Bidirectional cross metathesis and ring-closing metathesis/ring opening of a C2-symmetric building block: a strategy for the synthesis of decanolide natural products
and
Bernd Schmidt
Institut für Chemie, Organische Synthesechemie, Universität Potsdam, Karl-Liebknecht-Straße 24-25, 14476 Potsdam-Golm, Germany
Guest Editor: J. S. Dickschat Beilstein J. Org. Chem.2013,9, 2544–2555.https://doi.org/10.3762/bjoc.9.289 Received 14 Aug 2013,
Accepted 29 Oct 2013,
Published 18 Nov 2013
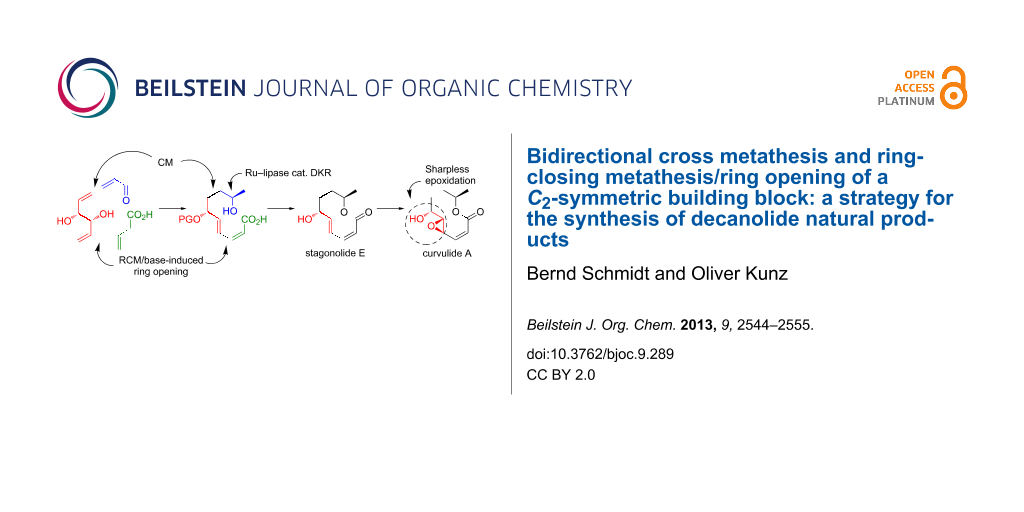
Starting from the conveniently available ex-chiral pool building block (R,R)-hexa-1,5-diene-3,4-diol, the ten-membered ring lactones stagonolide E and curvulide A were synthesized using a bidirectional olefin-metathesis functionalization of the terminal double bonds. Key steps are (i) a site-selective cross metathesis, (ii) a highly diastereoselective extended tethered RCM to furnish a (Z,E)-configured dienyl carboxylic acid and (iii) a Ru–lipase-catalyzed dynamic kinetic resolution to establish the desired configuration at C9. Ring closure was accomplished by macrolactonization. Curvulide A was synthesized from stagonolide E through Sharpless epoxidation.
Bidirectional synthesis through termini differentiation of meso, C2-symmetric or unsymmetric building blocks has emerged as an important strategy in natural product synthesis over the past two decades [1]. Early on, enantiomerically pure C2-symmetric compounds were identified as particularly useful starting materials, because their termini are homotopic. Therefore, desymmetrization can be accomplished by monofunctionalization, making elaborate reagent or catalyst-controlled transformations unnecessary [2]. In this regard, (R,R)-hexa-1,5-diene-3,4-diol (1) [3-6] and its enantiomer ent-1[7] have emerged as highly valuable starting points for target molecule syntheses which rely on dual olefin metathesis reactions. The two metathesis transformations may either be two identical CM [8,9] or RCM steps [10], yielding C2-symmetric products in which the newly formed double bonds remain homotopic, or two different CM or RCM steps, or a combination of one CM and one RCM transformation. The latter cases lead to C1-symmetric products and hence a differentiation of the C–C double bonds generated through metathesis. Examples for the utilization of these approaches in the synthesis of target molecules from 1 or ent-1 include sialic acids [11], cladospolide C [12], iriomoteolide 3a [13,14], thromboxane B2 [15], didemniserinolipid B [16], squamostolide [17], muricatacine [18], rollicosin [19], phomopsolide C [7] and both enantiomers of seimatopolide A [20].
Over the past few years the development and application of one-flask sequences comprising at least one metathesis step has attracted increasing attention [21-23]. Such sequences provide rapid access to constitutional isomers or functionalized derivatives of the actual metathesis products in just one step. An example recently published by us combines RCM of butenoates 2 with a base-induced highly stereoselective ring opening of the transient metathesis products 4, furnishing exclusively Z,E-dienes 3[24]. We assume that the reaction proceeds via formation of an enolate 5, followed by electrocyclic ring opening to carboxylates 6[25], although a non-concerted pathway can not be excluded (Scheme 1).
To the best of our knowledge, metathesis/non-metathesis one flask sequences have not been used before for the two directional elaboration or desymmetrization of C2-symmetric building blocks. In this contribution, we demonstrate that this combination can be advantageously used for the synthesis of decanolides. These natural products share a ten-membered lactone structure and are normally isolated from fungi. Diverse bioactivities have been reported, such as phytotoxicity, cytotoxicity, antimalarial and antibacterial activity, which are a strong motivation for total synthesis [26,27]. In addition, several ambiguities in the structural assignment of some of these natural products still exist, and chemical synthesis has been proven to be a powerful and reliable tool for completing the structure elucidation and for correcting erroneous assignments. In particular if ex-chiral pool starting materials with well established absolute configurations are used, such as D-mannitol-derived 1 or L-tartrate-derived ent-1, highly reliable structural assignments become possible. Two decanolides, for which the absolute configuration was only assigned based on analogy to related natural products are stagonolide E [28,29] and curvulide A [30]. Stagonolide E is a secondary metabolite of Stagonospora cirsii, which is a fungal pathogen of the weed Cirsium arvense[28]. It has also been isolated from the fungus Curvularia sp. PSU-F22 [29]. Curvulide A was identified as a metabolite of a different strain of Curvularia sp. [30]. In this case, the absolute configuration at C9 was assigned as 9R based on a comparison of its CD spectrum with that of a structurally related compound, whereas the configuration at C6 could not be clarified. For the epoxide moiety of curvulide A, only the relative configurations at C4 and C5 were elucidated based on H,H-coupling constants (Figure 1) [30].
Figure 1:
Structures and numbering scheme for stagonolide E and curvulide A.
Figure 1:
Structures and numbering scheme for stagonolide E and curvulide A.
So far, two syntheses of stagonolide E have been published, which both rely on asymmetric synthesis for establishing both stereocenters. The crucial (2Z,4E)-configuration of the diene moiety was constructed via Still–Gennari olefination [31] or via RCM of an acrylate with an E-configured diene at the opposite terminus [32]. Curvulide A has, to the best of our knowledge, not been synthesized previously.
Results and Discussion
We planned to use a macrolactonization of precursor 7 as the cyclization step. For the synthesis of 7, a cross metathesis of 1 (or a protected derivative) with methyl vinyl ketone (8) was envisaged as the first step, followed by an esterification of the more distant OH group with vinylacetic acid (9). This would provide a precursor 2 (Scheme 1) for the RCM-ring opening sequence (Scheme 2).
Our synthesis of stagonolide E started with a selective protection of one hydroxy group of diol 1 to furnish the known TBS-ether 10[6,19], because we knew from previous experience that a desymmetrization based solely on cross metathesis would most likely be inefficient and yield substantial amounts of double functionalized product (Scheme 3) [9]. We tested methyl vinyl ketone (8) as a cross-metathesis partner, and the recently described Umicore M51 initiator (A) [20,33,34], as well as Grubbs’ second generation carbene complex B[35] as precatalysts (Table 1). Phosphine-free precatalysts with a hemilabile alkoxy ligand are supposed to be well-suited for cross metathesis reactions [36,37]. With a moderate catalyst loading of 2.0 mol % of A, we isolated the expected cross metathesis product 11 in an acceptable yield of 76% (Table 1, entry 1). Surprisingly, the yield decreased considerably to 51% with an increased catalyst loading of 5.0 mol %, and formation of a byproduct with an Rf value very similar to that of the cross metathesis product 11 was observed by TLC (Table 1, entry 2). Inspection of the 1H NMR spectrum of the crude reaction mixture revealed that two singlets at 2.35 ppm and 6.75 ppm in a 1:3 ratio were present, which matches the NMR data previously reported for (E)-hex-3-ene-2,5-dione [38]. Obviously, catalyst A is sufficiently active to initiate the self metathesis of methyl vinyl ketone (8) to a considerable extent at a catalyst loading of 5.0 mol %, and consequently lower amounts of catalyst were tested in the following experiments. With 0.5 mol %, the isolated yield was 67% under otherwise identical conditions (Table 1, entry 3), and 85% of 11 were obtained with 1.0 mol % of A (Table 1, entry 4). With these catalyst loadings self metathesis of 8 is largely suppressed and the chromatographic isolation of 11 is facilitated, which might explain the improved yields under these conditions. With second generation Grubbs’ catalyst B, the self metathesis of the supposedly less reactive CM partner occurs only to a minor extent, even at elevated temperatures and catalyst loadings. When we used 2 mol % of B (Table 1, entry 5) the yield was 61%, but could be improved to 78% by adding phenol (Table 1, entry 6). The beneficial effect of phenol in cross metathesis reactions is known and has been attributed to a stabilization of the reactive 14-electron species [39]. A very high yield of 93% was eventually obtained by using 5 mol % of B in refluxing dichloromethane (Table 1, entry 7).
Scheme 3:
Synthesis of RCM/ring opening precursor 14.
Scheme 3:
Synthesis of RCM/ring opening precursor 14.
Table 1:
Optimization of conditions for CM of 10 and methyl vinyl ketone (8).a
entry
catalyst (mol %)
solvent
T
yield of 11
1
A (2.0)
CH2Cl2
40 °C
76%
2b
A (5.0)
CH2Cl2
40 °C
51%
3
A (0.5)
CH2Cl2
40 °C
67%
4
A (1.0)
CH2Cl2
40 °C
85%
5
B (2.0)
toluene
80 °C
61%
6c
B (2.0)
toluene
80 °C
78%
7
B (5.0)
CH2Cl2
40 °C
93%
aGeneral conditions: 8.0 equiv of 8, initial substrate concentration: c = 0.5 M; bformation of (E)-hex-3-ene-2,5-dione observed in the 1H NMR spectrum of the crude reaction mixture. cWith phenol (0.5 equiv) as additive.
In the next step, the remaining hydroxy group was protected as a MOM ether 12, which was then selectively reduced at the electron-deficient double bond, using polymethylhydrosiloxane (PMHS) [40] as a reducing agent and a BDP–Cu hydride catalyst. This variant of Stryker’s reagent [41] has more recently been described by Lipshutz et al. [42] and was found to furnish ketone 13 in 74% yield. Desilylation of 13 could be accomplished quantitatively using TBAF at elevated temperature. However, an unidentified byproduct presumably a six-membered lacol was formed, which could not be separated by chromatography. Therefore the mixture was subjected to the second step, a Steglich esterification [43] with vinylacetic acid, to give the desired precursor 14 in pure form in 71% yield. To avoid the formation of the inseparable byproduct, we investigated a reversed order of steps. To this end, 12 was first desilylated to allyl alcohol 15, which was then converted to butenoate 16, again via Steglich esterification. For the selective reduction of the enoate 16, the Stryker–Lipshutz protocol was again the method of choice and optimized conditions eventually furnished 14 in 87% yield (Scheme 3).
For the Stryker–Lipshutz reduction of 16 slightly different conditions were used than for the reduction of 12. In particular, tert-butanol was omitted as a co-solvent, and TBAF was added to the reaction mixture after completed reduction. This modification was the result of an optimization study based on mechanistic considerations (Table 2) [44].
Table 2:
Optimization of Cu–H-catalysed reduction of 16.
entry
Cu(OAc)2·H2O (mol %)
BDP (mol %)
PMHS (equiv)
solvent
yield of 14
1
5
1
2
toluene/t-BuOH (5:1)
72%
2
5
1
2
toluene/t-BuOH (2:1)
78%
3
1
0.5
1.2
toluene/t-BuOH (2:1)
67%
4a
5
1
2
toluene
87%
aTBAF (2 equiv) added after complete consumption of starting material.
The conditions previously used for the reduction of enoate 12 involved the use of tert-butanol as a co-solvent, together with toluene. Under these conditions, reproducible yields in the range between 67% and 78% were obtained (Table 2, entries 1–3). The alcohol is believed to protonate the Cu-enolate formed upon conjugate addition, resulting in the ketone and a Cu-alkoxide, which is then reduced with silane to regenerate the Cu-hydride. Alternatively, the Cu-enolate might enter a competing catalytic cycle by reacting with silane, furnishing a silyl enol ether and the catalytically active Cu-hydride species. The silyl enol ether is inert to protonation by tert-butanol, and therefore the competing secondary cycle will result in a decreased yield of reduction product. This reasoning prompted us to run the reaction in toluene without any protic co-solvent, which should exclusively lead to the silyl enol ether, and add TBAF as a desilylating agent after complete consumption of the starting material. The reduced product 14 was isolated under these conditions in 87% yield (Table 2, entry 4). With ketone 14 in hands, we decided to establish the required configuration at C9 in the next step. To this end, a CBS reduction [45,46] catalysed by the oxazaborolidine 17 was tested first (Table 3).
Table 3:
Investigation of CBS reduction of ketone 14.
entry
catalyst (mol %)
reducing agent
T
dra
1
17 (10)
H3B·SMe2
−78 °C
no conversion
2
17 (20)
H3B·THF
20 °C
complex mixture
3
17 (20)
H3B·THF
−50 °C
1:1
4
17 (20)
catechol borane
−78 °C
3:2
aDetermined from 1H NMR spectra of the crude reaction mixtures.
With borane–dimethylsulfide complex as the reductant and 10 mol % of catalyst, no conversion was observed at −78 °C (Table 3, entry 1), whereas attempted reduction at ambient temperature (Table 3, entry 2) resulted in the formation of a complex mixture, presumably due to competing hydroboration of the alkenes. With borane–THF at −50 °C the reduction proceeded to completion, but gave a 1:1 mixture of diastereomers (Table 3, entry 3). With catechol borane at −78 °C conversion was again complete, but the diastereoselectivity was far from being synthetically useful (Table 3, entry 4). Due to these rather discouraging results we did not pursue enantioselective reduction methods further to establish the required 9R-configuration, but considered a resolution approach. Ketone 14 was first reduced with NaBH4 to the expected diastereomeric mixture of alcohols 18, which were then subjected to the conditions of the RCM/base-induced ring-opening sequence. Unfortunately, the expected macrolactonization precursor 19 was not obtained, but an inseparable mixture of products. To access the intended substrate for the resolution, secondary alcohol 19, we investigated an inverted sequence of steps: ketone 14 was first converted to the 9-oxodienoic acid 20 under RCM/ring-opening conditions, followed by a reduction of the ketone with DIBAl-H to furnish 19. Unfortunately, the yields obtained via this two-step sequence were only moderate and most likely to low to provide sufficient amounts of material for an efficient resolution (Scheme 4).
Scheme 4:
Synthesis of a substrate 19 for “late stage” resolution.
Scheme 4:
Synthesis of a substrate 19 for “late stage” resolution.
These unsuccessful attempts to establish the correct configuration at C9 led to a revision of the synthetic strategy. We decided to investigate a dynamic kinetic resolution (DKR) approach at an earlier stage of the synthesis and identified the secondary alcohol 21 as a promising starting point for this approach (Scheme 5).
Scheme 5:
Synthesis of substrate 21 for “early stage” resolution.
Scheme 5:
Synthesis of substrate 21 for “early stage” resolution.
Compound 21 was obtained via two alternate routes, either by reduction of ketone 13 (Scheme 3) with NaBH4 or from ester 25 via one-flask reduction to the corresponding aldehyde and addition of methylmagnesium chloride. Ester 25 was in turn synthesized in three steps from monoprotected dienediol 10 via cross metathesis with methyl acrylate (22) [47] using a comparatively low loading of phosphine-free catalyst A, followed by MOM protection and Stryker–Lipshutz reduction of 24. Notably the latter step proceeds significantly more efficient in a toluene/tert-butanol solvent mixture than the analogous enone reductions outlined in Scheme 3 and Table 2. Compared to these reactions, the saturated ester 25 was obtained in a nearly quantitative yield using half the amount of Cu precatalyst and BDP ligand.
In order to obtain enantiomerically pure 21, an enzyme/transition metal-catalysed approach was investigated [48,49]. In this regard, the combination of Ru complexes such as Shvo’s catalyst (C) [50], the amino-Cp catalyst D[51], or [Ru(CO)2Cl(η5C5Ph5)] [52], and the lipase novozym 435 has emerged as particularly useful [53,54]. We tested Ru catalysts C and D under a variety of conditions (Table 4). In the absence of a Ru catalyst, a kinetic resolution occurs and 26 and the resolved alcohol (2S)-21 were isolated in similar yields (Table 4, entry 1). Upon addition of Shvo’s catalyst C, only minor amounts of the desired acetate 26 and no resolved alcohol were obtained. Instead, the dehydrogenation product 13 was the predominant product (Table 4, entry 2). Addition of the base Na2CO3 led only to a small improvement (Table 4, entry 3). Ketone formation has previously been described in attempted DKR’s of secondary alcohols when catalyst C was used in combination with isopropenyl or vinyl acetate as acylating agents [54]. For this reason, the aminocyclopentadienyl–Ru complex D was evaluated next. Very similar results were obtained with a catalytic amount of KOt-Bu in the presence or absence of a stoichiometric amount of Na2CO3 as a base, at ambient temperature and a reaction time of one week (Table 4, entries 4 and 5). A slightly elevated temperature led to a significantly improved yield of 67% of 26 and 31% of (2S)-21. Both compounds were obtained in a diastereomeric ratio higher than 19:1, as judged from the 1H NMR spectrum (Table 4, entry 6). In an attempt to further improve the yield of 26, the amount of catalyst D was increased to 5 mol %. This resulted indeed in an increased amount of 26, but at the expense of diastereoselectivity, which dropped to 6:1 (Table 4, entry 7). A prolonged reaction time of 14 d led, under otherwise identical conditions, to a slightly improved yield, but at the same time to an even more dramatic erosion of diastereoselectivity (dr = 3:1, Table 4, entry 8). Thus, the conditions listed in Table 4, entry 6 were identified as the optimum. As the alcohol (2S)-21 could also be isolated in a diastereomeric ratio higher than 19:1, it was converted to 26 via Mitsunobu inversion [55] with acetic acid as the nucleophile.
Table 4:
Optimization of conditions for Ru–lipase-catalysed DKR of 21.
entry
conditionsa
26
(2S)-21b,c
13c
1d
Novozym 435, iPPA (1.0 equiv), toluene, 20 °C, 24 h
49%
44%
n. d.
2d
C (2 mol %), Novozym 435, iPPA (10.0 equiv), toluene, 70 °C, 72 h
17%
n. d.
65%
3d
C (1 mol %), Novozym 435, iPPA (10.0 equiv), Na2CO3 (1.0 equiv), toluene, 70 °C, 24 h
30%
n. d.
30%
4d
D (2 mol %), Novozym 435, iPPA (1.5 equiv), Na2CO3 (1.0 equiv); t-BuOK (5 mol %), toluene, 20 °C, 7 d
50%
38%
n. d.
5d
D (2 mol %); Novozym 435, iPPA (1.5 equiv), t-BuOK (5 mol %), toluene, 20 °C, 7 d
50%
n. i.
n. d.
6d
D (2 mol %), Novozym 435, iPPA (3.0 equiv), Na2CO3 (1.0 equiv), t-BuOK (3 mol %), toluene, 30 °C, 7 d
67%
31%
n. d.
7e
D (5 mol %), Novozym 435, iPPA (1.5 equiv), Na2CO3 (1.0 equiv), t-BuOK (6 mol %), toluene, 30 °C, 5 d
76%
20%
n. d.
8f
D (5 mol %), Novozym 435, iPPA (3.0 equiv), Na2CO3 (1.0 equiv), t-BuOK (6 mol %), toluene, 30 °C, 14 d
80%
n. i.
n. d.
aiPPA: isopropenyl acetate; bn. d.: not determined; cn. i.: not isolated; ddr’s of 26 and (2S)-21 >19:1; edr of 26 = 6:1; fdr of 26 = 3:1.
The synthesis of stagonolide E commenced with the desilylation of 26 and Steglich esterification of the resulting allyl alcohol 27. One-flask reaction of 28 with catalyst B, followed by treatment with NaH, resulted in the expected RCM/ring-opening sequence, but also in a partial deacetylation. For this reason, the crude reaction mixture was subsequently treated with aqueous NaOH to complete the ester cleavage, giving the macrolactonization precursor 29[31] in 81% yield (Scheme 6).
Scheme 6:
Synthesis of macrolactonization precursor 29.
Scheme 6:
Synthesis of macrolactonization precursor 29.
In a previous study [24], we had investigated the macrolactonization of the 6-deoxygenated derivative of 29, which is itself a natural product named curvulalic acid (35) [29], and experienced enormous difficulties. No conversion to the expected cyclization product, another naturally occurring decanolide named fusanolide A (36) [56], was observed using the Steglich [43], Mukaiyama [57], Yamaguchi [58] or Shiina method [59] under their published standard conditions. For these reasons, we decided to investigate whether the macrolactonization of (2Z,4E)-9-hydroxy-2,4-dienoic acids is generally hampered, which might be caused by the build-up of ring strain. We started this investigation with the simple derivative 33, which was synthesized from 30[60] via a sequence of three steps. For the macrolactonization of 33 we chose Yamaguchi’s method, but applied significantly more forcing conditions by using increased amounts of reagents and in particular a large excess of DMAP, in combination with higher dilution and elevated reaction temperatures. This led indeed to the formation of the desired lactone 34, which could be isolated in a moderate yield of 27% (Scheme 7).
Scheme 7:
Synthesis of (2Z,4E)-9-hydroxy-2,4-dienoic acid (33) and its macrolactonization.
Scheme 7:
Synthesis of (2Z,4E)-9-hydroxy-2,4-dienoic acid (33) and its macrolactonization.
With this result in hand, we reinvestigated the cyclization of 35[24] to fusanolide A (36) under the conditions outlined above. Gratifyingly, 36 was obtained in a yield of 53%, which allowed us to compare its analytical data with those reported for natural fusanolide A [56]. This comparison confirmed our previously suggested revision of the ten-membered lactone structure originally assigned to fusanolide A, as the spectroscopic data obtained for synthetic 36 differ significantly from those reported for the natural product. As we mentioned in our previous publication describing the synthesis of curvulalic acid (35) [24], all spectroscopic data obtained for this compound match those reported for fusanolide A [56] perfectly, suggesting that curvulalic acid and fusanolide A are probably identical. It should, however, be noted that 36 might well be a natural product which has not yet been isolated from a natural source (Scheme 8).
Scheme 8:
Synthesis of published structure of fusanolide A (36).
Scheme 8:
Synthesis of published structure of fusanolide A (36).
To complete the synthesis of stagonolide E, the MOM-protected precursor 29 and the deprotected derivative 37 were subjected to the Yamaguchi conditions that were found to be successful for the synthesis of 34 and 36 (Scheme 9). While the attempted Yamaguchi lactonization of 37 failed completely and resulted only in the quantitative recovery of unreacted starting material, the 6-MOM-protected precursor 29 underwent cyclization to the protected decanolide 38[31] in 67% yield. Deprotection of 38 was accomplished with TFA in dichloromethane at ambient temperature without noticeable epimerization or elimination of water. Stagonolide E was isolated in 90% yield and its analytical data were identical to those reported for the natural product [28].
Only few examples for the macrolactonization of ω-hydroxy-2Z,4E-dienoic acids such as 29, 33 and 34 have been described in the literature, and we are not aware of another study which describes the cyclization of differently substituted derivatives under identical conditions. Notably, the yield of macrolactones is significantly affected by the substitution pattern and increases from 27% for the unsubstituted lactone 34 (Scheme 7) to 53% for the 9-methyl-substituted derivative 36 (Scheme 8) and to 67% for the 6,9-disubstituted compound 38 (Scheme 9). The presence of substituents and their relative configuration may have severe conformational effects on transition states, activation barriers and product stability [61,62]. An example for which a dramatically increased yield was reported upon incorporation of substituents has been reported in the course of an octalactin synthesis [61].
Having established a reliable route to stagonolide E, we investigated its epoxidation under Sharpless conditions [63]. We expected that this transformation would give either curvulide A [30] or one of its diastereomers, and help to resolve the remaining structural ambiguities, i.e. the absolute configurations at C4, C5 and C6. Based on the transition-state model for the Sharpless epoxidation of allylic alcohols bearing a stereogenic centre in the allylic position [64], we expected that levorotatory stagonolide E and L-(+)-diethyl tartrate (DET) should form the mismatched pair, while the matched pair would result with D-(−)-DET (Scheme 10).
Scheme 10:
Transition-state models for the Sharpless epoxidation of stagonolide E with L-(+)-DET (left) and D-(−)-DET (right).
Scheme 10:
Transition-state models for the Sharpless epoxidation of stagonolide E with L-(+)-DET (left) and D-...
We subjected (−)-stagonolide E to the conditions of a Sharpless epoxidation, using both L-(+)-DET and D-(−)-DET. As expected on the basis of the transition-state model, no reaction occurred after 2 d with L-(+)-DET, and the starting material could be recovered nearly quantitatively. In contrast, the use of D-(−)-DET led to the formation of an epoxide 39b in 58% yield. A comparison of the analytical data of 39b with those reported for curvulide A revealed that the NMR spectroscopic data are identical, and the value for the specific rotation of 39b is reasonably close to the value reported for the natural product ([α]D23 +133) [30]. Therefore, we conclude that the Sharpless-epoxidation product of stagonolide E is identical with curvulide A and suggest the (4R,5R,6R,9R)-configuration shown for 39b (Scheme 11).
Scheme 11:
Synthesis of 39b (curvulide A) from stagonolide E.
Scheme 11:
Synthesis of 39b (curvulide A) from stagonolide E.
While the R-configuration assigned to C6 and C9 is unequivocally established, because these stereocenters originate from stagonolide E, there still remains an uncertainty for the absolute configurations at C4 and C5. While the relative trans-configuration at these stereocenters is evident from a small 3J(H4–H5) value of 2.2 Hz and from the E-configuration of the precursor, the relative configuration of C6 and C5, and hence the absolute configurations at C4 and C5, can not be assigned with absolute reliability. However, a comparatively large coupling constant 3J(H5–H6) of 8.2 Hz is pointing towards a trans-orientation of these protons with a large dihedral angle. Unfortunately, we could not obtain the (4S,5S,6R,9R)-configured 39a and compare the crucial 3J(H5–H6) coupling constants of the two diastereoisomers. However, the calculated energy-minimized structures of 39a and 39b suggest that the H5–H6 dihedral angles should differ substantially (Figure 2). For 39a, this angle should be close to 90°, which is not in agreement with a coupling constant of 8.2 Hz. In contrast, the same dihedral angle can be expected to be approximately 170° in the case of the diastereomeric epoxide 39b, and this value fits well to the observed 3J(H5–H6) value (Figure 2) [65].
Figure 2:
MM2 energy-minimized structures of 39a and 39b.
Figure 2:
MM2 energy-minimized structures of 39a and 39b.
In summary, we synthesized the naturally occurring ten-membered lactones stagonolide E and curvulide A, starting from the ex-chiral pool building block (R,R)-hexa-1,5-diene-3,4-diol. Key elements of the stagonolide E synthesis are the two-directional functionalization of the enantiopure, C2-symmetrical starting material through cross metathesis and a one-flask ring-closing metathesis/base-induced ring-opening sequence, a Ru–lipase-catalyzed dynamic kinetic resolution to establish the stereochemistry at C6, and a Yamaguchi macrolactonization. The first synthesis of curvulide A was accomplished by Sharpless epoxidation of stagonolide E. While the previously assigned absolute and relative configurations of stagonolide E could be confirmed by the synthesis described herein, we were able to provide additional information concerning missing structural assignments for curvulide A, which is most likely (4R,5R,6R,9R)-configured.
Supporting Information
Supporting Information File 1:
Experimental procedures, characterization data and copies of 1H and 13C NMR spectra.
Generous financial support by the Deutsche Forschungsgemeinschaft (DFG grant Schm 1095/6-2) is gratefully acknowledged. We thank Evonik Oxeno for generous donations of solvents, and Umicore for generous donations of metathesis catalysts.
References
Poss, C. S.; Schreiber, S. L. Acc. Chem. Res.1994,27, 9–17. doi:10.1021/ar00037a002
Return to citation in text:
[1]
Rama Rao, A. V.; Mysorekar, S. V.; Gurjar, M. K.; Yadav, J. S. Tetrahedron Lett.1987,28, 2183–2186. doi:10.1016/S0040-4039(00)96076-3
Return to citation in text:
[1]
Crombez-Robert, C.; Benazza, M.; Fréchou, C.; Demailly, G. Carbohydr. Res.1997,303, 359–365. doi:10.1016/S0008-6215(97)00151-1
Return to citation in text:
[1]
Burke, S. D.; Sametz, G. M. Org. Lett.1999,1, 71–74. doi:10.1021/ol9905506
Return to citation in text:
[1]
Schmidt, B.; Nave, S. Adv. Synth. Catal.2007,349, 215–230. doi:10.1002/adsc.200600473
Return to citation in text:
[1]
[2]
Michaelis, S.; Blechert, S. Org. Lett.2005,7, 5513–5516. doi:10.1021/ol052332k
Return to citation in text:
[1]
[2]
Purser, S.; Claridge, T. D. W.; Odell, B.; Moore, P. R.; Gouverneur, V. Org. Lett.2008,10, 4263–4266. doi:10.1021/ol8017402
Return to citation in text:
[1]
Schmidt, B.; Staude, L.; Kelling, A.; Schilde, U. Eur. J. Org. Chem.2011, 1721–1727. doi:10.1002/ejoc.201001528
Return to citation in text:
[1]
[2]
Schmidt, B.; Nave, S. Chem. Commun.2006, 2489–2491. doi:10.1039/b604359c
Return to citation in text:
[1]
Voight, E. A.; Rein, C.; Burke, S. D. J. Org. Chem.2002,67, 8489–8499. doi:10.1021/jo0263014
Return to citation in text:
[1]
Chou, C.-Y.; Hou, D.-R. J. Org. Chem.2006,71, 9887–9890. doi:10.1021/jo061767y
Return to citation in text:
[1]
Cribiú, R.; Jäger, C.; Nevado, C. Angew. Chem., Int. Ed.2009,48, 8780–8783. doi:10.1002/anie.200903379
Return to citation in text:
[1]
Reddy, C. R.; Dharmapuri, G.; Rao, N. N. Org. Lett.2009,11, 5730–5733. doi:10.1021/ol9025183
Return to citation in text:
[1]
Marvin, C. C.; Clemens, A. J. L.; Burke, S. D. Org. Lett.2007,9, 5353–5356. doi:10.1021/ol7021214
Return to citation in text:
[1]
Marvin, C. C.; Voight, E. A.; Burke, S. D. Org. Lett.2007,9, 5357–5359. doi:10.1021/ol702454m
Return to citation in text:
[1]
Quinn, K. J.; Smith, A. G.; Cammarano, C. M. Tetrahedron2007,63, 4881–4886. doi:10.1016/j.tet.2007.03.151
Return to citation in text:
[1]
Quinn, K. J.; Isaacs, A. K.; Arvary, R. A. Org. Lett.2004,6, 4143–4145. doi:10.1021/ol040047f
Return to citation in text:
[1]
Quinn, K. J.; Isaacs, A. K.; DeChristopher, B. A.; Szklarz, S. C.; Arvary, R. A. Org. Lett.2005,7, 1243–1245. doi:10.1021/ol047352l
Return to citation in text:
[1]
[2]
Schmidt, B.; Kunz, O.; Petersen, M. H. J. Org. Chem.2012,77, 10897–10906. doi:10.1021/jo302359h
Return to citation in text:
[1]
[2]
Dräger, G.; Kirschning, A.; Thiericke, R.; Zerlin, M. Nat. Prod. Rep.1996,13, 365–375. doi:10.1039/np9961300365
Return to citation in text:
[1]
Sun, P.; Lu, S.; Ree, T. V.; Krohn, K.; Li, L.; Zhang, W. Curr. Med. Chem.2012,19, 3417–3455. doi:10.2174/092986712801215874
Return to citation in text:
[1]
Evidente, A.; Cimmino, A.; Berestetskiy, A.; Mitina, G.; Andolfi, A.; Motta, A. J. Nat. Prod.2008,71, 31–34. doi:10.1021/np0703038
Return to citation in text:
[1]
[2]
[3]
Trisuwan, K.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Arch. Pharmacal Res.2011,34, 709–714. doi:10.1007/s12272-011-0502-8
Return to citation in text:
[1]
[2]
[3]
Greve, H.; Schupp, P. J.; Eguereva, E.; Kehraus, S.; König, G. M. J. Nat. Prod.2008,71, 1651–1653. doi:10.1021/np8003326
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Sabitha, G.; Padmaja, P.; Reddy, P. N.; Jadav, S. S.; Yadav, J. S. Tetrahedron Lett.2010,51, 6166–6168. doi:10.1016/j.tetlet.2010.09.072
Return to citation in text:
[1]
[2]
[3]
Arlt, D.; Bieniek, M.; Karch, R. Novel Metathesis Catalysts. WO 2008/034552 A1, March 27, 2008.
Return to citation in text:
[1]
Mutlu, H.; Montero de Espinosa, L.; Türünç, O.; Meier, M. A. R. Beilstein J. Org. Chem.2010,6, 1149–1158. doi:10.3762/bjoc.6.131
Return to citation in text:
[1]
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett.1999,1, 953–956. doi:10.1021/ol990909q
Return to citation in text:
[1]
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed.2003,42, 1900–1923. doi:10.1002/anie.200200556
Return to citation in text:
[1]
Connon, S. J.; Blechert, S. Top. Organomet. Chem.2004,11, 93–124. doi:10.1007/b94650
Return to citation in text:
[1]
Bach, J.; Berenguer, R.; Garcia, J.; López, M.; Manzanal, J.; Vilarrasa, J. Tetrahedron1998,54, 14947–14962. doi:10.1016/S0040-4020(98)00936-3
Return to citation in text:
[1]
Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics2005,24, 4528–4542. doi:10.1021/om0503848
Return to citation in text:
[1]
Lawrence, N. J.; Drew, M. D.; Bushell, S. M. J. Chem. Soc., Perkin Trans. 11999, 3381–3391. doi:10.1039/A903662H
Return to citation in text:
[1]
Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc.1988,110, 291–293. doi:10.1021/ja00209a048
Return to citation in text:
[1]
Baker, B. A.; Bošković, Ž. V.; Lipshutz, B. H. Org. Lett.2008,10, 289–292. doi:10.1021/ol702689v
Return to citation in text:
[1]
Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl.1978,17, 522–524. doi:10.1002/anie.197805221
Return to citation in text:
[1]
[2]
Deutsch, C.; Krause, N.; Lipshutz, B. H. Chem. Rev.2008,108, 2916–2927. doi:10.1021/cr0684321
Return to citation in text:
[1]
Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc.1987,109, 5551–5553. doi:10.1021/ja00252a056
Return to citation in text:
[1]
Lu, K.-J.; Chen, C.-H.; Hou, D.-R. Tetrahedron2009,65, 225–231. doi:10.1016/j.tet.2008.10.059
Return to citation in text:
[1]
Stürmer, R. Angew. Chem., Int. Ed. Engl.1997,36, 1173–1174. doi:10.1002/anie.199711731
Return to citation in text:
[1]
Pàmies, O.; Bäckvall, J.-E. Chem. Rev.2003,103, 3247–3262. doi:10.1021/cr020029g
Return to citation in text:
[1]
Shvo, Y.; Czarkie, D.; Rahamim, Y.; Chodosh, D. F. J. Am. Chem. Soc.1986,108, 7400–7402. doi:10.1021/ja00283a041
Return to citation in text:
[1]
Choi, J. H.; Choi, Y. K.; Kim, Y. H.; Park, E. S.; Kim, E. J.; Kim, M.-J.; Park, J. J. Org. Chem.2004,69, 1972–1977. doi:10.1021/jo0355799
Return to citation in text:
[1]
Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J.-E. J. Am. Chem. Soc.2005,127, 8817–8825. doi:10.1021/ja051576x
Return to citation in text:
[1]
Larsson, A. L. E.; Persson, B. A.; Bäckvall, J.-E. Angew. Chem., Int. Ed. Engl.1997,36, 1211–1212. doi:10.1002/anie.199712111
Return to citation in text:
[1]
Persson, B. A.; Larsson, A. L. E.; Le Ray, M.; Bäckvall, J.-E. J. Am. Chem. Soc.1999,121, 1645–1650. doi:10.1021/ja983819+
Return to citation in text:
[1]
[2]
Shimada, A.; Kusano, M.; Matsumoto, K.; Nishibe, M.; Kawano, T.; Kimura, Y. Z. Naturforsch., B2002,57b, 239–242.
Return to citation in text:
[1]
[2]
[3]
Mukaiyama, T.; Usui, M.; Saigo, K. Chem. Lett.1976,5, 49–50. doi:10.1246/cl.1976.49
Return to citation in text:
[1]
Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn.1979,52, 1989–1993. doi:10.1246/bcsj.52.1989
Return to citation in text:
[1]
Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem.2004,69, 1822–1830. doi:10.1021/jo030367x
Return to citation in text:
[1]
Enders, D.; Nguyen, D. Synthesis2000, 2092–2098. doi:10.1055/s-2000-8732
Return to citation in text:
[1]
Johnson, R. B.; Sharpless, K. B. Catalytic Asymmetric Epoxidation of Allylic Alcohols. In Catalytic Asymmetric Synthesis; Ojima, I., Ed.; Wiley-VCH: Weinheim, Germany, 1993; pp 103–158.
Return to citation in text:
[1]
Karplus, M. J. Chem. Phys.1959,30, 11–15. doi:10.1063/1.1729860
Return to citation in text:
[1]
Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics2005,24, 4528–4542. doi:10.1021/om0503848
Johnson, R. B.; Sharpless, K. B. Catalytic Asymmetric Epoxidation of Allylic Alcohols. In Catalytic Asymmetric Synthesis; Ojima, I., Ed.; Wiley-VCH: Weinheim, Germany, 1993; pp 103–158.


![[1860-5397-9-289-i1]](/bjoc/content/inline/1860-5397-9-289-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-1]](/bjoc/content/figures/1860-5397-9-289-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i2]](/bjoc/content/inline/1860-5397-9-289-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i3]](/bjoc/content/inline/1860-5397-9-289-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-289-i12.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-289-i13.svg?max-width=637&scale=1.0)
![[1860-5397-9-289-i4]](/bjoc/content/inline/1860-5397-9-289-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i5]](/bjoc/content/inline/1860-5397-9-289-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i6]](/bjoc/content/inline/1860-5397-9-289-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i7]](/bjoc/content/inline/1860-5397-9-289-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i8]](/bjoc/content/inline/1860-5397-9-289-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i9]](/bjoc/content/inline/1860-5397-9-289-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i10]](/bjoc/content/inline/1860-5397-9-289-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-i11]](/bjoc/content/inline/1860-5397-9-289-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-289-2]](/bjoc/content/figures/1860-5397-9-289-2.png?scale=2.0&max-width=1024&background=FFFFFF)