, R.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2011, 353, 3301–3306. doi:10.1002/adsc.201100522
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w
Return to citation in text: [1] -
Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094
Return to citation in text: [1] -
Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h
Return to citation in text: [1] -
Wang, B.; Comany, X.; Li, J.; Moyano, A.; Rios, R. Tetrahedron Lett. 2012, 53, 4124–4129. doi:10.1016/j.tetlet.2012.05.121
Return to citation in text: [1] [2] -
Jiang, Z.; Pan, Y.; Zhao, Y.; Ma, T.; Lee, R.; Yuang, Y.; Huang, K.-W.; Wong, M. W.; Tan, C.-H. Angew. Chem., Int. Ed. 2009, 48, 3627–3631. doi:10.1002/anie.200900964
Return to citation in text: [1] -
Pan, Y.; Zhao, Y.; Ma, T.; Yang, Y.; Liu, H.; Jiang, Z.; Tan, C.-H. Chem.–Eur. J. 2010, 16, 779–782. doi:10.1002/chem.200902830
Return to citation in text: [1]
| 20. | Cho, C.-W.; Kong, J.-R.; Krische, M. J. Org. Lett. 2004, 6, 1337–1339. doi:10.1021/ol049600j |
| 21. | Park, H.; Cho, C.-W.; Krische, M. J. J. Org. Chem. 2006, 71, 7892–7894. doi:10.1021/jo061218s |
| 22. | Cui, H.-L.; Feng, X.; Peng, J.; Lei, J.; Jiang, K.; Chen, Y.-C. Angew. Chem., Int. Ed. 2009, 48, 5737–5740. doi:10.1002/anie.200902093 |
| 23. | Huang, J.-R.; Cui, H.-L.; Lei, J.; Sun, X.-H.; Chen, Y.-C. Chem. Commun. 2011, 47, 4784–4786. doi:10.1039/c0cc05616b |
| 24. | Deng, H.-P.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 1956–1960. doi:10.1002/ejoc.201001660 |
| 25. | Lin, A.; Mao, H.; Zhu, X.; Ge, H.; Tan, R.; Zhu, C.; Cheng, Y. Chem.–Eur. J. 2011, 17, 13676–13679. doi:10.1002/chem.201102522 |
| 38. | Wang, B.; Comany, X.; Li, J.; Moyano, A.; Rios, R. Tetrahedron Lett. 2012, 53, 4124–4129. doi:10.1016/j.tetlet.2012.05.121 |
| 7. | Cho, C.-W.; Krische, M. J. Angew. Chem., Int. Ed. 2004, 43, 6689–6691. doi:10.1002/anie.200461381 |
| 8. | van Steenis, D. J. V. C.; Marcelli, T.; Lutz, M.; Spek, A. L.; van Maarseveen, J. H.; Hiemstra, H. Adv. Synth. Catal. 2007, 349, 281–286. doi:10.1002/adsc.200600467 |
| 9. | Jiang, Y.-Q.; Shi, Y.-L.; Shi, M. J. Am. Chem. Soc. 2008, 130, 7202–7203. doi:10.1021/ja802422d |
| 10. | Cui, H.-L.; Peng, J.; Feng, X.; Du, W.; Jiang, K.; Chen, Y.-C. Chem.–Eur. J. 2009, 15, 1574–1577. doi:10.1002/chem.200802534 |
| 11. | Jiang, K.; Peng, J.; Cui, H.-L.; Chen, Y.-C. Chem. Commun. 2009, 45, 3955–3957. doi:10.1039/b905177e |
| 12. | Cui, H.-L.; Huang, J.-R.; Lei, J.; Wang, Z.-F.; Chen, S.; Wu, L.; Chen, Y.-C. Org. Lett. 2010, 12, 720–723. doi:10.1021/ol100014m |
| 13. | Furukawa, T.; Nishimine, T.; Tokunaga, E.; Hasegawa, K.; Shiro, M.; Shibata, N. Org. Lett. 2011, 13, 3972–3975. doi:10.1021/ol201490e |
| 14. | Li, Y.; Liang, F.; Li, Q.; Xu, Y.-C.; Wang, Q.-R.; Jiang, L. Org. Lett. 2011, 13, 6082–56085. doi:10.1021/ol202572u |
| 15. | Yang, W.; Wei, X.; Pan, Y.; Lee, R.; Zhu, B.; Liu, H.; Yan, L.; Huang, K.-W.; Jiang, Z.; Tan, C.-H. Chem.–Eur. J. 2011, 17, 8066–8070. doi:10.1002/chem.201100929 |
| 16. | Furukawa, T.; Kawazoe, J.; Zhang, W.; Nishimine, T.; Tojunaga, E.; Matsumoto, T.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 9684–9688. doi:10.1002/anie.201103748 |
| 17. | Company, X.; Valero, G.; Ceban, V.; Calvet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Org. Biomol. Chem. 2011, 9, 7986–7989. doi:10.1039/C1OB06308A |
| 18. | Yang, W.; Tan, D.; Li, L.; Han, Z.; Yan, L.; Huang, K.-W.; Tan, C.-H.; Jiang, Z. J. Org. Chem. 2012, 77, 6600–6607. doi:10.1021/jo3012539 |
| 19. | Tong, G.; Zhu, B.; Lee, R.; Yang, W.; Tan, D.; Yang, C.; Han, Z.; Yan, L.; Huang, K.-W.; Jiang, Z. J. Org. Chem. 2013, 78, 5067–5072. doi:10.1021/jo400496z |
| 38. | Wang, B.; Comany, X.; Li, J.; Moyano, A.; Rios, R. Tetrahedron Lett. 2012, 53, 4124–4129. doi:10.1016/j.tetlet.2012.05.121 |
| 39. | Jiang, Z.; Pan, Y.; Zhao, Y.; Ma, T.; Lee, R.; Yuang, Y.; Huang, K.-W.; Wong, M. W.; Tan, C.-H. Angew. Chem., Int. Ed. 2009, 48, 3627–3631. doi:10.1002/anie.200900964 |
| 40. | Pan, Y.; Zhao, Y.; Ma, T.; Yang, Y.; Liu, H.; Jiang, Z.; Tan, C.-H. Chem.–Eur. J. 2010, 16, 779–782. doi:10.1002/chem.200902830 |
| 5. | Rios, R. Catal. Sci. Technol. 2012, 2, 267–278. doi:10.1039/c1cy00387a |
| 6. | Liu, T.-Y.; Xie, M.; Chen, Y.-C. Chem. Soc. Rev. 2012, 41, 4101–4112. doi:10.1039/c2cs35017c |
| 16. | Furukawa, T.; Kawazoe, J.; Zhang, W.; Nishimine, T.; Tojunaga, E.; Matsumoto, T.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 9684–9688. doi:10.1002/anie.201103748 |
| 2. | Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer: Berlin, 2000. doi:10.1007/978-3-662-04164-2 |
| 3. | Uneyama, K. Organo-fluorine Chemisry; Blackwell: Oxford, 2006. |
| 4. | Filler, R.; Kobayashi, Y.; Yagupolskii, L. M., Eds. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications; Elservier, 1993. |
| 17. | Company, X.; Valero, G.; Ceban, V.; Calvet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Org. Biomol. Chem. 2011, 9, 7986–7989. doi:10.1039/C1OB06308A |
| 35. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w |
| 36. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 37. | Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h |
| 14. | Li, Y.; Liang, F.; Li, Q.; Xu, Y.-C.; Wang, Q.-R.; Jiang, L. Org. Lett. 2011, 13, 6082–56085. doi:10.1021/ol202572u |
| 34. | Lin, A.; Mao, H.; Zhu, X.; Ge, H.; Tan, R.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2011, 353, 3301–3306. doi:10.1002/adsc.201100522 |
| 15. | Yang, W.; Wei, X.; Pan, Y.; Lee, R.; Zhu, B.; Liu, H.; Yan, L.; Huang, K.-W.; Jiang, Z.; Tan, C.-H. Chem.–Eur. J. 2011, 17, 8066–8070. doi:10.1002/chem.201100929 |
| 31. | Hong, L.; Sun, W.; Liu, C.; Zhao, D.; Wang, R. Chem. Commun. 2010, 46, 2856–2858. doi:10.1039/b926037d |
| 32. | Sun, W.; Hong, L.; Liu, C.; Wang, R. Org. Lett. 2010, 12, 3914–3917. doi:10.1021/ol101601d |
| 33. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 26. | Trost, B. M.; Tsui, H.-C.; Toste, F. D. J. Am. Chem. Soc. 2000, 122, 3534–3535. doi:10.1021/ja994326n |
| 27. | Kim, J. N.; Lee, H. J.; Gong, J. H. Tetrahedron Lett. 2002, 43, 9141–9146. doi:10.1016/S0040-4039(02)02274-8 |
| 28. | Feng, X.; Yuan, Y.-Q.; Jiang, K.; Chen, Y.-C. Org. Biomol. Chem. 2009, 7, 3660–3662. doi:10.1039/b912110b |
| 29. | Hu, Z.; Cui, H.; Jiang, K.; Chen, Y. Sci. China, Ser. B: Chem. 2009, 52, 1309–1313. doi:10.1007/s11426-009-0187-8 |
| 30. | Zhu, B.; Yan, L.; Pan, Y.; Lee, R.; Liu, H.; Han, Z.; Huang, K.-W.; Tan, C.-H.; Jiang, Z. J. Org. Chem. 2011, 76, 6894–6900. doi:10.1021/jo201096e |
| 13. | Furukawa, T.; Nishimine, T.; Tokunaga, E.; Hasegawa, K.; Shiro, M.; Shibata, N. Org. Lett. 2011, 13, 3972–3975. doi:10.1021/ol201490e |
© 2013 Yan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)