Abstract
The regioselective carbon–carbon bond formation was studied using 5,5,5-trifluoro-1-phenylpent-3-en-1-yne as a model substrate, and predominant acceptance of electrophiles β to a CF3 group as well as a deuterium trap experiment of the lithiated species led to the conclusion that the obtained regioselectivity is kinetically determined for the reactions with electrophiles, under equilibration of the possible two anionic species.

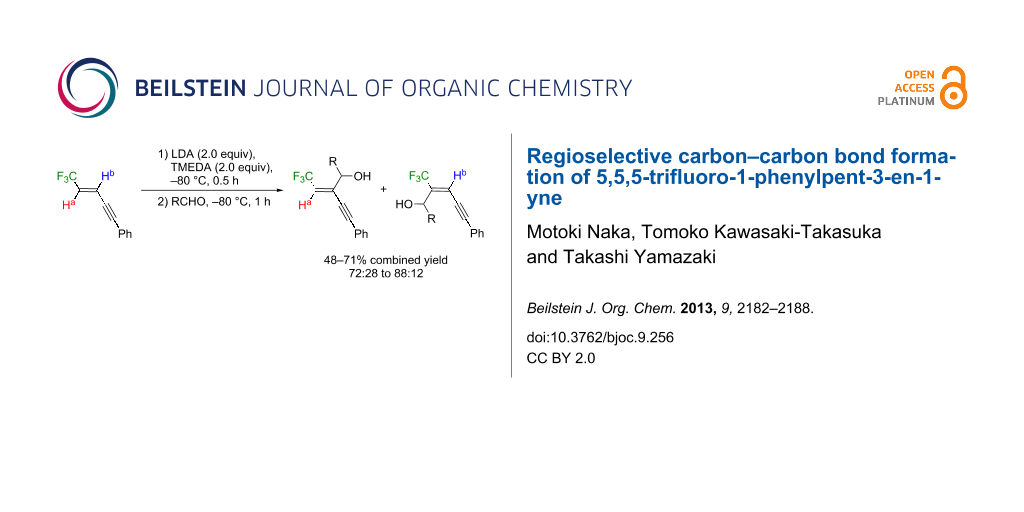
Graphical Abstract
Introduction
We have previously reported [1] the interesting behavior of (E)-1-chloro-3,3,3-trifluoropropene ((E)-1) [2-4] towards MeLi, where the proportion of two possible products, propargylic alcohols 2 and allylic alcohols 3, was proved to be significantly dependent on the equivalents of MeLi used. Thus, as shown in Scheme 1, under the action of up to 1.6 equiv of MeLi, 2 was obtained as a sole product probably by initial Hb abstraction from (E)-1 and the resultant Int-1 was stabilized by the energetically favorable 5-membered intramolecular Li···F chelation [5]. This intermediate Int-1 experienced Fritsch–Buttenberg-Wiechell (FBW) rearrangement [6,7] to give 3,3,3-trifluoropropyne, and Int-2 derived from this alkyne eventually captured appropriate aldehydes to afford CF3-containing propargylic alcohols 2 [8-12]. Alternatively, stereospecific and exclusive construction of the corresponding allylic alcohols 3 was attained by utilization of greater than 1.7 equiv of MeLi where the stabilized intermediate Int-3 formed by complexation of Int-1 with MeLi might play an important role. It turned out that the isomeric (Z)-1 as a substrate furnished only 2 [13] even by the addition of 1.7 equiv of MeLi, presumably as a result of regioselective deprotonation of Ha, followed by smooth elimination [14,15] of the trans-disposed chlorine atom. However, it is also likely that the anionic intermediate produced after Hb abstraction would prefer the reaction course to 2 by way of FBW rearrangement because of its lower stability than Int-1 with loss of the possibility for Li···F chelation.
![[1860-5397-9-256-i1]](/bjoc/content/inline/1860-5397-9-256-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed reaction mechanism between 1 and MeLi.
Scheme 1: Proposed reaction mechanism between 1 and MeLi.
It was not only MeLi but also LDA which demonstrated this unique base-amount dependent product selectivity of (E)-1 and this substrate was successfully converted to 2 or a mixture of 2:3 = 25:75 by the action of 1.2 or 2.2 equiv of LDA, respectively. Although these phenomena are quite interesting, issues remain to be solved for complete mechanistic understanding of the process depicted in Scheme 1.
For clarification of the reactivity of (E)-1 on deprotonation, enyne 4 was used as a model because, after trapping the vinylic anionic species by appropriate electrophiles, comparison of the yields of the resultant regioisomeric 5 and 6 would give us a hint for solving this puzzling question. Moreover, we also expected that such data as well as the additional deuterium trap experiment would offer deeper insight to the actual mechanism.
Results and Discussion
Investigation of reaction conditions was initially carried out in Et2O. A complete recovery of 4 was observed irrespective of the base used (Table 1, entries 1 and 2).
Table 1: Investigation of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-256-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Base | Additive | Temp. | 19F NMR Yield (%) | Recov.a | |||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | (equiv) | (equiv) | (°C) | 5a | 6a | (%) |
| 1 | Et2O | n-BuLi (1.0) | — | −80 | 0 | 0 | 100 |
| 2 | MeLi (1.0) | — | −80 | 0 | 0 | 100 | |
| 3 | THF | n-BuLi (1.0) | — | −80 | 0 | 0 | 5 |
| 4 | MeLi (1.0) | — | −80 | 0 | 0 | 17 | |
| 5 | LDA (1.0) | — | −80 | 24 | 6 | 20 | |
| 6 | PhMgBr (1.0) | — | −80 | 0 | 0 | 17 | |
| 7 | LHMDS (1.0) | — | −80 | 0 | 0 | 17 | |
| 8 | LDA (2.0) | — | −80 | 41 | 10 | 15 | |
| 9 | LDA (3.0) | — | −80 | 44 | 8 | 32 | |
| 10 | LDA (1.0) | HMPA (1.0) | −80 | —b | |||
| 11 | LDA (1.0) | DMPU (1.0) | −80 | —b | |||
| 12 | LDA (1.0) | TMEDA (1.0) | −80 | 34 | 2 | 11 | |
| 13 | LDA (2.0) | TMEDA (2.0) | −80 | 68 | 11 | 15 | |
| 14 | LDA (3.0) | TMEDA (3.0) | −80 | 64 | 19 | 14 | |
| 15 | LDA (2.0) | TMEDA (2.0) | −100 | 19 | 5 | 76 | |
| 16 | LDA (2.0) | TMEDA (2.0) | −60 | 50 | 17 | 1 | |
| 17 | LDA (2.0) | TMEDA (2.0) | −40 | —b | 43 | ||
| 18 | LDA (2.0) | TMEDA (2.0) | −80c | 54 | 17 | 8 | |
aRecovered starting material. bAlmost no fluorinated products were detected by 19F NMR. cAfter addition of PhCHO, stirring was continued for 1 h at −80 °C, followed by 3 h at 0 °C.
Neither were 5a and 6a detected in THF when MeLi or n-BuLi were employed and this system produced only a complex mixture (Table 1, entries 3 and 4). A survey of bases established that, in spite of failures with PhMgBr and LHMDS, the use of LDA led to the formation of 5a and 6a in 24 and 6% yields, respectively, after trapping the resultant anionic intermediates by PhCHO (Table 1, entries 5 to 7). An increase in LDA concentration to 2 equiv improved the yield of product to 41% (5a) and 10% (6a), but additional LDA was not effective (Table 1, entries 8 and 9). Subsequent to determining that LDA was the base of choice, a brief study of the effect of additives was performed. The addition of HMPA or DMPU was detrimental and no trace amount of 4, 5a, nor 6a was detected (Table 1, entries 10 and 11). However, TMEDA affected this process to some extent (Table 1, entries 5 vs 12) [16-18], and the addition of 2 equiv each of LDA and TMEDA produced the regioisomeric 5a and 6a in better yields of 68% and 11%, respectively (Table 1, entry 13). The recovery of 15% of the substrate 4 under these conditions prompted further raise in their amount to 3 equiv, but again, no significant improvement was noticed (Table 1, entries 13 vs 14). The effect of temperature on the reaction was crucial. Thus, efficient deprotonation was not occurred at −100 °C (Table 1, entry 15) with the consequence that decomposition of the vinylic anions at −40 °C led to formation of different intermediates which would further consume LDA, leaving a larger amount of 4 unreacted (Table 1, entry 17). In conclusion, the reaction conditions in entry 13 in Table 1 were selected to be optimal.
With optimized conditions in hand, the scope and limitation of this procedure were investigated. A number of carbonyl compounds were employed as electrophiles for the anions generated from the enyne 4. Benzaldehydes with electron-donating (Table 2, entries 2 and 3) as well as -withdrawing (Table 2, entry 4) substituents at the para position were nicely accepted as electrophiles. The allylic alcohols 5 were formed as the major product without exception in a similar 5/(5+6) ratio of 70–75% (Table 2, entries 1 to 5). An inseparable mixture of the compounds 5 and 6 was also synthesized from aliphatic aldehydes as shown in entries 5 and 6 in Table 2. Their proportion seemed to be affected by the bulky substituent and pivalaldehyde attained the higher ratio of 88%. Although 5 and 6 were obtained in 50 to 70% combined yields by the reaction with aldehydes, lower reactivity was displayed by the less electrophilic and more hindered acetophenone. Products 5g and 6g were formed in 15% and 8% yields, respectively, while there was a 52% recovery of 4 under these conditions detected by 19F NMR (Table 2, entry 7).
Table 2: Scope and limitation of the present reactions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-256-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Isolated yielda (%) | 5/(5+6)b | Recov.c | |||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | 5 | 6 | (%) | (%) |
| 1 | Ph- | H | a | 50 (65) | 21 (25) | 72 | 15 |
| 2 | p-Me-C6H4- | H | b | 49 (52) | 17 (17) | 75 | 18 |
| 3 | p-MeO-C6H4- | H | c | 49 (43) | 15 (17) | 72 | 16 |
| 4 | p-F3C-C6H4- | H | d | 36 (43) | 12 (17) | 72 | 32 |
| 5 | Et- | H | e | 47 (57) | 16 (19) | 75 | 26 |
| 6 | t-Bu- | H | f | 54 (59) | 10 (8) | 88 | 23 |
| 7 | Ph- | Me- | g | (15) | (8) | 65 | 52 |
aIn the parenthesis were shown the yields determined by 19F NMR. bThese ratios were determined by 19F NMR for the crude materials. cRecovered starting material.
Compounds 5 and 6 were readily characterized by 19F NMR spectra. The former material showed clear 3J couplings of 7 to 9 Hz between F and Ha, but with the latter 4JF-Hb was usually not observed. Moreover, the fact that isolation of the furan 7a in 51% yield was realized by subjection of a 70:30 mixture of inseparable 5a and 6a to the already reported Pd-catalyzed cyclization conditions [19] suggested that the major isomer should be 5a, not 6a (Scheme 2). In the case of 5 and 6, electrophiles were incorporated without stereochemical contamination in all instances.
![[1860-5397-9-256-i2]](/bjoc/content/inline/1860-5397-9-256-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Furan synthesis from a mixture of 5a and 6a.
Scheme 2: Furan synthesis from a mixture of 5a and 6a.
For obtaining further mechanistic proofs for the present reaction, we have planned to capture the intermediary anionic species with the aid of the usual deuteration technique. Thus, a large excess amount (26 equiv) of CD3OD was introduced to a solution containing the anionic species which was prepared from 4 by the standard conditions, which expected us to obtain a mixture of 4-d1 and 4-d2 or either of them predominantly (Scheme 3).
![[1860-5397-9-256-i3]](/bjoc/content/inline/1860-5397-9-256-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deuteration of anionic species from 4.
Scheme 3: Deuteration of anionic species from 4.
Observation of the well-resolved two sets of peaks was possible for the two vinylic protons Ha and Hb in the enyne 4 at δ 6.15 (qd, J = 6.6, 15.9 Hz) and 6.48 (qd, J = 2.4, 15.9 Hz), respectively (Figure 1 and Supporting Information File 1).
![[1860-5397-9-256-1]](/bjoc/content/figures/1860-5397-9-256-1.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 1: A part of 1H NMR chart of 4 and its deuterated mixture.
Figure 1: A part of 1H NMR chart of 4 and its deuterated mixture.
On the other hand, relatively complex peaks were detected from the crude deuterated mixture due to incomplete quench of the reactive anionic species by CD3OD, but new resonance peaks (indicated by ■ and▲) undoubtedly appeared with overlapping the ones of the original Ha (![[Graphic 3]](/bjoc/content/inline/1860-5397-9-256-i8.png?max-width=637&scale=0.472728) ) and Hb (●). At the lower field (around 6.5 ppm) area, peaks indicated by ▲ are considered as a part of quartet with a coupling constant of 2.3 Hz, which is close to the 4JH-F coupling value of 4 shown above. Moreover, disappearance of the large 3JH-H constant typical for (E)-alkenes suggested that a deuterium atom should be incorporated at the site where Ha was originally situated and that 4-d2 was actually produced. It is also clear that the Ha region also contains a couple of peaks (■) with the 3JH-F coupling constant of 6.6 Hz basically identical to the one of 4, which anticipated us the simultaneous formation of the regioisomeric 4-d1 on the basis of the similar consideration.
) and Hb (●). At the lower field (around 6.5 ppm) area, peaks indicated by ▲ are considered as a part of quartet with a coupling constant of 2.3 Hz, which is close to the 4JH-F coupling value of 4 shown above. Moreover, disappearance of the large 3JH-H constant typical for (E)-alkenes suggested that a deuterium atom should be incorporated at the site where Ha was originally situated and that 4-d2 was actually produced. It is also clear that the Ha region also contains a couple of peaks (■) with the 3JH-F coupling constant of 6.6 Hz basically identical to the one of 4, which anticipated us the simultaneous formation of the regioisomeric 4-d1 on the basis of the similar consideration.
Thus, lithiation of 4 and the following quench with CD3OD furnished a mixture of 4-d1 and 4-d2, which directly proved that the both lithiated species 4-Li1 and 4-Li2 were actually generated (Scheme 4), although it is quite unfortunate not to be able to quantitatively discuss the proportion of 4-d1 to 4-d2. On the other hand, as apparent from Table 2, aldehydes preferentially yielded 5 with qualitatively better ratio of 5/6 than the one of 4-d1/4-d2. Our computation of 4-Li1 and 4-Li2 by Gaussian 09W [20] using the B3LYP/6-31+G* level of theory uncovered that the former was energetically more favorable than the latter by 5.34 (3.67) kcal/mol under vacuum (in THF [21]), which would indicate that intramolecular Li···F chelation is contributed to the stability more significantly rather than the electron-withdrawing stabilization by the CF3 group. Thus, with this computational information in hand, the process shown in Scheme 4 would be elucidated as follows: deuteration would quickly occur to afford 4-d1 and 4-d2 whose proportion would reflect the ratio of 4-Li1 and 4-Li2. On the other hand, less reactive aldehydes should be captured more slowly and the product preference of 5 to 6 would be kinetically determined by their activation energy difference with equilibration between these two lithiated species in the presence of such a proton source as iPr2NH [22].
![[1860-5397-9-256-i4]](/bjoc/content/inline/1860-5397-9-256-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Lithiation of 4 and the following electrophilic reactions.
Scheme 4: Lithiation of 4 and the following electrophilic reactions.
This interpretation also led to the reconsideration of the proposed mechanism shown in Scheme 1. If deprotonation of both Ha and Hb in (E)-1 was possible, lithiated species Int-4 and Int-1 was obtained, respectively (Scheme 5). Due to slow elimination of a LiCl molecule from Int-4 due to the cis relationship of Li and Cl, Int-4 would act as a base to abstract a terminal proton of 3,3,3-trifluoropropyne via FBW rearrangement of Int-1. The regenerated (E)-1 would be converted to a mixture of Int-1 and Int-4 again to eventually yield the propargylic alcohols 2. However, a larger amount of MeLi would allow constructing the complex Int-3 presumably with possessing better stability and the allylic alcohols 3 were obtained. In the case of the stereoisomeric (Z)-1, situation was totally different, and Int-5 without intramolecular chelation should accelerate the elimination of LiCl and the ideal trans disposition of Li and Cl in Int-6 also favored the route to trifluoropropyne, and as a result, propargylic alcohols 2 were selectively afforded.
![[1860-5397-9-256-i5]](/bjoc/content/inline/1860-5397-9-256-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Alternative reaction mechanism between 1 and MeLi.
Scheme 5: Alternative reaction mechanism between 1 and MeLi.
Conclusion
By using the representative model substrate 4, we have reached to a conclusion that its initial lithiation would occur at both vinylic sites and the following reactions with appropriate electrophiles would proceed under kinetic control where equilibration of the resultant lithiated species might be effective if the capture of electrophiles was slow. This important information allowed us to reconsider our previously proposed mechanism as shown in Scheme 5.
Supporting Information
| Supporting Information File 1: Experimental. | ||
| Format: PDF | Size: 170.6 KB | Download |
References
-
Miyagawa, A.; Naka, M.; Yamazaki, T.; Kawasaki-Takasuka, T. Eur. J. Org. Chem. 2009, 4395–4399. doi:10.1002/ejoc.200900370
Return to citation in text: [1] -
Taguchi, T.; Tomizawa, G.; Nakajima, M.; Kobayashi, Y. Chem. Pharm. Bull. 1985, 33, 4077–4080. doi:10.1248/cpb.33.4077
Return to citation in text: [1] -
Quan, H.-D.; Yang, H.-E.; Tamura, M.; Sekiya, A. J. Fluorine Chem. 2007, 128, 190–195. doi:10.1016/j.jfluchem.2006.12.004
Return to citation in text: [1] -
Komata, T.; Akiba, S.; Hosoi, K.; Ogura, K. J. Fluorine Chem. 2008, 129, 35–39. doi:10.1016/j.jfluchem.2007.08.007
Return to citation in text: [1] -
Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013
Return to citation in text: [1] -
Jahnke, E.; Tykwinski, R. R. Chem. Commun. 2010, 46, 3235–3249. doi:10.1039/c003170d
Return to citation in text: [1] -
Knorr, R. Chem. Rev. 2004, 104, 3795–3850. doi:10.1021/cr030616h
Return to citation in text: [1] -
Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/S0022-1139(00)80446-7
Return to citation in text: [1] -
Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/C39950000051
Return to citation in text: [1] -
Katrizky, A. R.; Qi, M.; Wells, A. P. J. Fluorine Chem. 1996, 80, 145–147. doi:10.1016/S0022-1139(96)03494-X
Return to citation in text: [1] -
Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h
Return to citation in text: [1] -
Shimizu, M.; Higashi, M.; Takeda, Y.; Murai, M.; Jiang, G.; Asai, Y.; Nakao, Y.; Shirakawa, E.; Hiyama, T. Future Med. Chem. 2009, 1, 921–945. doi:10.4155/fmc.09.69
Return to citation in text: [1] -
Eymery, F.; Iorga, B.; Savignac, P. Synthesis 2000, 185–213. doi:10.1055/s-2000-6241
Return to citation in text: [1] -
Cristol, S. J.; Begoon, A.; Norris, W. P.; Ramey, P. S. J. Am. Chem. Soc. 1954, 76, 4558–4561. doi:10.1021/ja01647a015
Return to citation in text: [1] -
Behr, O. M.; Eglinton, G.; Lardy, I. A.; Raphael, R. A. J. Chem. Soc. 1964, 1151–1154. doi:10.1039/jr9640001151
Return to citation in text: [1] -
Kacem, Y.; Hassine, B. B. Tetrahedron Lett. 2013, 54, 4023–4025. doi:10.1016/j.tetlet.2013.05.082
Return to citation in text: [1] -
Riggs, J. C.; Singh, K. J.; Yun, M.; Collum, D. B. J. Am. Chem. Soc. 2008, 130, 13709–13717. doi:10.1021/ja804087r
Return to citation in text: [1] -
Grant, B. J.; Kramp, C. R.; Knight, J. D.; Meierhoefer, M. A.; Vella, J. H.; Sober, C. L.; Jones, S. S.; Metz, C. R.; Beam, C. F.; Pennington, W. T.; Vanderveer, D. G.; Camper, N. D. J. Heterocycl. Chem. 2007, 44, 627–632. doi:10.1002/jhet.5570440318
Return to citation in text: [1] -
Qing, F.-L.; Gao, W.-Z.; Ying, J. J. Org. Chem. 2000, 65, 2003–2006. doi:10.1021/jo991463u
Return to citation in text: [1] -
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] -
The integral equation formalism polarizable continuum model (IEF-PCM) was used for consideration of the solvent effect of THF.
Return to citation in text: [1] -
Mongin, F.; Tognini, A.; Cottet, F.; Schlosser, M. Tetrahedron Lett. 1998, 39, 1749–1752. doi:10.1016/S0040-4039(98)00028-8
Return to citation in text: [1]
| 1. | Miyagawa, A.; Naka, M.; Yamazaki, T.; Kawasaki-Takasuka, T. Eur. J. Org. Chem. 2009, 4395–4399. doi:10.1002/ejoc.200900370 |
| 8. | Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/S0022-1139(00)80446-7 |
| 9. | Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/C39950000051 |
| 10. | Katrizky, A. R.; Qi, M.; Wells, A. P. J. Fluorine Chem. 1996, 80, 145–147. doi:10.1016/S0022-1139(96)03494-X |
| 11. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h |
| 12. | Shimizu, M.; Higashi, M.; Takeda, Y.; Murai, M.; Jiang, G.; Asai, Y.; Nakao, Y.; Shirakawa, E.; Hiyama, T. Future Med. Chem. 2009, 1, 921–945. doi:10.4155/fmc.09.69 |
| 6. | Jahnke, E.; Tykwinski, R. R. Chem. Commun. 2010, 46, 3235–3249. doi:10.1039/c003170d |
| 7. | Knorr, R. Chem. Rev. 2004, 104, 3795–3850. doi:10.1021/cr030616h |
| 5. | Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013 |
| 2. | Taguchi, T.; Tomizawa, G.; Nakajima, M.; Kobayashi, Y. Chem. Pharm. Bull. 1985, 33, 4077–4080. doi:10.1248/cpb.33.4077 |
| 3. | Quan, H.-D.; Yang, H.-E.; Tamura, M.; Sekiya, A. J. Fluorine Chem. 2007, 128, 190–195. doi:10.1016/j.jfluchem.2006.12.004 |
| 4. | Komata, T.; Akiba, S.; Hosoi, K.; Ogura, K. J. Fluorine Chem. 2008, 129, 35–39. doi:10.1016/j.jfluchem.2007.08.007 |
| 19. | Qing, F.-L.; Gao, W.-Z.; Ying, J. J. Org. Chem. 2000, 65, 2003–2006. doi:10.1021/jo991463u |
| 21. | The integral equation formalism polarizable continuum model (IEF-PCM) was used for consideration of the solvent effect of THF. |
| 16. | Kacem, Y.; Hassine, B. B. Tetrahedron Lett. 2013, 54, 4023–4025. doi:10.1016/j.tetlet.2013.05.082 |
| 17. | Riggs, J. C.; Singh, K. J.; Yun, M.; Collum, D. B. J. Am. Chem. Soc. 2008, 130, 13709–13717. doi:10.1021/ja804087r |
| 18. | Grant, B. J.; Kramp, C. R.; Knight, J. D.; Meierhoefer, M. A.; Vella, J. H.; Sober, C. L.; Jones, S. S.; Metz, C. R.; Beam, C. F.; Pennington, W. T.; Vanderveer, D. G.; Camper, N. D. J. Heterocycl. Chem. 2007, 44, 627–632. doi:10.1002/jhet.5570440318 |
| 22. | Mongin, F.; Tognini, A.; Cottet, F.; Schlosser, M. Tetrahedron Lett. 1998, 39, 1749–1752. doi:10.1016/S0040-4039(98)00028-8 |
| 14. | Cristol, S. J.; Begoon, A.; Norris, W. P.; Ramey, P. S. J. Am. Chem. Soc. 1954, 76, 4558–4561. doi:10.1021/ja01647a015 |
| 15. | Behr, O. M.; Eglinton, G.; Lardy, I. A.; Raphael, R. A. J. Chem. Soc. 1964, 1151–1154. doi:10.1039/jr9640001151 |
| 13. | Eymery, F.; Iorga, B.; Savignac, P. Synthesis 2000, 185–213. doi:10.1055/s-2000-6241 |
© 2013 Naka et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)