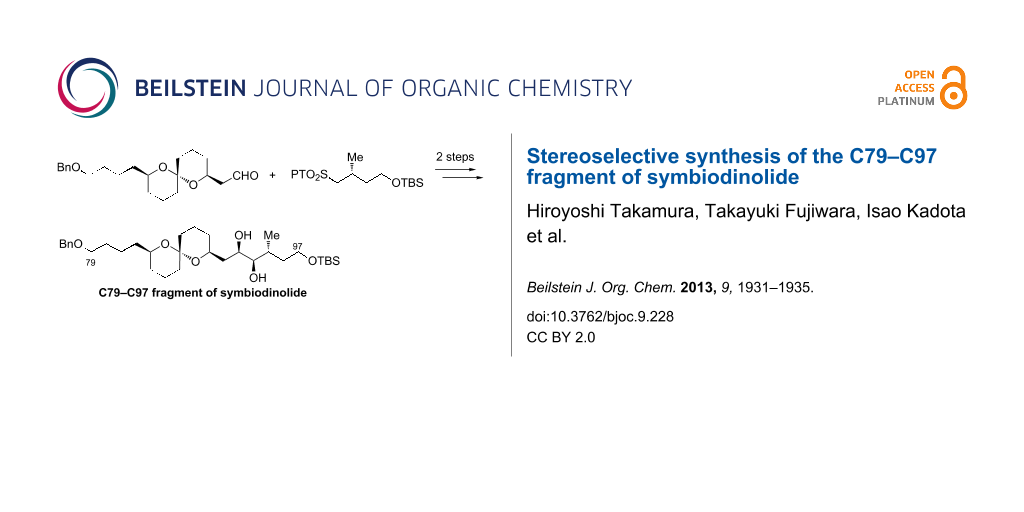
Abstract
Symbiodinolide is a polyol marine natural product with a molecular weight of 2860. Herein, a streamlined synthesis of the C79–C97 fragment of symbiodinolide is described. In the synthetic route, a spiroacetalization, a Julia–Kocienski olefination, and a Sharpless asymmetric dihydroxylation were utilized as the key transformations.

Graphical Abstract
Findings
A 62-membered polyol marine natural product, symbiodinolide (1, Figure 1), was isolated from the 80% aqueous ethanol extract of the cultured symbiotic dinoflagellate Symbiodinium sp. in 2007 [1]. Symbiodinolide shows voltage-dependent N-type Ca2+ channel-opening activity at 7 nmol/L and COX-1 inhibitory effect at 2 μmol/L (65% inhibition). Furthermore, 1 ruptures the tissue surface of the acoel flatworm Amphiscolops sp. at 2.5 μmol/L. The entire planar structure of 1 was established by the detailed 2D NMR spectroscopic analysis. However, the complete stereostructural determination of 1 with its 61 chirality centres and a molar mass of 2860 has been an unsolved issue. Therefore, in order to complete the configurational elucidation of 1, we are now investigating its chemical degradation [1-3] and chemical synthesis of the fragments [4-11].
![[1860-5397-9-228-1]](/bjoc/content/figures/1860-5397-9-228-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of symbiodinolide (1).
Figure 1: Structure of symbiodinolide (1).
Previously, we reported the stereoselective synthesis of the spiroacetal C79–C96 fragment [4], which is summarized in Scheme 1. Triflate 2 was reacted with the lithium acetylide prepared from alkyne 3 to give the desired coupling product 4. The TBDPS ether 4 was transformed to TIPS ether 5 because of the lability of the TBDPS protecting group under the following Birch conditions. The alkyne 5 was subjected to the Birch reduction to afford the trans-alkene 6, wherein the benzyl moiety was deprotected. The alkene 6 was derivatized to the spiroacetal C79–C96 fragment 7 in four steps including the benzyl protection and Sharpless asymmetric dihydroxylation (AD). Although the desired spiroacetal fragment 7 was synthesized stereoselectively, the transformation starting with the coupling between 2 and 3 to the final product 7 needed eight steps. Therefore, we decided to examine the more efficient synthesis of the spiroacetal fragment. Herein, we report the improved synthesis of the spiroacetal fragment by utilizing Julia–Kocienski olefination as the coupling reaction.
![[1860-5397-9-228-i1]](/bjoc/content/inline/1860-5397-9-228-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our previous synthesis of the C79–C96 fragment 7.
Scheme 1: Our previous synthesis of the C79–C96 fragment 7.
The new retrosynthetic analysis of the C79–C97 fragment 8 is described in Scheme 2. We envisaged that the diol 8 could be synthesized by the Julia–Kocienski olefination [12-14] between aldehyde 9 and 1-phenyl-1H-tetrazol-5-yl (PT)-sulfone 10 and subsequent Sharpless AD [15], wherein the target molecule 8 could be prepared in two steps from the coupling. The carbon framework of 9 could be constructed through the stereoselective spiroacetalization of dihydroxyketone 11.
![[1860-5397-9-228-i2]](/bjoc/content/inline/1860-5397-9-228-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of the C79–C97 fragment 8.
Scheme 2: Retrosynthetic analysis of the C79–C97 fragment 8.
First, we commenced the stereocontrolled synthesis of aldehyde 20 (Scheme 3). Treatment of epoxide 13, which was prepared from L-aspartic acid (12) by the known procedure [8], with 3-butenylmagnesium bromide/CuI [16] provided the corresponding secondary alcohol. Protection of the alcohol with TBSCl afforded TBS ether 14 in 91% yield in two steps. Alkene 14 was reacted with m-CPBA to produce epoxide 15 as a 1:1 diastereomeric mixture. Epoxide 15 was coupled with alkyne 16 [4] in the presence of n-BuLi/BF3∙OEt2 [17] to give the desired product 17 in 92% yield from 15. Hydrogenation of the alkyne moiety of 17 followed by TPAP oxidation [18] yielded ketone 18. Removal of the three TBS protecting groups and subsequent stereoselective spiroacetalization were performed in one-pot with CSA in MeOH to provide spiroacetal 19 as a single stereoisomer [19,20]. The stereochemistry of 19 was elucidated by the observed NOE correlations between H-83 and H-91 as indicated by an arrow. The plausible rationale for the stereoselective formation of 19 is the thermodynamic stability due to the double anomeric effect. Oxidation of the alcohol 19 with SO3∙pyr/Et3N/DMSO [21] afforded aldehyde 20.
![[1860-5397-9-228-i3]](/bjoc/content/inline/1860-5397-9-228-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, we carried out the synthesis of PT-sulfones 23 and 24 which were the coupling partners of the aldehyde 20 (Scheme 4). The synthesis started from commercially available methyl (S)-3-hydroxy-2-methylpropanoate (21), which was converted to alcohol 22 by the known method [22]. Alcohol 22 was treated with 1-phenyl-1H-tetrazole-5-thiol/DEAD/PPh3 to furnish the corresponding PT-sulfide, which was oxidized with H2O2/Mo(VI) [23] to yield PT-sulfone 23. The TBDPS protecting group of 23 was transformed to the TBS group in two steps to provide PT-sulfone 24.
![[1860-5397-9-228-i4]](/bjoc/content/inline/1860-5397-9-228-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of PT-sulfones 23 and 24.
Scheme 4: Synthesis of PT-sulfones 23 and 24.
With the coupling precursors aldehyde 20 and PT-sulfones 23 and 24 in hand, we next examined the Julia–Kocienski olefination [12-14] of these compounds (Table 1). Deprotonation of the PT-sulfone 23 with KHMDS, followed by addition of the aldehyde 20, gave rise to the desired coupling product (E)-25 along with (Z)-25 in 27% combined yield at a 3.5:1 diastereomeric ratio (Table 1, entry 1). When NaHMDS was used as the base, the chemical yield was improved to 77%, however, the E/Z ratio was decreased to 1.3:1 (Table 1, entry 2). When LDA was used as the base, the chemical yield and diastereomeric ratio were increased to 98% and 2.6:1, respectively (Table 1, entry 3). Reaction of PT-sulfone 24 using LDA gave the coupling products (E)- and (Z)-26 in 86% yield, wherein the diastereomeric ratio was increased to 5.0:1 (Table 1, entry 4). The configurations of the coupling products were elucidated by their coupling constants between H-93 and H-94 (15.3 Hz in (E)-25 and (E)-26, 10.7 Hz in (Z)-25 and (Z)-26). Finally, the desired alkene (E)-26 was subjected to the Sharpless AD [15] with AD-mix-β to furnish the C79–C97 fragment 27 in 72% yield as a single diastereomer (Scheme 5). The configuration of the resulting two vicinal hydroxy groups at C93 and C94 of 27 were unambiguously confirmed by the modified Mosher method, respectively (see Supporting Information File 1).
Table 1: Julia–Kocienski olefination between aldehyde 20 and PT-sulfones 23 and 24.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-228-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | PT-Sulfone | Base | Yield (%)a | Ratio (E:Z)b |
|---|---|---|---|---|
| 1 | 23 | KHMDS | 27 | 3.5:1 |
| 2 | 23 | NaHMDS | 77 | 1.3:1 |
| 3 | 23 | LDA | 98 | 2.6:1 |
| 4 | 24 | LDA | 86 | 5.0:1 |
aIsolated yield from 20. bDetermined by 1H NMR spectroscopy.
![[1860-5397-9-228-i5]](/bjoc/content/inline/1860-5397-9-228-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the C79–C97 fragment 27.
Scheme 5: Synthesis of the C79–C97 fragment 27.
In conclusion, we have achieved the stereoselective synthesis of the C79–C97 fragment. The synthetic route has featured a stereoselective spiroacetalization, a Julia–Kocienski olefination, and a Sharpless asymmetric dihydroxylation. This synthesis of the spiroacetal fragment, wherein the two-step sequence was conducted and the overall yield was 52% from the coupling, has been improved over the previous synthesis wherein the eight-step transformation was needed and the overall yield was 31% from the coupling. Further synthetic effort of symbiodinolide toward the complete structural elucidation is currently underway and will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectroscopic data, and NMR spectra of all new compounds. | ||
| Format: PDF | Size: 5.0 MB | Download |
Acknowledgements
We are grateful to Mr. Yohei Kawakubo (Okayama University) for valuable discussions. We acknowledge The Kurata Memorial Hitachi Science and Technology Foundation, The Research Foundation for Pharmaceutical Sciences, and The Society of Synthetic Organic Chemistry, Japan (Shionogi Award in Synthetic Organic Chemistry, Japan) for financial supports. This research was supported by a Grant-in-Aid for Scientific Research (No. 24710250) from the Japan Society for the Promotion of Science (JSPS).
References
-
Kita, M.; Ohishi, N.; Konishi, K.; Kondo, M.; Koyama, T.; Kitamura, M.; Yamada, K.; Uemura, D. Tetrahedron 2007, 63, 6241–6251. doi:10.1016/j.tet.2007.02.093
Return to citation in text: [1] [2] -
Han, C.; Uemura, D. Tetrahedron Lett. 2008, 49, 6988–6990. doi:10.1016/j.tetlet.2008.09.118
Return to citation in text: [1] -
Han, C.; Yamano, Y.; Kita, M.; Takamura, H.; Uemura, D. Tetrahedron Lett. 2009, 50, 5280–5282. doi:10.1016/j.tetlet.2009.07.014
Return to citation in text: [1] -
Takamura, H.; Ando, J.; Abe, T.; Murata, T.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2008, 49, 4626–4629. doi:10.1016/j.tetlet.2008.05.078
Return to citation in text: [1] [2] [3] -
Murata, T.; Sano, M.; Takamura, H.; Kadota, I.; Uemura, D. J. Org. Chem. 2009, 74, 4797–4803. doi:10.1021/jo900546k
Return to citation in text: [1] -
Takamura, H.; Murata, T.; Asai, T.; Kadota, I.; Uemura, D. J. Org. Chem. 2009, 74, 6658–6666. doi:10.1021/jo901162v
Return to citation in text: [1] -
Takamura, H.; Kadonaga, Y.; Yamano, Y.; Han, C.; Aoyama, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2009, 50, 863–866. doi:10.1016/j.tetlet.2008.11.056
Return to citation in text: [1] -
Takamura, H.; Kadonaga, Y.; Yamano, Y.; Han, C.; Kadota, I.; Uemura, D. Tetrahedron 2009, 65, 7449–7456. doi:10.1016/j.tet.2009.07.019
Return to citation in text: [1] [2] -
Takamura, H.; Kadonaga, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2010, 51, 2603–2605. doi:10.1016/j.tetlet.2010.03.014
Return to citation in text: [1] -
Takamura, H.; Kadonaga, Y.; Kadota, I.; Uemura, D. Tetrahedron 2010, 66, 7569–7576. doi:10.1016/j.tet.2010.07.045
Return to citation in text: [1] -
Takamura, H.; Tsuda, K.; Kawakubo, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2012, 53, 4317–4319. doi:10.1016/j.tetlet.2012.06.005
Return to citation in text: [1] -
Blakemore, P. R.; Cole, W. J.; Kocieński, P. J.; Morley, A. Synlett 1998, 26–28. doi:10.1055/s-1998-1570
Return to citation in text: [1] [2] -
Aïssa, C. Eur. J. Org. Chem. 2009, 1831–1844. doi:10.1002/ejoc.200801117
Return to citation in text: [1] [2] -
Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H
Return to citation in text: [1] [2] -
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009
Return to citation in text: [1] [2] -
Paterson, I.; Anderson, E. A.; Dalby, S. M.; Lim, J. H.; Maltas, P.; Loiseleur, O.; Genovino, J.; Moessner, C. Org. Biomol. Chem. 2012, 10, 5861–5872. doi:10.1039/c2ob25100k
Return to citation in text: [1] -
Yamaguchi, M.; Hirao, I. Tetrahedron Lett. 1983, 24, 391–394. doi:10.1016/S0040-4039(00)81416-1
Return to citation in text: [1] -
Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis 1994, 639–666. doi:10.1055/s-1994-25538
Return to citation in text: [1] -
Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015
Return to citation in text: [1] -
Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n
Return to citation in text: [1] -
Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505–5507. doi:10.1021/ja00997a067
Return to citation in text: [1] -
Kurosawa, S.; Mori, K. Eur. J. Org. Chem. 2000, 955–962. doi:10.1002/(SICI)1099-0690(200003)2000:6<955::AID-EJOC955>3.0.CO;2-Y
Return to citation in text: [1] -
Schultz, H. S.; Freyermuth, H. B.; Buc, S. R. J. Org. Chem. 1963, 28, 1140–1142. doi:10.1021/jo01039a512
Return to citation in text: [1]
| 15. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009 |
| 1. | Kita, M.; Ohishi, N.; Konishi, K.; Kondo, M.; Koyama, T.; Kitamura, M.; Yamada, K.; Uemura, D. Tetrahedron 2007, 63, 6241–6251. doi:10.1016/j.tet.2007.02.093 |
| 12. | Blakemore, P. R.; Cole, W. J.; Kocieński, P. J.; Morley, A. Synlett 1998, 26–28. doi:10.1055/s-1998-1570 |
| 13. | Aïssa, C. Eur. J. Org. Chem. 2009, 1831–1844. doi:10.1002/ejoc.200801117 |
| 14. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H |
| 23. | Schultz, H. S.; Freyermuth, H. B.; Buc, S. R. J. Org. Chem. 1963, 28, 1140–1142. doi:10.1021/jo01039a512 |
| 4. | Takamura, H.; Ando, J.; Abe, T.; Murata, T.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2008, 49, 4626–4629. doi:10.1016/j.tetlet.2008.05.078 |
| 12. | Blakemore, P. R.; Cole, W. J.; Kocieński, P. J.; Morley, A. Synlett 1998, 26–28. doi:10.1055/s-1998-1570 |
| 13. | Aïssa, C. Eur. J. Org. Chem. 2009, 1831–1844. doi:10.1002/ejoc.200801117 |
| 14. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H |
| 4. | Takamura, H.; Ando, J.; Abe, T.; Murata, T.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2008, 49, 4626–4629. doi:10.1016/j.tetlet.2008.05.078 |
| 5. | Murata, T.; Sano, M.; Takamura, H.; Kadota, I.; Uemura, D. J. Org. Chem. 2009, 74, 4797–4803. doi:10.1021/jo900546k |
| 6. | Takamura, H.; Murata, T.; Asai, T.; Kadota, I.; Uemura, D. J. Org. Chem. 2009, 74, 6658–6666. doi:10.1021/jo901162v |
| 7. | Takamura, H.; Kadonaga, Y.; Yamano, Y.; Han, C.; Aoyama, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2009, 50, 863–866. doi:10.1016/j.tetlet.2008.11.056 |
| 8. | Takamura, H.; Kadonaga, Y.; Yamano, Y.; Han, C.; Kadota, I.; Uemura, D. Tetrahedron 2009, 65, 7449–7456. doi:10.1016/j.tet.2009.07.019 |
| 9. | Takamura, H.; Kadonaga, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2010, 51, 2603–2605. doi:10.1016/j.tetlet.2010.03.014 |
| 10. | Takamura, H.; Kadonaga, Y.; Kadota, I.; Uemura, D. Tetrahedron 2010, 66, 7569–7576. doi:10.1016/j.tet.2010.07.045 |
| 11. | Takamura, H.; Tsuda, K.; Kawakubo, Y.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2012, 53, 4317–4319. doi:10.1016/j.tetlet.2012.06.005 |
| 21. | Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505–5507. doi:10.1021/ja00997a067 |
| 1. | Kita, M.; Ohishi, N.; Konishi, K.; Kondo, M.; Koyama, T.; Kitamura, M.; Yamada, K.; Uemura, D. Tetrahedron 2007, 63, 6241–6251. doi:10.1016/j.tet.2007.02.093 |
| 2. | Han, C.; Uemura, D. Tetrahedron Lett. 2008, 49, 6988–6990. doi:10.1016/j.tetlet.2008.09.118 |
| 3. | Han, C.; Yamano, Y.; Kita, M.; Takamura, H.; Uemura, D. Tetrahedron Lett. 2009, 50, 5280–5282. doi:10.1016/j.tetlet.2009.07.014 |
| 22. | Kurosawa, S.; Mori, K. Eur. J. Org. Chem. 2000, 955–962. doi:10.1002/(SICI)1099-0690(200003)2000:6<955::AID-EJOC955>3.0.CO;2-Y |
| 4. | Takamura, H.; Ando, J.; Abe, T.; Murata, T.; Kadota, I.; Uemura, D. Tetrahedron Lett. 2008, 49, 4626–4629. doi:10.1016/j.tetlet.2008.05.078 |
| 18. | Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis 1994, 639–666. doi:10.1055/s-1994-25538 |
| 16. | Paterson, I.; Anderson, E. A.; Dalby, S. M.; Lim, J. H.; Maltas, P.; Loiseleur, O.; Genovino, J.; Moessner, C. Org. Biomol. Chem. 2012, 10, 5861–5872. doi:10.1039/c2ob25100k |
| 19. | Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015 |
| 20. | Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n |
| 8. | Takamura, H.; Kadonaga, Y.; Yamano, Y.; Han, C.; Kadota, I.; Uemura, D. Tetrahedron 2009, 65, 7449–7456. doi:10.1016/j.tet.2009.07.019 |
| 15. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009 |
| 17. | Yamaguchi, M.; Hirao, I. Tetrahedron Lett. 1983, 24, 391–394. doi:10.1016/S0040-4039(00)81416-1 |
© 2013 Takamura et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)