Abstract
2-Chloro-3-amino-4-picoline (CAPIC) is a strategic building block for the preparation of nevirapine, a widely-prescribed non-nucleosidic reverse transcriptase inhibitor for the treatment of HIV-infected patients. A continuous synthesis to the bromo derivative of a CAPIC intermediate, 2-bromo-4-methylnicotinonitrile, that terminates in a dead-end crystallization is described. The route uses inexpensive, acyclic commodity-based raw materials and has the potential to enable lower cost production of nevirapine as well as other value added structures that contain complex pyridines. The route terminates in a batch crystallization yielding high purity CAPIC. This outcome is expected to facilitate regulatory implementation of the overall process.
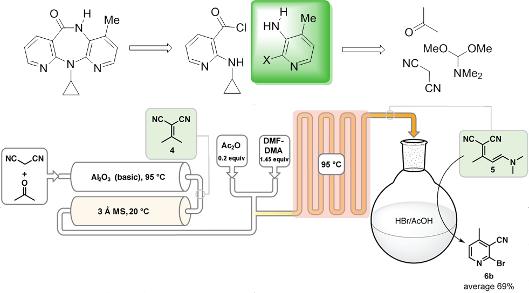
Graphical Abstract
Introduction
Nevirapine (3) was the first commercially available non-nucleoside reverse transcriptase inhibitor (NNRTI), and has remained an important medicine in the management of human immunodeficiency virus (HIV) [1,2]. Nevirapine combined with lamivudine (3TC) and azidothymidine (AZT) or tenofovir (TDF) is one of the preferred first-line combination drug therapies recommended by the World Health Organization (WHO) [3-5]. WHO initiatives are expected to increase the demand for nevirapine over the next 10 years (Figure 1) [6]. Although several viable NNRT substitutes for nevirapine are available, nevirapine manufacturing requirements will remain high because clinicians are reluctant to change treatment once a successful combination therapy is identified and many remain healthy with the nevirapine based combinations. Furthermore, the recent development of an extended release dosage form of nevirapine that enables once a day administration is expected to further increase market demand [6-8]. The high demand coupled with the financial burden associated with long-term HIV treatments has resulted in shortages and patients opting to reduce dosing which increases development of resistant strains [9-12]. This confluence of increased demand and cost provides an opportunity to reevaluate both the chemistry as well as the manufacturing platforms by which this drug can be produced.
![[1860-5397-9-292-1]](/bjoc/content/figures/1860-5397-9-292-1.png?scale=1.7&max-width=1024&background=FFFFFF)
Figure 1: The estimated demand of for nevirapine until 2015 [6].
Figure 1: The estimated demand of for nevirapine until 2015 [6].
The two key Food and Drug Administration (FDA) registered starting materials in the commercial nevirapine process are 2-chloro-3-amino-4-picoline (CAPIC) (1a) and 2-cyclopropylaminonicotinic acid (2-CAN) (2) (Scheme 1) [13]. The CAPIC process comprises approximately 64% of the total production cost. Based on our previous experience with the development of the current commercial batch processes for nevirapine [13] and its pyridine precursors [14], we have started a program to define lower costs nevirapine processes. After a cost of goods analysis, we have come to the conclusion that the most promising cost saving path forward is through the use of acyclic, commodity-based starting materials in the assembly of the active pharmaceutical ingredient (API) (analysis will be included on future publications). We hypothesized that by both reducing the cost of goods via chemistry changes and reducing the unit operations the most significant cost reduction could be achieved. Herein, we demonstrate a proof of concept flow synthesis of the key intermediate used to produce the bromo derivative of the CAPIC precursor, 2-bromo-4-methylnicotinonitrile (6b). The synthesis telescopes three steps using substantially less expensive starting materials.
![[1860-5397-9-292-i1]](/bjoc/content/inline/1860-5397-9-292-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Commercial building blocks to nevirapine.
Scheme 1: Commercial building blocks to nevirapine.
Flow or continuous chemistry is alternative to batch chemistry where reactions are performed by passing reagents through devices containing small-dimensional channels as opposed to using batch reactors [15-17]. Flow reactors are particularly advantageous in multistep syntheses where telescoping steps avoids isolation of dangerous and/or unstable intermediates and reduces solvent usage and waste production incurred through intermediate purifications [18-27]. The large surface to volume ratios found in the small channels allow for more efficient mixing and heat transfer often resulting in shorter contact times [28,29]. Consequently, flow chemistry allows chemists to expand their window of process operability by working at elevated temperatures and pressures to increase reaction rates and decrease catalyst loadings [30-33]. Unlike scaling-up batch reactions, which requires additional optimization, scaling-up flow processes only requires implementing multiple reactors to work in parallel [29].
We have recently developed a method to synthesize polysubstituted 2-halonicotinonitriles in high yields via enamine intermediates 5 by reacting alkylidene malononitriles in the presence of acetic anhydride with N,N-dimethylformamide dimethyl acetal (DMF-DMA) [34]. Previous attempts to synthesize the nicotinonitriles via enamines resulted in poor yields due to dimerization of the starting alkylidene malononitrile [35-37]. A high yield enamine approach allows us to begin the synthesis from the commodity chemicals (acetone and malononitrile) and bypass the pyridone intermediate used in the original CAPIC synthesis (Scheme 2a) by effecting the ring closure under Pinner reaction conditions (Scheme 2b) [14].
![[1860-5397-9-292-i2]](/bjoc/content/inline/1860-5397-9-292-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: a) Current commercial process to CAPIC and b) newly developed batch synthesis to CAPIC and its bromo derivative.
Scheme 2: a) Current commercial process to CAPIC and b) newly developed batch synthesis to CAPIC and its brom...
We set out to investigate the possibility of performing a continuous synthesis of 2-bromo-4-methylnicotinonitrile starting from acetone and malononitrile (Scheme 3) using the Vapourtec R series reactor system [38]. The batch synthesis commences with a Knoevenagel reaction condensing malononitrile and acetone catalyzed by aluminum oxide producing isopropylidenemalononitrile (4) [39,40]. The penultimate enamine 5 results by treating 4 with DMF-DMA in the presence of acetic anhydride, and ultimately 2-bromo-4-methylnicotinonitrile (6b) is produced after 5 is treated with HBr in acetic acid. Transferring the batch synthesis into a semi-continuous process requires one to consider solvent exchanges and byproducts that might complicate downstream operations.
![[1860-5397-9-292-i3]](/bjoc/content/inline/1860-5397-9-292-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed synthesis to 2-bromo-4-methylnicotinonitrile using a continuous approach with the considerations for each reaction. i. Knoevenagel condensation to produce the isopropylidenemalononitrile (4). ii. Reaction of the isopropylidenemalononitrile with DMF-DMA to produce an enamine (5). iii. Dead end cyclization to the desired 2-bromo-4-methylnicotinonitrile (6b) using HBr.
Scheme 3: Proposed synthesis to 2-bromo-4-methylnicotinonitrile using a continuous approach with the consider...
We immediately recognized the water formed in the Knoevenagel condensation would quench the DMF-DMA in the second step. In addition, our batch Knoevenagel condensation was base catalyzed and we discovered that base increased dimer byproducts in the enamine step. Therefore, we chose to employ a solid basic reagent that would simultaneously catalyze the Knoevenagel reaction and confine the reagent to the first step, as well as a solid desiccant to remove the water. Previously we have used solid catalysts and/or solid reagents in a number of continuous processes [41-44]. Another challenge was the need to increase the rate of enamine 5 formation. Under some conditions, the enamine step required up to 24 hours [34]. Factoring these and other considerations, we designed the process shown in Scheme 3 with a summary of considerations for each step.
Results and Discussion
We began our investigation by optimizing the enamine formation (5, Figure 2) because we predicted that success with this central step would help define the flanking reactions. Initial batch studies revealed that the reaction occurred rapidly (1 h) in toluene (1.0 M) heated to 45 °C to produce 4 in 94% yield [34], but the product precipitated and these conditions were rejected to avoid reactor clogging. Based on literature precedent and our own screening, we discovered that DCM solubilized 4, 5, and 6b (Scheme 3). In batch, use of DCM would require non-traditional glassware because temperatures exceeding the standard boiling point at atmospheric pressure were required to avoid unwanted dimer formation unless low reaction concentrations (0.10 M) were used. A flow reactor is ideal for performing reactions well outside of normal operating conditions and we pushed forward seeking high temperature conditions using DCM [45].
![[1860-5397-9-292-2]](/bjoc/content/figures/1860-5397-9-292-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) Flow scheme to produce the enamine intermediate 5 from isopropylidenemalononitrile (4) (see Supporting Information File 1 for details). b) Coil temperature effect on yield at a 0.10 M reaction concentration with a 2 min residence time.
Figure 2: a) Flow scheme to produce the enamine intermediate 5 from isopropylidenemalononitrile (4) (see Supporting Information File 1 for...
When screening conditions, we initially investigated the effect temperature had on the reaction at a 0.10 M concentration (Figure 2b). Placing backpressure regulators after the heated coil allowed the temperature of the reactor coil to be raised far above the boiling point of DCM. As shown in Figure 2b, increasing the temperature to 80 °C provided 67% yield with a 2 min residence time. We then examined reaction concentrations to reduce the volume of DCM (Table 1). We were not only able to increase the concentration to 1.0 M by heating to 95 °C, but were also able to increase yields to >93% with a 2 min residence time. In comparison, our batch method with similar concentration conditions in toluene was complete in 1 h with 94% yield [34]. These flow conditions were high yielding in one thirtieth of the reaction time. This example underscores the benefit of operating outside of normal process windows [45]. Attempts to increase the reaction concentration beyond 1.0 M led to reactor clogging due to the limited of solubility of 5.
Table 1: Concentration screen for enamine formation.
| Entry | Reaction concentration (M) | Residence time (min) | Coil temperature (°C) | Yield (%)a |
|---|---|---|---|---|
| 1 | 0.10 | 2 | 100 | 68 |
| 2 | 0.20 | 2 | 95 | 93 |
| 3 | 0.40 | 2 | 95 | 96 |
| 4 | 0.60 | 2 | 95 | 98 |
| 5 | 0.80 | 2 | 95 | 97 |
| 6 | 0.98 | 2 | 95 | 97 |
aDetermined by GC analysis using mesitylene as an internal standard. See Figure 2a for flow scheme.
We proceeded to develop a continuous process by coupling the enamine step with the Knoevenagel condensation (Scheme 4). To achieve this, we included two columns: a packed-bed of Al2O3 to catalyze the reaction and a packed bed of 3 Å molecular sieves to absorb water before the addition of DMF-DMA (Scheme 4). The mass of each solid used (2.00 g of Al2O3 and 1.50 g molecular sieves) was chosen based on the size of the available columns. Assuming that the Knoevenagel condensation occurred primarily in the Al2O3 column, we only varied the temperature of the Al2O3 column. The Al2O3 column temperature was initially set to 25 °C which yielded 91% of 5 from acetone and malononitrile (Table 2, entry 2). We observed that the Al2O3 column reactor temperature increased during the reaction and thus examined the use of temperatures below 25 °C. Cooling the column did not provide any observable improvement (Table 2, entries 3 and 4) prompting us to examine higher temperatures. Heating the column past 25 °C increased the byproduct formation which lowered the yield (Table 2, entries 5–8). Increasing the residence time through the alumina column had no positive impact on yield (Table 2, entry 9).
![[1860-5397-9-292-i4]](/bjoc/content/inline/1860-5397-9-292-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Flow scheme to produce the enamine 5 starting from acetone and malononitrile (See Supporting Information File 1 for details).
Scheme 4: Flow scheme to produce the enamine 5 starting from acetone and malononitrile (See Supporting Information File 1 for details).
Table 2: Screened conditions for formation of 5 starting from acetone and malononitrile.
| Entry | Al2O3 column temperature (°C) | 3 Å MS column temperature (°C) | Residence time of coil (min)a | Yield (%)b |
|---|---|---|---|---|
| 1 | 25 | 20 | 2 | NAc |
| 2 | 25 | 20 | 4 | 91 |
| 3 | 20 | 25 | 4 | 91 |
| 4 | 10 | 25 | 4 | 92 |
| 5 | 35 | 20 | 4 | 88 |
| 6 | 50 | 20 | 4 | 88 |
| 7 | 75 | 20 | 4 | 84 |
| 8 | 95 | 20 | 4 | 81 |
| 9 | 95 | 20 | 6 | 81 |
aCoil temperature was 95 °C. bDetermined by GC analysis using mesitylene as an internal standard. cThe fast flow rates needed for this residence time caused the pressure of the system to exceed the maximum limit. See Scheme 4 for flow scheme.
The successful combination of the Knoevenagel/enamine steps prompted us to evaluate the stability of this two-step system. We often find that when multisteps are combined the system stability can become an issue. To measure the stability, we ran the reaction under the optimized conditions and monitored the product distribution. When the aforementioned optimized column temperatures were used (20 °C Al2O3 column, 25 °C 3 Å MS column), the alumina column begins to fail (Figure 3). At a collection time of 12 min (Table 2, entry 3), the yield is at its maximum at 91%. However, by ~17 min, the yield drops to 48%. We speculate that at high reactant concentrations the water produced fouls the Al2O3 column. This conjecture is supported by the increased production of 7 when excess malononitrile reacts with DMF-DMA (Scheme 5). Heating the column to 95 °C allowed the Al2O3 column to remain activated longer. Despite the fact that higher alumina column temperatures result in less than optimal yields of the enamine, we examined the system stability at 95 °C. As can be seen in Figure 3, increasing the alumina column temperature provides improved stability compared to 25 °C; however, the column performance exhibits shallow decline over the first 37 min and then fails rapidly beyond 37 min. The fact that we can resurrect the column performance somewhat suggests that this stability issue can be addressed when and if this process is implemented on scale. To demonstrate that further gains in stability are possible, we examined the impact of reaction concentration on column stability.
![[1860-5397-9-292-3]](/bjoc/content/figures/1860-5397-9-292-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Comparing the long-term stability of the Al2O3 and 3 Å MS columns when the Al2O3 column temperature (20 °C and 95 °C) and Knoevenagel reaction concentration (2.0 M and 0.50 M) are varied. The time between 0 and 13 min was the equilibration period.
Figure 3: Comparing the long-term stability of the Al2O3 and 3 Å MS columns when the Al2O3 column temperature...
![[1860-5397-9-292-i5]](/bjoc/content/inline/1860-5397-9-292-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Several byproducts were observed when producing 5 starting from acetone and malononitrile. 7 is formed from excess malononitrile when the Knoevenagel reaction does not go to completion. The formation of dimers 8a and 8b can begin at any point during the reaction.
Scheme 5: Several byproducts were observed when producing 5 starting from acetone and malononitrile. 7 is for...
Process chemists often seek the highest operating concentrations to reduce solvent costs. Recognizing this aspiration, we performed the aforementioned Knoevenagel reaction at 2.0 M. Considering that many process reactions run at 0.20 M, this starting concentration was high. The high starting concentration also allowed us to realize a 1.0 M reaction concentration once addition of the acetic anhydride and DMF-DMA (Scheme 4). While the higher the concentration the better, our prior efforts have revealed that packed-bed catalyst stability can rapidly decline at high concentrations while at lower concentrations can run for an extended length of time [44]. With this in mind, we lowered the Knoevenagel concentration to 0.50 M. This setup also resulted in the residence time in the Al2O3 column and 3 Å MS to reduce from 2.88 min and 2.72 min to 0.90 min and 0.85 min respectively. The faster residence time could account for less water absorption in the Al2O3 column. Because we can still hold the acetic anhydride/DMF-DMA concentrations high lowering the Knoevenagel concentration only results in the enamine concentration decreasing by factor of 1.25 (0.40 M). As shown in Figure 3, reducing the Knoevenagel to 0.50 M and heating the alumina column to 95 °C results in a dramatic improvement in system stability. The output of the enamine, however, remains similar. The reaction at a 1 M concentration would produce approximately 7 g of enamine product within the 24 min window and at a 0.4 M concentration approximately 5.8 g in the 42 min window. While we are bolstered by these improvements, we suggest that a commercial version of this process must address how to achieve long-term stability at the higher concentrations.
Creating a chemical process for an active pharmaceutical ingredient is a careful integration of chemical and regulatory challenges. While from an academic standpoint a completely continuous process provides the opportunity to advance process chemistry/technology, a new process can often require significant investment for regulatory validation. We wish to implement our technology as quickly as feasible and to do so we want to avoid potential regulatory problems. Therefore, we have opted to carry out the Pinner cyclization as terminal cyclization/crystallization step where the already validated material could be collected. To optimize the cyclization step, we combined only the enamine/cyclization steps to reduce system complexity. When we subjected 25 mL of the enamine 5 output to a solution of HBr in AcOH for 45 min (55 °C) the desired nicotinonitrile 6b crystallized out of solution in 81% overall yield (2-steps, Scheme 6). While our intent was not to create a completely continuous process at this time, commercially available reactors that can handle strong acid are available and this step could easily be achieved in flow (for an example of an strong acid resistant reactor, see reference [38]).
![[1860-5397-9-292-i6]](/bjoc/content/inline/1860-5397-9-292-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Flow scheme to produce 2-bromo-4-methylnicotinonitrile (6b) in 81% yield from 4 with a 1 M concentration and 2 min residence time in the coil (See Supporting Information File 1 for further details).
Scheme 6: Flow scheme to produce 2-bromo-4-methylnicotinonitrile (6b) in 81% yield from 4 with a 1 M concentr...
We completed the process by integrating all three steps together using the lower concentration Knoevenagel condensation (0.50 M) with the heated Al2O3 column at 95 °C due to its long-term stability (Scheme 7). We ran 100 mL of material through the process and after simple trituration with water 69% (5.4 g for 100 mL) of the desired 2-bromo-4-methylnicotinonitrile (6b) was obtained and was analytically pure as determined by elemental analysis. We have also performed the enamine cyclization using HCl instead of HBr and have produced the registered chloride in 81% yield [34].
![[1860-5397-9-292-i7]](/bjoc/content/inline/1860-5397-9-292-i7.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reactor scheme for the continuous synthesis of 2-bromo-4-methylnicotinonitrile (6b) with an average of 69% yield. The reaction concentration within the columns was 0.50 M, while the reaction concentration in the coil was 0.40 M. The residence time in the Al2O3 column was 0.90 min, in the 3 Å MS column 0.85 min, in the coil 4 min, and the reaction time for the cyclization to occur to produce 6b was 45 min (See Supporting Information File 1 for further details).
Scheme 7: Reactor scheme for the continuous synthesis of 2-bromo-4-methylnicotinonitrile (6b) with an average...
Conclusion
Here, we have demonstrated the semi-continuous synthesis of 2-bromo-4-methylnicotinonitrile starting from acetone and malononitrile by utilizing solid Al2O3 and 3 Å MS columns and decreasing the reaction time of the enamine formation to a matter of minutes using DCM conditions outside of normal process windows. The cyclization under Pinner conditions using the crude output from the Knoevenagel/enamine steps provides an overall yield of 69% (>88% yield per step). While the Al2O3 column is stable for a limited time (between 24 and 42 min depending on the reaction concentration used), the current solution would be to simply replace the columns throughout the production process or implement larger columns. At the current state, the amount of Al2O3 and molecular sieves needed for every one gram of 2-bromo-4-methylnicotinonitrile is 0.37 g and 0.28 g respectively. Despite the limited stability, the amount of Al2O3 used in flow to produce 5.4 g of 2-bromo-4-methylnicotinonitrile is less than the amount of Al2O3 required for the batch process (about 0.98 g is needed per 1 g of product [34]). We also predict that by replacing the alumina columns with a soluble base and a water/base separation a stable, higher concentration process can be easily achieved from our preliminary results [22,46]. Finally, we have demonstrated that from a simple acyclic precursor, 2-bromo-4-methylnicotinonitrile can be achieved in a three-unit operation process yielding high purity crystalline materials. The chloride product is already registered and suggests that this strategy could be implemented in existing nevirapine processes.
Supporting Information
The Supporting Information describes synthesis and characterization data of all substances given in this article, reactor setup, operational details and screening conditions.
| Supporting Information File 1: Experimantal section. | ||
| Format: PDF | Size: 292.6 KB | Download |
References
-
De Clercq, E. J. Clin. Virol. 2004, 30, 115–133. doi:10.1016/j.jcv.2004.02.009
Return to citation in text: [1] -
De Clercq, E. Antiviral Res. 1998, 38, 153–179. doi:10.1016/S0166-3542(98)00025-4
Return to citation in text: [1] -
World Health Organization. Antiretroviral therapy for HIV infection in adults and adolescents: Recommendations for a public health approach. 2010 revision. http://www.who.int/hiv/pub/arv/adult2010/en/index.html (accessed Oct 31, 2012).
Return to citation in text: [1] -
Doherty, T.; Sanders, D.; Goga, A.; Jackson, D. Implications of the new WHO guidelines on HIV and infant feeding for child survival in South Africa. http://www.who.int/bulletin/volumes/89/1/10-079798/en/index.html (accessed Oct 31, 2012).
Return to citation in text: [1] -
van Leth, F.; Phanuphak, P.; Ruxrungtham, K.; Baraldi, E.; Miller, S.; Gazzard, B.; Cahn, P.; Lalloo, U. G.; van der Westhuizen, I. P.; Malan, D. R.; Johnson, M. A.; Santos, B. R.; Mulcahy, F.; Wood, R.; Levi, G. C.; Reboredo, G.; Squires, K.; Cassetti, I.; Petit, D.; Raffi, F.; Katlama, C.; Murphy, R. L.; Horban, A.; Dam, J. P.; Hassink, E.; van Leeuwen, R.; Robinson, P.; Wit, F. W.; Lange, J. M. A. Lancet 2004, 363, 1253–1263. doi:10.1016/S0140-6736(04)15997-7
Return to citation in text: [1] -
Proceedings of "Joint WHO/UNAIDS annual consultation with pharmaceutical companies: Global forecasts of antiretroviral demand 2012-2015". November 5-6, 2012, Geneva, Switzerland. http://www.who.int/hiv/amds/forecasting_meeting2012/en/ (accessed June 6, 2013).
Return to citation in text: [1] [2] [3] -
Renaud-Théry, F.; Avila-Figueroa, C.; Stover, J.; Thierry, S.; Vitoria, M.; Habiyambere, V.; Souteyrand, Y. AIDS Res. Treat. 2011, No. 749041. doi:10.1155/2011/749041
Return to citation in text: [1] -
Arribas, J. R.; Eron, J. Curr. Opin. HIV AIDS 2013, 8, 341–349.
Return to citation in text: [1] -
Pasquet, A.; Messou, E.; Gabillard, D.; Minga, A.; Depoulosky, A.; Deuffic-Burban, S.; Losina, E.; Freedberg, K. A.; Danel, C.; Anglaret, X.; Yazdanpanah, Y. PLoS One 2010, 5, e13414. doi:10.1371/journal.pone.0013414
Return to citation in text: [1] -
Paredes, R.; Marconi, V. C.; Lockman, S.; Abrams, E. J.; Kuhn, L. J. Infect. Dis. 2013, 207 (Suppl. 2), S93–S100. doi:10.1093/infdis/jit110
Return to citation in text: [1] -
WHO Progress Report 2011: Global HIV/AIDS Response. http://www.who.int/hiv/pub/progress_report2011/en/index.html (accessed June 19, 2013).
Return to citation in text: [1] -
Hirsch, M. S.; Günthard, H. F.; Schapiro, J. M.; Vézinet, F. B.; Clotet, B.; Hammer, S. M.; Johnson, V. A.; Kuritzkes, D. R.; Mellors, J. W.; Pillay, D.; Yeni, P. G.; Jacobsen, D. M.; Richman, D. D. Clin. Infect. Dis. 2008, 47, 266–285. doi:10.1086/589297
Return to citation in text: [1] -
Boswell, R. F.; Gupton, B. F.; Lo, Y. S. Method for Making Nevirapine. U.S. Patent 6,680,383, Jan 20, 2004.
Return to citation in text: [1] [2] -
Gupton, B. F. Process for making 3-amino-2-chloro-4-methylpyridine. U.S. Patent 6,399,781, June 4, 2002.
Return to citation in text: [1] [2] -
Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583–4592. doi:10.1039/C0CC05060A
Return to citation in text: [1] -
Wiles, C.; Watts, P. Chem. Commun. 2011, 47, 6512–6535. doi:10.1039/C1CC00089F
Return to citation in text: [1] -
Yoshida, J.-I. Chem. Rec. 2010, 10, 332–341. doi:10.1002/tcr.201000020
Return to citation in text: [1] -
Bogdan, A. R.; Poe, S. L.; Kubis, D. C.; Broadwater, S. J.; McQuade, D. T. Angew. Chem., Int. Ed. 2009, 48, 8547–8550. doi:10.1002/anie.200903055
Return to citation in text: [1] -
Lévesque, F.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 1706–1709. doi:10.1002/anie.201107446
Return to citation in text: [1] -
Booker-Milburn, K. Nat. Chem. 2012, 4, 433–435. doi:10.1038/nchem.1356
Return to citation in text: [1] -
Smith, C. J.; Smith, C. D.; Nikbin, N.; Ley, S. V.; Baxendale, I. R. Org. Biomol. Chem. 2011, 9, 1927–1937. doi:10.1039/c0ob00813c
Return to citation in text: [1] -
Sahoo, H. R.; Kralj, J. G.; Jensen, K. F. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. doi:10.1002/anie.200701434
Return to citation in text: [1] [2] -
Baxendale, I. R. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. doi:10.1002/jctb.4012
Return to citation in text: [1] -
Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/c2ob27002a
Return to citation in text: [1] -
Polyzos, A.; O'Brien, M.; Petersen, T. P.; Baxendale, I. R.; Ley, S. V. Angew. Chem., Int. Ed. 2011, 50, 1190–1193. doi:10.1002/anie.201006618
Return to citation in text: [1] -
Baxendale, I. R.; Ley, S. V.; Mansfield, A. C.; Smith, C. D. Angew. Chem., Int. Ed. 2009, 48, 4017–4021. doi:10.1002/anie.200900970
Return to citation in text: [1] -
Kupracz, L.; Hartwig, J.; Wegner, J.; Ceylan, S.; Kirschning, A. Beilstein J. Org. Chem. 2011, 7, 1441–1448. doi:10.3762/bjoc.7.168
Return to citation in text: [1] -
Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. doi:10.1002/anie.201004637
Return to citation in text: [1] -
Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c
Return to citation in text: [1] [2] -
Hessel, V. Chem. Eng. Technol. 2009, 32, 1655–1681. doi:10.1002/ceat.200900474
Return to citation in text: [1] -
Pieber, B.; Cantillo, D.; Kappe, C. O. Chem.–Eur. J. 2012, 18, 5047–5055. doi:10.1002/chem.201103748
Return to citation in text: [1] -
Reichart, B.; Tekautz, G.; Kappe, C. O. Org. Process Res. Dev. 2013, 17, 152–157. doi:10.1021/op300273u
Return to citation in text: [1] -
Reichart, B.; Kappe, C. O. Tetrahedron Lett. 2012, 53, 952–955. doi:10.1016/j.tetlet.2011.12.043
Return to citation in text: [1] -
Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Baldwin, J. J.; Raab, A. W.; Ponticello, G. S. J. Org. Chem. 1978, 43, 2529–2535. doi:10.1021/jo00406a052
Return to citation in text: [1] -
Cope, A. C.; Hoyle, K. E. J. Am. Chem. Soc. 1941, 63, 733–736. doi:10.1021/ja01848a025
Return to citation in text: [1] -
Williams, J. K. J. Org. Chem. 1963, 28, 1054–1059. doi:10.1021/jo01039a045
Return to citation in text: [1] -
Vapourtec Flow Chemistry Equipment, UK. http://www.vapourtec.co.uk (accessed June 21, 2013).
Return to citation in text: [1] [2] -
Texier-Boullet, F.; Foucaud, A. Tetrahedron Lett. 1982, 23, 4927–4928. doi:10.1016/S0040-4039(00)85749-4
Return to citation in text: [1] -
Broman, S. L.; Petersen, A. U.; Tortzen, C. G.; Vibenholt, J.; Bond, A. D.; Nielsen, M. B. Org. Lett. 2012, 14, 318–321. doi:10.1021/ol2030586
Return to citation in text: [1] -
Bogdan, A. R.; Mason, B. P.; Sylvester, K. T.; McQuade, D. T. Angew. Chem., Int. Ed. 2007, 46, 1698–1701. doi:10.1002/anie.200603854
Return to citation in text: [1] -
Bogdan, A.; McQuade, D. T. Beilstein J. Org. Chem. 2009, 5, No. 17. doi:10.3762/bjoc.5.17
Return to citation in text: [1] -
Opalka, S. M.; Longstreet, A. R.; McQuade, D. T. Beilstein J. Org. Chem. 2011, 7, 1671–1679. doi:10.3762/bjoc.7.197
Return to citation in text: [1] -
Opalka, S. M.; Park, J. K.; Longstreet, A. R.; McQuade, D. T. Org. Lett. 2013, 15, 996–999. doi:10.1021/ol303442m
Return to citation in text: [1] [2] -
Razzaq, T.; Kappe, C. O. Chem.–Asian J. 2010, 5, 1274–1289. doi:10.1002/asia.201000010
Return to citation in text: [1] [2] -
Noël, T.; Kuhn, S.; Musacchio, A. J.; Jensen, K. F.; Buchwald, S. L. Angew. Chem., Int. Ed. 2011, 50, 5943–5946. doi:10.1002/anie.201101480
Return to citation in text: [1]
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
| 22. | Sahoo, H. R.; Kralj, J. G.; Jensen, K. F. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. doi:10.1002/anie.200701434 |
| 46. | Noël, T.; Kuhn, S.; Musacchio, A. J.; Jensen, K. F.; Buchwald, S. L. Angew. Chem., Int. Ed. 2011, 50, 5943–5946. doi:10.1002/anie.201101480 |
| 1. | De Clercq, E. J. Clin. Virol. 2004, 30, 115–133. doi:10.1016/j.jcv.2004.02.009 |
| 2. | De Clercq, E. Antiviral Res. 1998, 38, 153–179. doi:10.1016/S0166-3542(98)00025-4 |
| 9. | Pasquet, A.; Messou, E.; Gabillard, D.; Minga, A.; Depoulosky, A.; Deuffic-Burban, S.; Losina, E.; Freedberg, K. A.; Danel, C.; Anglaret, X.; Yazdanpanah, Y. PLoS One 2010, 5, e13414. doi:10.1371/journal.pone.0013414 |
| 10. | Paredes, R.; Marconi, V. C.; Lockman, S.; Abrams, E. J.; Kuhn, L. J. Infect. Dis. 2013, 207 (Suppl. 2), S93–S100. doi:10.1093/infdis/jit110 |
| 11. | WHO Progress Report 2011: Global HIV/AIDS Response. http://www.who.int/hiv/pub/progress_report2011/en/index.html (accessed June 19, 2013). |
| 12. | Hirsch, M. S.; Günthard, H. F.; Schapiro, J. M.; Vézinet, F. B.; Clotet, B.; Hammer, S. M.; Johnson, V. A.; Kuritzkes, D. R.; Mellors, J. W.; Pillay, D.; Yeni, P. G.; Jacobsen, D. M.; Richman, D. D. Clin. Infect. Dis. 2008, 47, 266–285. doi:10.1086/589297 |
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
| 6. | Proceedings of "Joint WHO/UNAIDS annual consultation with pharmaceutical companies: Global forecasts of antiretroviral demand 2012-2015". November 5-6, 2012, Geneva, Switzerland. http://www.who.int/hiv/amds/forecasting_meeting2012/en/ (accessed June 6, 2013). |
| 7. | Renaud-Théry, F.; Avila-Figueroa, C.; Stover, J.; Thierry, S.; Vitoria, M.; Habiyambere, V.; Souteyrand, Y. AIDS Res. Treat. 2011, No. 749041. doi:10.1155/2011/749041 |
| 8. | Arribas, J. R.; Eron, J. Curr. Opin. HIV AIDS 2013, 8, 341–349. |
| 35. | Baldwin, J. J.; Raab, A. W.; Ponticello, G. S. J. Org. Chem. 1978, 43, 2529–2535. doi:10.1021/jo00406a052 |
| 36. | Cope, A. C.; Hoyle, K. E. J. Am. Chem. Soc. 1941, 63, 733–736. doi:10.1021/ja01848a025 |
| 37. | Williams, J. K. J. Org. Chem. 1963, 28, 1054–1059. doi:10.1021/jo01039a045 |
| 6. | Proceedings of "Joint WHO/UNAIDS annual consultation with pharmaceutical companies: Global forecasts of antiretroviral demand 2012-2015". November 5-6, 2012, Geneva, Switzerland. http://www.who.int/hiv/amds/forecasting_meeting2012/en/ (accessed June 6, 2013). |
| 30. | Hessel, V. Chem. Eng. Technol. 2009, 32, 1655–1681. doi:10.1002/ceat.200900474 |
| 31. | Pieber, B.; Cantillo, D.; Kappe, C. O. Chem.–Eur. J. 2012, 18, 5047–5055. doi:10.1002/chem.201103748 |
| 32. | Reichart, B.; Tekautz, G.; Kappe, C. O. Org. Process Res. Dev. 2013, 17, 152–157. doi:10.1021/op300273u |
| 33. | Reichart, B.; Kappe, C. O. Tetrahedron Lett. 2012, 53, 952–955. doi:10.1016/j.tetlet.2011.12.043 |
| 3. | World Health Organization. Antiretroviral therapy for HIV infection in adults and adolescents: Recommendations for a public health approach. 2010 revision. http://www.who.int/hiv/pub/arv/adult2010/en/index.html (accessed Oct 31, 2012). |
| 4. | Doherty, T.; Sanders, D.; Goga, A.; Jackson, D. Implications of the new WHO guidelines on HIV and infant feeding for child survival in South Africa. http://www.who.int/bulletin/volumes/89/1/10-079798/en/index.html (accessed Oct 31, 2012). |
| 5. | van Leth, F.; Phanuphak, P.; Ruxrungtham, K.; Baraldi, E.; Miller, S.; Gazzard, B.; Cahn, P.; Lalloo, U. G.; van der Westhuizen, I. P.; Malan, D. R.; Johnson, M. A.; Santos, B. R.; Mulcahy, F.; Wood, R.; Levi, G. C.; Reboredo, G.; Squires, K.; Cassetti, I.; Petit, D.; Raffi, F.; Katlama, C.; Murphy, R. L.; Horban, A.; Dam, J. P.; Hassink, E.; van Leeuwen, R.; Robinson, P.; Wit, F. W.; Lange, J. M. A. Lancet 2004, 363, 1253–1263. doi:10.1016/S0140-6736(04)15997-7 |
| 29. | Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c |
| 14. | Gupton, B. F. Process for making 3-amino-2-chloro-4-methylpyridine. U.S. Patent 6,399,781, June 4, 2002. |
| 18. | Bogdan, A. R.; Poe, S. L.; Kubis, D. C.; Broadwater, S. J.; McQuade, D. T. Angew. Chem., Int. Ed. 2009, 48, 8547–8550. doi:10.1002/anie.200903055 |
| 19. | Lévesque, F.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 1706–1709. doi:10.1002/anie.201107446 |
| 20. | Booker-Milburn, K. Nat. Chem. 2012, 4, 433–435. doi:10.1038/nchem.1356 |
| 21. | Smith, C. J.; Smith, C. D.; Nikbin, N.; Ley, S. V.; Baxendale, I. R. Org. Biomol. Chem. 2011, 9, 1927–1937. doi:10.1039/c0ob00813c |
| 22. | Sahoo, H. R.; Kralj, J. G.; Jensen, K. F. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. doi:10.1002/anie.200701434 |
| 23. | Baxendale, I. R. J. Chem. Technol. Biotechnol. 2013, 88, 519–552. doi:10.1002/jctb.4012 |
| 24. | Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/c2ob27002a |
| 25. | Polyzos, A.; O'Brien, M.; Petersen, T. P.; Baxendale, I. R.; Ley, S. V. Angew. Chem., Int. Ed. 2011, 50, 1190–1193. doi:10.1002/anie.201006618 |
| 26. | Baxendale, I. R.; Ley, S. V.; Mansfield, A. C.; Smith, C. D. Angew. Chem., Int. Ed. 2009, 48, 4017–4021. doi:10.1002/anie.200900970 |
| 27. | Kupracz, L.; Hartwig, J.; Wegner, J.; Ceylan, S.; Kirschning, A. Beilstein J. Org. Chem. 2011, 7, 1441–1448. doi:10.3762/bjoc.7.168 |
| 13. | Boswell, R. F.; Gupton, B. F.; Lo, Y. S. Method for Making Nevirapine. U.S. Patent 6,680,383, Jan 20, 2004. |
| 28. | Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. doi:10.1002/anie.201004637 |
| 29. | Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c |
| 13. | Boswell, R. F.; Gupton, B. F.; Lo, Y. S. Method for Making Nevirapine. U.S. Patent 6,680,383, Jan 20, 2004. |
| 6. | Proceedings of "Joint WHO/UNAIDS annual consultation with pharmaceutical companies: Global forecasts of antiretroviral demand 2012-2015". November 5-6, 2012, Geneva, Switzerland. http://www.who.int/hiv/amds/forecasting_meeting2012/en/ (accessed June 6, 2013). |
| 15. | Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583–4592. doi:10.1039/C0CC05060A |
| 16. | Wiles, C.; Watts, P. Chem. Commun. 2011, 47, 6512–6535. doi:10.1039/C1CC00089F |
| 17. | Yoshida, J.-I. Chem. Rec. 2010, 10, 332–341. doi:10.1002/tcr.201000020 |
| 39. | Texier-Boullet, F.; Foucaud, A. Tetrahedron Lett. 1982, 23, 4927–4928. doi:10.1016/S0040-4039(00)85749-4 |
| 40. | Broman, S. L.; Petersen, A. U.; Tortzen, C. G.; Vibenholt, J.; Bond, A. D.; Nielsen, M. B. Org. Lett. 2012, 14, 318–321. doi:10.1021/ol2030586 |
| 14. | Gupton, B. F. Process for making 3-amino-2-chloro-4-methylpyridine. U.S. Patent 6,399,781, June 4, 2002. |
| 38. | Vapourtec Flow Chemistry Equipment, UK. http://www.vapourtec.co.uk (accessed June 21, 2013). |
| 44. | Opalka, S. M.; Park, J. K.; Longstreet, A. R.; McQuade, D. T. Org. Lett. 2013, 15, 996–999. doi:10.1021/ol303442m |
| 38. | Vapourtec Flow Chemistry Equipment, UK. http://www.vapourtec.co.uk (accessed June 21, 2013). |
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
| 45. | Razzaq, T.; Kappe, C. O. Chem.–Asian J. 2010, 5, 1274–1289. doi:10.1002/asia.201000010 |
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
| 45. | Razzaq, T.; Kappe, C. O. Chem.–Asian J. 2010, 5, 1274–1289. doi:10.1002/asia.201000010 |
| 41. | Bogdan, A. R.; Mason, B. P.; Sylvester, K. T.; McQuade, D. T. Angew. Chem., Int. Ed. 2007, 46, 1698–1701. doi:10.1002/anie.200603854 |
| 42. | Bogdan, A.; McQuade, D. T. Beilstein J. Org. Chem. 2009, 5, No. 17. doi:10.3762/bjoc.5.17 |
| 43. | Opalka, S. M.; Longstreet, A. R.; McQuade, D. T. Beilstein J. Org. Chem. 2011, 7, 1671–1679. doi:10.3762/bjoc.7.197 |
| 44. | Opalka, S. M.; Park, J. K.; Longstreet, A. R.; McQuade, D. T. Org. Lett. 2013, 15, 996–999. doi:10.1021/ol303442m |
| 34. | Longstreet, A. R.; Campbell, B. S.; Gupton, B. F.; McQuade, D. T. Org. Lett. 2013, 15, 5298–5301. doi:10.1021/ol4025265 |
© 2013 Longstreet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)