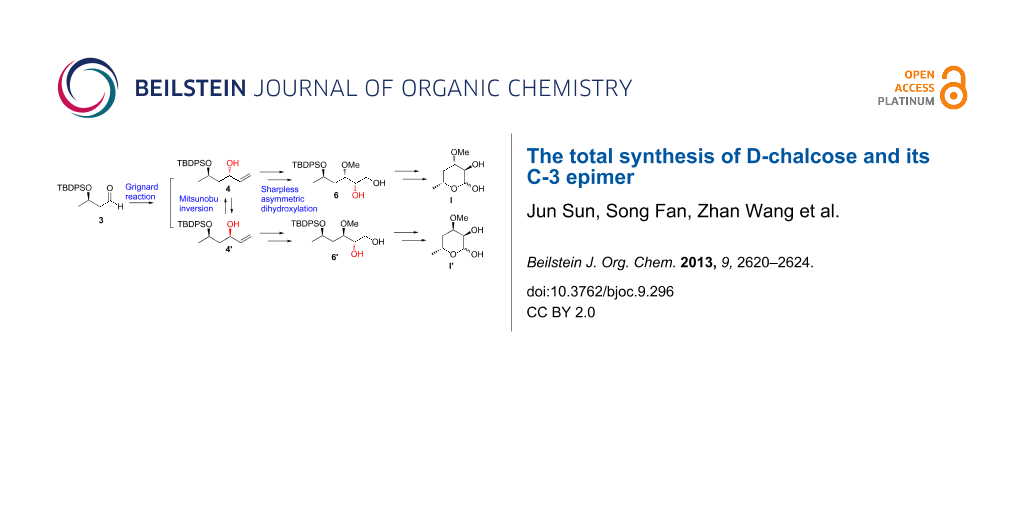
Abstract
We completed a new and efficient synthesis of D-chalcose (I) and the first synthesis of its C-3 epimer (I′) in nine steps with overall yields of 23% and 24%, respectively. The key steps in the sequence were the formation of the stereocenter on C3 via Grignard reaction, the introduction of the stereogenic center on C2 by Sharpless asymmetric dihydroxylation, the protection of the C1 and C2 hydroxy groups with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf), and the selective cleavage of the primary OTBS ether using catalytic DL-10-camphorsulfonic acid (CSA) in MeOH.

Graphical Abstract
Introduction
Chalcose (4,6-dideoxy-3-O-methyl-D-xylo-hexose, I [1,2]) is a structural component of many macrolide antibiotics, such as chalcomycin [3], neutramycin [4], and lankamycin [5] (Scheme 1). After its structure was determined using chemical degradation and spectroscopic analyses, several syntheses of chalcose were reported. The conversion of desosamine into D-chalcose was described by Westwood’s group [6]. A small number of stereospecific syntheses beginning from carbohydrate precursors have been reported [7-10], but the route developed by Usov et al. [11] was especially significant because it demonstrated a facile method to introduce the deoxy functionalities. Acrolein dimer [12], methyl 2-cis-5-hexadienoate [13], and trans-1-methoxy-3-(trimethylsilyloxy)-1,3-butadiene [14] have been utilized as non-carbohydrate precursors during the synthesis of DL-chalcose, but no attempt was made to resolve the racemate. D- and L-chalcose were prepared from racemic or meso-divinylglycols by Schmidt et al. [15,16], however, this method had drawbacks, such as non-commercially sourced material and unsatisfactory overall yield. As part of our study concerning the structure–activity relationships and chemistry of new antibiotics, we sought to develop a reliable and efficient synthetic route starting from inexpensive, commercial sources to provide access to chalcose. We describe a new and efficient 9-step synthesis of D-chalcose (I) with a 23% overall yield. In addition its efficiency, the route enabled facile preparation of chalcose’s C-3 epimer (4,6-dideoxy-3-O-methyl-D-ribo-hexose, I′); the C-3 epimer has not been synthesized previously.
Results and Discussion
The retrosynthetic analysis of I and I′ is presented in Scheme 1. Diol II and II′ arose from a Sharpless asymmetric dihydroxylation that form the C2 stereogenic center. The installation of the C3 stereocenter on vinyl ether III was proposed to utilize a Grignard reaction followed by chromatographic separation. Aldehyde IV would be generated from commercial ethyl (R)-3-hydroxybutyrate (1) via reduction.
![[1860-5397-9-296-i1]](/bjoc/content/inline/1860-5397-9-296-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of I and I'. PG = protecting group; protecting groups may vary independently.
Scheme 1: Retrosynthesis of I and I'. PG = protecting group; protecting groups may vary independently.
The synthesis began by protecting the commercial ethyl (R)-3-hydroxybutyrate (1) using a nearly quantitative silylation of the free hydroxy group under classical conditions (TBDPSCl, imidazole, DMF, rt) [17] to form the corresponding ester 2 (Scheme 2). The reduction of 2 using DIBAL-H in anhydrous CH2Cl2 at −78 °C was quenched with methanol at −30 °C to provide aldehyde 3 [18]. Subsequently, treating aldehyde 3 with vinylmagnesium bromide in the presence of CuI at −78 °C generated alcohol 4 and 4′ as a 4:5 mixture of diastereomers [19]. This mixture was separated using silica gel chromatography to achieve a 78% overall yield over three steps. The unwanted syn-diastereomer 4' was converted into the required anti-diastereomer 4 via Mitsunobu inversion followed by removal of the benzoate group under basic conditions [20]. Similarly, alcohol 4 could be converted to 4′.
![[1860-5397-9-296-i2]](/bjoc/content/inline/1860-5397-9-296-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (a) TBDPSCl, DMAP, imidazole, CH2Cl2, rt, 99%; (b) DIBAL-H, CH2Cl2, −78 °C, 93%; (c) vinylmagnesium bromide, CuI, Et2O, −78 °C, 85%; (d) 1) DIAD, PPh3, PhCOOH, THF, 40 °C; 2) 10% NaOH, THF, rt, 90% in two steps; (e) DEAD, PPh3, PhCOOH, THF, 40 °C; 2) 10% NaOH, THF, rt, 91% in two steps. TBDPS = t-butyldiphenylsilyl, DMAP = N,N-4-dimethylaminopyridine, DIBAL-H = diisobutylaluminum hydride, DIAD = diisopropyl azodicarboxylate, DEAD = diethyl azodicarboxylate.
Scheme 2: Reagents and conditions: (a) TBDPSCl, DMAP, imidazole, CH2Cl2, rt, 99%; (b) DIBAL-H, CH2Cl2, −78 °C...
Alcohol 4 was methylated using MeI and t-BuOK to produce 5 in nearly quantitative yield (Scheme 3). Sharpless asymmetric dihydroxylation [21] of olefin 5 using AD-mix-β in a 1:1 mixture of t-BuOH/H2O at 0 °C over four days afforded diol 6 with S configuration in 80% yield.
![[1860-5397-9-296-i3]](/bjoc/content/inline/1860-5397-9-296-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (a) MeI, t-BuOK,THF, rt, 96%; (b) AD-mix-β, t-BuOH/H2O, 0 °C, 80%; (c) TEMPO, NaClO, CH2Cl2, −5 °C. AD = asymmetric dihydroxylation, TEMPO = 2,2,6,6-tetramethyl-1-piperidinyloxy, free radical.
Scheme 3: Reagents and conditions: (a) MeI, t-BuOK,THF, rt, 96%; (b) AD-mix-β, t-BuOH/H2O, 0 °C, 80%; (c) TEM...
Next, to selectively oxidize the primary alcohol into an aldehyde, we utilized TEMPO/NaClO [22] to oxidize diol 6. However, the reaction failed to deliver aldehyde 7 (Scheme 3). Therefore, we proposed that diol 6 should be protected and the primary OH group could then be selectively deprotected and oxidized to yield the corresponding aldehyde. Our first step toward this goal was to select appropriate protective groups. Diol 6 was initially converted into benzylidene derivative 8 (α,α-dimethoxytoluene/PPTS/CH2Cl2); 8 was subjected to a regioselective reductive ring-opening reaction [23] with DIBAL-H in CH2Cl2 to afford a 1:1 mixture of free primary alcohol 9 and undesired secondary alcohol 10 (Scheme 4).
![[1860-5397-9-296-i4]](/bjoc/content/inline/1860-5397-9-296-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: (a) PhCH(OCH3)2, PPTS, CH2Cl2, rt; (b) DIBAL-H, CH2Cl2, 0 °C, 76% in two steps. PPTS = pyridinium p-toluenesulfonate.
Scheme 4: Reagents and conditions: (a) PhCH(OCH3)2, PPTS, CH2Cl2, rt; (b) DIBAL-H, CH2Cl2, 0 °C, 76% in two s...
Consequently, we attempted to treat diol 6 with 2.5 equiv TBSOTf in anhydrous CH2Cl2 in the presence of 2,6-lutidine for 12 h at room temperature, providing compound 11 in 90% yield [24]. Subsequently, we examined the deprotection of 11 using the various conditions listed in Table 1. Compound 11 was initially treated with PPTS/MeOH [25] (Table 1, entry 1 and 2) to form alcohol 12 as the major expected product. Unfortunately, this reaction was very low yielding and afforded diol 6 as the major product. In addition, we used HF·pyridine/THF [26] (Table 1, entry 3 and 4) with similar effect. Treating 11 with AcOH/H2O/THF (13:7:3) [27] (Table 1, entry 5 and 6) very slowly afforded alcohol 12 in low yield. Eventually, acid-catalyzed deprotection conditions were examined using CSA [28] (Table 1, entry 7–11) in various solvents. After several attempts, we discovered that treating 11 with CSA (0.1 equiv) in methanol at 0 °C for 45 min provided alcohol 12 in the highest yield (Table 1, entry 10). Additionally, the combined yield increased with increasing reaction times, but, the amount of the desired alcohol 12 decreased (Table 1, entries 9 and 10). Moreover, using a protic solvent, such as methanol, was beneficial for the reaction (Table 1, entry 7). Consequently, controlling reaction time was crucial to avoid generating undesired diol 6. Fortunately, diol 6 could be recycled via TBSOTf to furnish 11, and 11 could be converted into 12 (Scheme 5).
Table 1: Deprotection of 11.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-296-i8.svg?max-width=637&scale=1.0)
|
|||
| Entry | Conditions | Yield (%)a | Ratio (12:6)b |
|---|---|---|---|
| 1 | PPTS (0.1 equiv), MeOH, 0 °C, 2 h | trace | |
| 2 | PPTS (0.1 equiv), MeOH, 25 °C, 5 h | 11 | 0.5:1 |
| 3 | HF-pyridine, THF, 0 °C, 1 h | trace | |
| 4 | HF-pyridine, THF, 25 °C, 1 h | 17 | 0.2:1 |
| 5 | AcOH:H2O:THF (13:7:3), 25 °C, 2 d | 16 | 1.2:1 |
| 6 | AcOH:H2O:THF (13:7:3), 40 °C, 20 h | 23 | 0.4:1 |
| 7 | CSA (0.1 equiv), CH2Cl2:MeOH (1:1), 0 °C, 1 h | 20 | 0.8:1 |
| 8 | CSA (0.1 equiv), MeOH, 0 °C,1 h | 74 | 2.4:1 |
| 9 | CSA (0.1 equiv), MeOH, 0 °C,1.2 h | 76 | 1.5:1 |
| 10 | CSA (0.1 equiv), MeOH, 0 °C, 45 min | 71 | 4.7:1 |
| 11 | CSA (0.1 equiv), MeOH, 0 °C, 30 min | 52 | 4.9:1 |
aCombined yields of 12 and 6. bThe ratios were determined after chromatographic separation.
![[1860-5397-9-296-i5]](/bjoc/content/inline/1860-5397-9-296-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reagents and conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2, rt, 90%; (b) CSA, MeOH, 0 °C, 59%. TBS = t-butyldimethylsilyl, Tf = trifluoromethanesulfonyl, CSA = DL-10-camphorsulfonic acid.
Scheme 5: Reagents and conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2, rt, 90%; (b) CSA, MeOH, 0 °C, 59%. TBS = ...
With requisite intermediate 12 in hand, our attention was directed toward preparing the final product, chalcose. Alcohol 12 was subjected to Dess–Martin periodinane [29] because it afforded aldehyde 13 in 86% yield, making it superior to the Swern oxidation. Subsequently, aldehyde 13 was efficiently deprotected using TBAF in THF at 0 °C for 1 h to form D-chalcose (I) in 84% yield. Finally, D-chalcose (I) converted into diacetate 14 via treatment with acetic anhydride to facilitate characterization (Scheme 6). The 1H NMR data for diacetate 14 was identical to the data reported for the natural product-derived D-chalcose diacetate [10].
![[1860-5397-9-296-i6]](/bjoc/content/inline/1860-5397-9-296-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, rt, 86%; (b) TBAF, THF, 0 °C, 84%; (c) Py, AcO2, rt, 80%. TBAF = tetra-n-butylammonium fluoride.
Scheme 6: Reagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, rt, 86%; (b) TBAF, THF, 0 °C, 84%; (c...
A nearly identical synthetic procedure was used to transform 4′ into the C-3 epimer of chalcose (I′); this procedure began with a methylation to form 5′ (Scheme 7). The stereoselective dihydroxylation of 5′ using AD-mix-β afforded desired diol 6′ in 82% yield. Subsequently, treating 6′ with TBSOTf afforded 7′. Selectively deprotecting the primary hydroxy group using CSA in methanol at 0 °C for 45 min provided alcohol 8′ in 60% yield, as well as a 15% yield of diol 6′ that could be recycled to form 7′ using TBSOTf. Alcohol 8′ was oxidized using Dess–Martin periodinane to give aldehyde 9′ in 86% yield. Finally, 9′ was efficiently deprotected with TBAF, leading to the C-3 epimer of chalcose (I′) in 83% yield. Epimer I′ was acetylated using acetic anhydride in pyridine, furnishing diacetate 10′ in 80% yield; in the major product, C1 was assigned a β-configuration based on the NMR spectroscopy data.
![[1860-5397-9-296-i7]](/bjoc/content/inline/1860-5397-9-296-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reagents and conditions: (a) MeI, t-BuOK,THF, rt, 96%; (b) AD-mix-β, t-BuOH/H2O, 0 °C, 82%; (c) TBSOTf, 2,6-lutidine, CH2Cl2, rt, 91%; (d) CSA, MeOH, 0 °C, 60%; (e) Dess–Martin periodinane, CH2Cl2, rt, 86%; (f) TBAF, THF, 0 °C, 83%; (g) Py, AcO2, rt, 80%.
Scheme 7: Reagents and conditions: (a) MeI, t-BuOK,THF, rt, 96%; (b) AD-mix-β, t-BuOH/H2O, 0 °C, 82%; (c) TBS...
Conclusion
In conclusion, we developed a new and efficient synthesis of D-chalcose (I), requiring nine steps and demonstrating a 23% overall yield. The first synthesis of its C-3 epimer (I′) was achieved using a similar route in 24% overall yield. Key epimeric intermediates 4 and 4′ could be interconverted via Mitsunobu reaction, and their absolute configurations were assigned after their transformation into D-chalcose (I) and its C-3 epimer (I′), respectively. Notably, the stereocenter on C3 was set using a Grignard reaction, and the hydroxy group on C2 could be introduced with the correct stereochemistry by performing a Sharpless asymmetric dihydroxylation on vinyl ether 5. In addition, diol 6 was converted to 11 by protection with TBSOTf followed by selective unveiling of the C1 hydroxy group via exposure to CSA/MeOH for 45 min at 0 °C; this process afforded alcohol 12 in good yield. The notable advantage of this strategy is its high degree of flexibility, rendering it applicable to the preparation of various C-3 analogues of chalcose and other related 4-deoxy sugars.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data of all products, and copies of MS and NMR spectra. | ||
| Format: PDF | Size: 4.8 MB | Download |
References
-
Woo, P. W. K.; Dion, H. W.; Bartz, Q. R. J. Am. Chem. Soc. 1961, 83, 3352–3353. doi:10.1021/ja01476a053
Return to citation in text: [1] -
Woo, P. W. K.; Dion, H. W.; Johnson, L. F. J. Am. Chem. Soc. 1962, 84, 1066–1067. doi:10.1021/ja00865a049
Return to citation in text: [1] -
Woo, P. W. K.; Dion, H. W.; Bartz, Q. R. J. Am. Chem. Soc. 1964, 86, 2726–2727. doi:10.1021/ja01067a047
Return to citation in text: [1] -
Kunstmann, M. P.; Mitscher, L. A. Experientia 1965, 21, 372–373. doi:10.1007/BF02139741
Return to citation in text: [1] -
Keller-Schierlein, W.; Roncari, G. Helv. Chim. Acta 1962, 45, 138–152. doi:10.1002/hlca.19620450118
Return to citation in text: [1] -
Foster, A. B.; Stacey, M.; Webber, J. M.; Westwood, J. H. J. Chem. Soc. 1965, 2318–2323. doi:10.1039/JR9650002318
Return to citation in text: [1] -
McNally, S.; Overend, W. G. Chem. Ind. (London) 1964, 49, 2021.
Return to citation in text: [1] -
Lawton, B. T.; Ward, D. J.; Szarek, W. A.; Jones, J. K. N. Can. J. Chem. 1969, 47, 2899–2901. doi:10.1139/v69-484
Return to citation in text: [1] -
Kefurt, K.; Kefurtová, Z.; Jarý, J. Collect. Czech. Chem. Commun. 1973, 38, 2627–2632. doi:10.1135/cccc19732627
Return to citation in text: [1] -
Redlich, H.; Roy, W. Carbohydr. Chem. 1979, 68, 275–285.
Return to citation in text: [1] [2] -
Kochetkov, N. K.; Usov, A. I. Tetrahedron Lett. 1963, 4, 519–521. doi:10.1016/S0040-4039(01)90664-1
Return to citation in text: [1] -
Srivastava, R. M.; Brown, R. K. Can. J. Chem. 1970, 48, 830–837. doi:10.1139/v70-133
Return to citation in text: [1] -
Torssell, K.; Tyagi, M. P. Acta Chem. Scand., Ser. B 1977, B31, 7–10. doi:10.3891/acta.chem.scand.31b-0007
Return to citation in text: [1] -
Danishefsky, S.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 1597–1598. doi:10.1021/jo00347a053
Return to citation in text: [1] -
Küfner, U.; Schmidt, R. R. Angew. Chem. 1986, 98, 90–91. doi:10.1002/ange.19860980116
Angew. Chem., Int. Ed. Engl. 1986, 25, 89 doi:10.1002/anie.198600891
Return to citation in text: [1] -
Küfner, U.; Schmidt, R. R. Carbohydr. Res. 1987, 161, 211–223. doi:10.1016/S0008-6215(00)90078-8
Return to citation in text: [1] -
Hanessian, S.; Lavallee, P. Can. J. Chem. 1975, 53, 2975–2977. doi:10.1139/v75-419
Return to citation in text: [1] -
Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006
Return to citation in text: [1] -
Elsworth, J. D.; Willis, C. L. Chem. Commun. 2008, 1587–1589. doi:10.1039/b717078e
Return to citation in text: [1] -
Jana, N.; Mahapatra, T.; Nanda, S. Tetrahedron: Asymmetry 2009, 20, 2622–2628. doi:10.1016/j.tetasy.2009.10.007
Return to citation in text: [1] -
Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispini, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M.; Xu, D.; Zhang, X.-L. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003
Return to citation in text: [1] -
de Nooy, A. E. J.; Besemer, A. C.; van Bekkum, H. Synthesis 1996, 10, 1153–1176. doi:10.1055/s-1996-4369
Return to citation in text: [1] -
Takano, S.; Akiyama, M.; Sato, S.; Ogasawara, K. Chem. Lett. 1983, 12, 1593–1596. doi:10.1246/cl.1983.1593
Return to citation in text: [1] -
Corey, E. J.; Cho, H.; Rücker, C.; Hua, D. H. Tetrahedron Lett. 1981, 22, 3455–3458. doi:10.1016/S0040-4039(01)81930-4
Return to citation in text: [1] -
Prakash, C.; Saleh, S.; Blair, I. A. Tetrahedron Lett. 1989, 30, 19–22. doi:10.1016/S0040-4039(01)80311-7
Return to citation in text: [1] -
Nicolaou, K. C.; Webber, S. E. Synthesis 1986, 453–461. doi:10.1055/s-1986-31673
Return to citation in text: [1] -
Kawai, A.; Hara, O.; Hamada, Y.; Shioiri, T. Tetrahedron Lett. 1988, 29, 6331–6334. doi:10.1016/S0040-4039(00)82339-4
Return to citation in text: [1] -
Hara, A.; Morimoto, R.; Iwasaki, Y.; Saitoh, T.; Ishikawa, Y.; Nishiyama, S. Angew. Chem., Int. Ed. 2012, 51, 9877–9880. doi:10.1002/anie.201204992
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070
Return to citation in text: [1]
| 1. | Woo, P. W. K.; Dion, H. W.; Bartz, Q. R. J. Am. Chem. Soc. 1961, 83, 3352–3353. doi:10.1021/ja01476a053 |
| 2. | Woo, P. W. K.; Dion, H. W.; Johnson, L. F. J. Am. Chem. Soc. 1962, 84, 1066–1067. doi:10.1021/ja00865a049 |
| 6. | Foster, A. B.; Stacey, M.; Webber, J. M.; Westwood, J. H. J. Chem. Soc. 1965, 2318–2323. doi:10.1039/JR9650002318 |
| 20. | Jana, N.; Mahapatra, T.; Nanda, S. Tetrahedron: Asymmetry 2009, 20, 2622–2628. doi:10.1016/j.tetasy.2009.10.007 |
| 5. | Keller-Schierlein, W.; Roncari, G. Helv. Chim. Acta 1962, 45, 138–152. doi:10.1002/hlca.19620450118 |
| 21. | Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispini, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M.; Xu, D.; Zhang, X.-L. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003 |
| 4. | Kunstmann, M. P.; Mitscher, L. A. Experientia 1965, 21, 372–373. doi:10.1007/BF02139741 |
| 18. | Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006 |
| 3. | Woo, P. W. K.; Dion, H. W.; Bartz, Q. R. J. Am. Chem. Soc. 1964, 86, 2726–2727. doi:10.1021/ja01067a047 |
| 19. | Elsworth, J. D.; Willis, C. L. Chem. Commun. 2008, 1587–1589. doi:10.1039/b717078e |
| 13. | Torssell, K.; Tyagi, M. P. Acta Chem. Scand., Ser. B 1977, B31, 7–10. doi:10.3891/acta.chem.scand.31b-0007 |
| 15. |
Küfner, U.; Schmidt, R. R. Angew. Chem. 1986, 98, 90–91. doi:10.1002/ange.19860980116
Angew. Chem., Int. Ed. Engl. 1986, 25, 89 doi:10.1002/anie.198600891 |
| 16. | Küfner, U.; Schmidt, R. R. Carbohydr. Res. 1987, 161, 211–223. doi:10.1016/S0008-6215(00)90078-8 |
| 12. | Srivastava, R. M.; Brown, R. K. Can. J. Chem. 1970, 48, 830–837. doi:10.1139/v70-133 |
| 17. | Hanessian, S.; Lavallee, P. Can. J. Chem. 1975, 53, 2975–2977. doi:10.1139/v75-419 |
| 11. | Kochetkov, N. K.; Usov, A. I. Tetrahedron Lett. 1963, 4, 519–521. doi:10.1016/S0040-4039(01)90664-1 |
| 7. | McNally, S.; Overend, W. G. Chem. Ind. (London) 1964, 49, 2021. |
| 8. | Lawton, B. T.; Ward, D. J.; Szarek, W. A.; Jones, J. K. N. Can. J. Chem. 1969, 47, 2899–2901. doi:10.1139/v69-484 |
| 9. | Kefurt, K.; Kefurtová, Z.; Jarý, J. Collect. Czech. Chem. Commun. 1973, 38, 2627–2632. doi:10.1135/cccc19732627 |
| 10. | Redlich, H.; Roy, W. Carbohydr. Chem. 1979, 68, 275–285. |
| 14. | Danishefsky, S.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 1597–1598. doi:10.1021/jo00347a053 |
| 24. | Corey, E. J.; Cho, H.; Rücker, C.; Hua, D. H. Tetrahedron Lett. 1981, 22, 3455–3458. doi:10.1016/S0040-4039(01)81930-4 |
| 22. | de Nooy, A. E. J.; Besemer, A. C.; van Bekkum, H. Synthesis 1996, 10, 1153–1176. doi:10.1055/s-1996-4369 |
| 23. | Takano, S.; Akiyama, M.; Sato, S.; Ogasawara, K. Chem. Lett. 1983, 12, 1593–1596. doi:10.1246/cl.1983.1593 |
| 29. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070 |
| 27. | Kawai, A.; Hara, O.; Hamada, Y.; Shioiri, T. Tetrahedron Lett. 1988, 29, 6331–6334. doi:10.1016/S0040-4039(00)82339-4 |
| 28. | Hara, A.; Morimoto, R.; Iwasaki, Y.; Saitoh, T.; Ishikawa, Y.; Nishiyama, S. Angew. Chem., Int. Ed. 2012, 51, 9877–9880. doi:10.1002/anie.201204992 |
| 25. | Prakash, C.; Saleh, S.; Blair, I. A. Tetrahedron Lett. 1989, 30, 19–22. doi:10.1016/S0040-4039(01)80311-7 |
| 26. | Nicolaou, K. C.; Webber, S. E. Synthesis 1986, 453–461. doi:10.1055/s-1986-31673 |
© 2013 Sun et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)