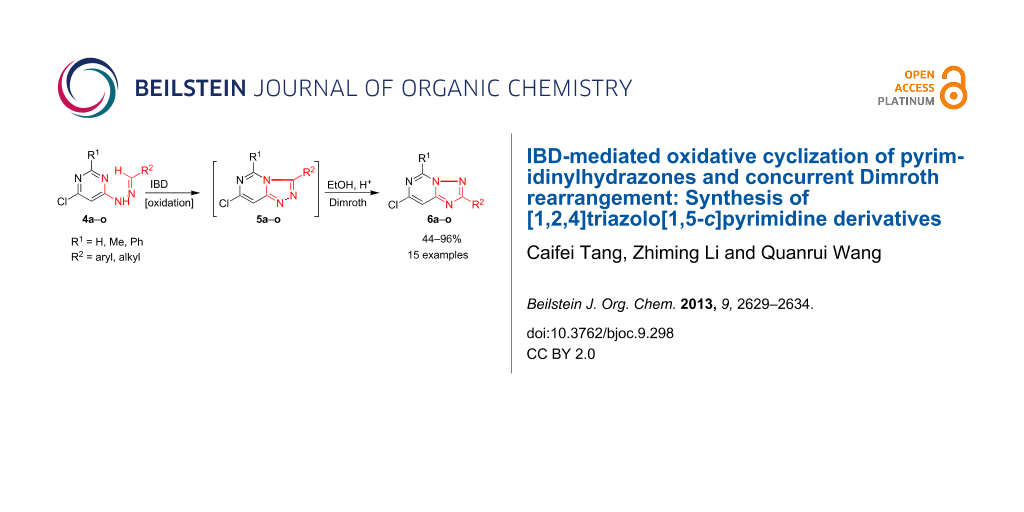
Abstract
Oxidative cyclization of 6-chloro-4-pyrimidinylhydrazones 4 with iodobenzene diacetate (IBD) in dichloromethane gives rise to [1,2,4]triazolo[4,3-c]pyrimidine derivatives 5a–o. These incipient products undergo feasible Dimroth rearrangement to furnish the isolated [1,2,4]triazolo[1,5-c]pyrimidines 6a–o in moderate to high yields.

Graphical Abstract
Introduction
The pyrimidine moiety is an important pharmacophore [1]. Especially, the fused bi- or tricyclic heterocyles containing a pyrimidine motif have received considerable interest in the design and discovery of new compounds for pharmaceutical and herbicidal applications [2,3]. For example, the pyrrolo[2,3-d]pyrimidine derivative, ruxolitinib (INCB018424), was discovered as a selective JAK1 and JAK2 inhibitor, which is currently under clinical investigation [4]. To date, a number of fused pyrimidine-type compounds have been successfully commercialized, such as the pyrazolo[3,4-d]pyrimidine allopurinol and the pyrazolo[4,3-d]pyrimidine sildenafil. In addition, trapidil is a [1,2,4]triazolo[1,5-a]pyrimidine compound, which is the first triazolopyrimidine registered as a drug possessing antiproliferative activity in glioma cells and vascular smooth muscle cells (VSMCs) and used as a coronary vasodilator in the therapy of syndromes that may be influenced by immunomodulators [5,6]. Interestingly, the first [1,2,4]triazolo[1,5-a]pyrimidine antibiotic essramycin has been isolated from natural sources (Figure 1) [7].
![[1860-5397-9-298-1]](/bjoc/content/figures/1860-5397-9-298-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structure of two representatives of [1,2,4]triazolopyrimidines.
Figure 1: The structure of two representatives of [1,2,4]triazolopyrimidines.
It has also been revealed that compounds carrying a [1,2,4]triazolo[1,5-c]pyrimidine nucleus behave as selective antagonists for human A2A and A3 adenosine receptor sub-types which offer great promise in the treatment of Parkinson’s disease, or as Hsup90 modulators [8]. Clarkson and co-workers have prepared a range of benzothieno-fused 1,2,4-triazolo[4,3-c]pyrimidines and 1,2,4-triazolo[1,5-c]pyrimidines by the oxidative cyclisation of benzothieno[2,3-d]pyrimidine hydrazones. The investigation has also suggested the important ability as inhibitors of Shiga toxin trafficking for protecting HeLa cells [9].
The wide range of biological activities shown by various triazolopyrimidines encouraged synthetic organic and medicinal chemists to synthesize new members and to study their applications [10,11]. Several strategies for constructing triazolopyrimidine rings are available. These include the pre-formation of 1-amino-6-imino-substituted pyrimidines followed by reaction with carboxylic acids or derivatives as the one-carbon cyclizing agent [12] and the dehydrative cyclization of carboxylic acid N'-(pyrimidin-4-yl)-hydrazide and ring rearrangement [13]. Recently, Thiel and coworkers have also established an efficient palladium-catalyzed intermolecular coupling of aldehyde-derived hydrazones with chloro-substituted pyridines and related heterocycles, from which the triazolopyridines and other related fused heterocycles can be obtained after oxidative cyclization [14]. In accordance with the significance shown by triazolopyrimidines, the development of complementary and simple synthetic methods for the efficient preparation of novel triazolopyrimidines is needed.
Recently, we have reported the synthesis of 8-bromo-7-chloro-[1,2,4]triazolo[1,5-c]pyrimidines from the reaction of pyrimidinylhydrazones with bromine followed by Dimroth rearrangement [15]. In the present paper, we wish to report an efficient synthesis of new [1,2,4]triazolo[1,5-c]pyrimidine derivatives from hypervalent iodine (IBD)-mediated oxidative cyclisation of aldehyde pyrimidinylhydrazones and consecutive Dimroth rearrangement in relatively good yields under very mild conditions.
Results and Discussion
The investigation commenced with the preparation of 1-(6-chloropyrimidin-4-yl)hydrazines 3. The required starting 4,6-dihydroxypyrimidines 1 can be prepared following the reported procedure by condensation of dimethyl malonate either with formamide (for 1a) or the respective formamidine salts (for 1b and 1c) [15].
As outlined in Scheme 1, the conversion of 4,6-dihydroxypyrimidines 1 to the dichloro derivatives 2 was accomplished by refluxing in phosphorus oxychloride for 4 h in the presence of triethylamine in yields ranging from 75 to 90% [16,17]. After screening a set of conditions, 1-(6-chloropyrimidin-4-yl)hydrazines 3 were then obtained in 90–95% yield through the reaction of compounds 2 with two equivalents of 50% hydrazine in ethanol at 45 °C for 2 h (Scheme 1).
![[1860-5397-9-298-i1]](/bjoc/content/inline/1860-5397-9-298-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 1-(6-chloropyrimidin-4-yl)hydrazines 3.
Scheme 1: Synthesis of 1-(6-chloropyrimidin-4-yl)hydrazines 3.
We next examined the synthesis of the corresponding hydrazones of various aldehydes. After screening a set of conditions, optimal conditions could be determined. Thus, 1-(6-chloropyrimidin-4-yl)hydrazine (3a) was condensed with 20% excess benzaldehyde to give 85% yield of the chloropyrimidinylhydrazone 4a in absolute ethanol at room temperature in about 1 h. As shown in Table 1, the reaction proceeded readily for most of the employed aldehydes, affording the corresponding hydrazones 4. In general, aromatic aldehydes provided higher yields than aliphatic aldehydes.
Table 1: Synthesis of the chloropyrimidinylhydrazones 4a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-298-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1 | R2 | Product | Yield [%]b |
|---|---|---|---|---|
| 1 | H | Ph | 4a | 85 |
| 2 | H | 2-ClC6H4 | 4b | 84 |
| 3 | H | 2-furanyl | 4c | 75 |
| 4 | H | 4-(MeO)C6H4 | 4d | 80 |
| 5 | H | Et | 4e | 66 |
| 6 | Me | Ph | 4f | 81 |
| 7 | Me | 2-ClC6H4 | 4g | 85 |
| 8 | Me | 2-furanyl | 4h | 83 |
| 9 | Me | 4-(MeO)C6H4 | 4i | 85 |
| 10 | Me | Et | 4j | 84 |
| 11 | Ph | Ph | 4k | 89 |
| 12 | Ph | 2-ClC6H4 | 4l | 87 |
| 13 | Ph | 2-furanyl | 4m | 90 |
| 14 | Ph | 4-(MeO)C6H4 | 4n | 85 |
| 15 | Ph | Et | 4o | 96 |
aReagents and conditions: chloropyriminylhydrazine 3 (3.0 mmol), EtOH (15 mL), aldehyde (3.6 mmol, 1.2 equiv), rt, 30–60 min. bIsolated yield after column chromatography.
In recent years, organo hypervalent iodine reagents have drawn considerable interests as versatile and environmentally benign oxidants with many applications in organic synthesis because of their low toxicity, stability, high reactivity, and availability. Efficient formation of new carbon–heteroatom bonds as well as carbon–carbon bonds can be achieved typically under mild reaction conditions with these metal-free reagents [18,19]. For example, Salgado has described the synthesis of novel [1,2,4]triazolo[1,5-a]pyrimidine derivatives by iodobenzenediacetate (IBD)-mediated oxidative cyclization of suitable N-benzylidene-N′-pyrimidin-2-yl hydrazine precursors, followed by a Dimroth rearrangement [20]. We envisioned that our aldehyde chloropyrimidinylhydrazones 4 would undergo feasible oxidative cyclisation to afford novel triazolopyrimidines.
We initiated our investigation by examining the oxidative cyclization of 4a with iodobenzenediacetate (IBD) as an oxidant (Table 2). Thus, 4a was treated with one equivalent of freshly prepared IBD in dichloromethane at room temperature. After completion of the reaction, the solvent was removed and the residue was dissolved in EtOH with a catalytic amount of conc. HCl. Usual workup afforded the [1,2,4]triazolo[1,5-c]pyrimidine 6a in 57% yield (Table 2, entry 1).
Table 2: Selected results of screening the optimal conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-298-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | IBD (equiv) | Additive | T (°C) | Time (h) | Solvent | Yield [%]a |
|---|---|---|---|---|---|---|
| 1 | 1.0 | – | 25 | 4 | CH2Cl2 | 57 |
| 2 | 1.1 | – | 25 | 4 | CH2Cl2 | 66 |
| 3 | 1.2 | – | 25 | 4 | CH2Cl2 | 75 |
| 4 | 1.3 | – | 25 | 4 | CH2Cl2 | 75 |
| 5 | 1.3 | – | 25 | 6 | CH2Cl2 | 81 |
| 6 | 1.3 | – | 25 | 24 | CH2Cl2 | 79 |
| 7 | 1.3 | – | 40 | 10 | CH2Cl2 | 72 |
| 8 | 1.3 | – | 25 | 6 | THF | 70 |
| 9 | 1.3 | – | 25 | 6 | EtOAc | 70 |
| 10 | 1.3 | – | 25 | 6 | H2O | 12b |
| 11 | 1.3 | – | 25 | 6 | EtOH | 25 |
| 12 | 1.3 | – | 25 | 6 | DMF | c |
| 13 | 1.3 | Bu4NId | 25 | 6 | CH2Cl2 | 75 |
| 14 | 1.3 | I2d | 25 | 6 | CH2Cl2 | 75 |
aIsolated yields of 6a. bPerformed under microwave irradiation. cComplex mixture of products. dAn amount of 10 mol % was used.
With this promising result in hand, the reaction efficiency was improved by optimizing the conditions, such as the amount of IBD, solvent, reaction time and purification method. Gratifyingly, 6a was formed in 81% yield when 1.3 equiv of IBD were used in dichloromethane for 6 h (Table 2, entry 5). Further extension of the reaction time to 24 h did not provide superior results (Table 2, entry 6). Meanwhile, reactions in THF or ethyl acetate resulted in marginally lower yields (Table 2, entries 8 and 9). Switching to water with assistance by microwave irradiation or performing the reaction in ethanol provided even unacceptable yields (Table 2, entries 10 and 11). Further, when replacing the solvent by DMF, a complex mixture of products was produced, and no pure desired product could be isolated (Table 2, entry 12). Employment of 10 mol % Bu4NI or I2 as an additive did not improve the result (Table 2, entries 13 and 14). It has also been observed that temperature elevation did not benefit the transformation (Table 2, entry 7). Thus, the use of IBD (1.3 equiv) at room temperature in dichloromethane for 6 h was found to be most efficient and therefore used as the standard conditions.
To probe the generality of the IBD-oxidative cyclization protocol, all of the other pyrimidinylhydrazones 4 with different R1 and R2 groups were examined under optimal conditions. As shown in Table 3, the protocol appeared to be quite general. In all cases studied, the respective [1,2,4]triazolo[1,5-c]pyrimidines 6 could be obtained. Various aryl substituents on the triazole ring (R2), including phenyl, electron-rich and electron-poor aryls, were compatible with this reaction, and high yields of the triazolopyrimidines 6 could generally be obtained. Substrates carrying a heteroaromatic group such as 2-furanyl also proceeded smoothly to give a good yield of the product. Aromatic aldehyde hydrazones bearing an electron-donating functional group on the para position of the aromatic ring seems to afford slightly lower yields (Table 3, entries 4, 9, 14).
Table 3: Synthesis of the triazolopyrimidines 6a.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-298-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1 | R2 | Product | Yield [%]b |
|---|---|---|---|---|
| 1 | H | Ph | 6a | 81 |
| 2 | H | 2-ClC6H4 | 6b | 80 |
| 3 | H | 2-furanyl | 6c | 81 |
| 4 | H | 4-(MeO)C6H4 | 6d | 69 |
| 5 | H | Et | 6e | 64 |
| 6 | Me | Ph | 6f | 81 |
| 7 | Me | 2-ClC6H4 | 6g | 86 |
| 8 | Me | 2-furanyl | 6h | 86 |
| 9 | Me | 4-(MeO)C6H4 | 6i | 71 |
| 10 | Me | Et | 6j | 34 |
| 11 | Ph | Ph | 6k | 84 |
| 12 | Ph | 2-ClC6H4 | 6l | 87 |
| 13 | Ph | 2-furanyl | 6m | 87 |
| 14 | Ph | 4-(MeO)C6H4 | 6n | 71 |
| 15 | Ph | Et | 6o | 36 |
aReagents and conditions: hydrazone 4 (1.0 mmol), IBD (1.3 mmol), DCM (6 mL) at rt, then refluxed in abs. EtOH for 1–3 h. bIsolated yield after column chromatography.
On the other hand, the reaction is less sensitive to substitution of 4 at the C-2 position. Although both aromatic and aliphatic aldehyde-derived hydrazones 4 participated well in the reaction, the latter furnished only low to moderate yields (Table 3, entries 5, 10 and 15).
Although the employed procedure was believed to afford initially [1,2,4]triazolo[4,3-c]pyrimidines 5, a formal Dimroth rearrangement occurred readily to give the isolated products that belong to the [1,5-c] series 6. This kind of rearrangement has been described previously [10,11,21]. The accepted mechanism of the Dimroth rearrangement might involve protonation of 5 to give I (5-H+), ring opening (II), tautomerization by H-shift (III) of the 1,2,4-triazole ring, ring closure (IV) and deprotonation to isomeric 1,2,4-triazolo[1,5-c] pyrimidine 6 (Scheme 2).
![[1860-5397-9-298-i2]](/bjoc/content/inline/1860-5397-9-298-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanism for the transformation of [1,2,4]triazolo[4,3-c]pyrimidines 5 to the [1,5-c] series 6.
Scheme 2: Plausible mechanism for the transformation of [1,2,4]triazolo[4,3-c]pyrimidines 5 to the [1,5-c] se...
Indeed, small quantities of the triazole 6 can be observed by 1H NMR and thin-layer chromatography in our original attempts to isolate pure 5. Although very slow, the rearrangement of the triazole 5 to 6 was found to occur spontaneously. The process can be catalyzed under catalytic amount of HCl. In one case, pure 1,2,4-triazolo[4,3-c]pyrimidine 5f was isolated and characterized (see Supporting Information File 1 for full experimental data), which was dissolved in ethanol and allowed to be stirred at room temperature. After one day, formation of 6f was observed. The isomerization was completed after 10 days as evidenced by the disappearance of the methyl signal at 2.39 ppm in 5f and fully shifting to downfield absorption at 3.04 ppm in 6f.
Since compounds 5 are generally unstable as compared with their [1,5-c]-counterparts, isolation of the initial [1,2,4]triazolo[4,3-c]pyrimidine intermediates proved to be of no preparative value. Instead, they were allowed directly to isomerize under HCl catalysis to provide the thermodynamically more stable title compounds through tandem ring opening and ring closing Dimroth reactions.
The structure of bicycles 6a–o was principally assigned from their spectral evidence and mass spectrometry analysis (see Supporting Information File 2 for full data). To confirm the structure of the regio-isomer of the cyclization rearrangement, colorless single crystals of compound 6h were obtained from CH2Cl2/n-hexane and its molecular structure was determined by X-ray crystallography, providing unequivocal evidence for the assignments [22].
Conclusion
In conclusion, we report herein a general and convenient method for the synthesis of novel [1,2,4]triazolo[1,5-c]pyrimidine derivatives with moderate to excellent yields. The protocol features initial oxidative cyclization of readily available chloropyrimidinylhydrazones followed by feasible Dimroth rearrangement. The procedure offers several advantages including good yields, operational simplicity, environmental friendliness and shorter reaction time along with broad substrate scope, which make it a useful and attractive process for the synthesis of structurally diverse triazolopyrimidines. Moreover, the 7-chloro-substitution would allow further manipulations through nucleophilic substitution [23] and metal-catalyzed cross-coupling reactions, which are under investigation in our group.
Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 21372045) and the Scientific Study Fund for Distinguished Doctor Candidates at Fudan University. Wang is grateful to the Hangzhou Government for Qian-Jiang scholar assistance. We are also indebted to Dr. Z. Chen for X-ray measurements and G. Tang for acquiring high-resolution mass spectrometry data.
References
-
Lagoja, I. M. Chem. Biodiversity 2005, 2, 1–50. doi:10.1002/cbdv.200490173
Return to citation in text: [1] -
Gribble, G. W.; Joule, J. A. Progress in Heterocyclic Chemistry; Elsevier: Oxford, 2007; Vol. 18.
Return to citation in text: [1] -
Yet, L. Five-membered ring systems: With more than one N atom in Progress in Heterocyclic Chemistry 2011, Vol. 23, pp 231–266.
Return to citation in text: [1] -
Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.-Y.; Liu, P.; Zhou, J. Org. Lett. 2009, 11, 1999–2002. doi:10.1021/ol900350k
Return to citation in text: [1] -
Cheng, Y.; Liu, P.; Chen, H.; Zeng, F. J. Cardiovasc. Pharm. 2000, 35, 1–6. doi:10.1097/00005344-200001000-00001
Return to citation in text: [1] -
Kenji, I.; Hisayo, F.; Hida, M.; Awazu, M.; Midori, A. Eur. J. Pharmacol. 2005, 515, 28–33. doi:10.1016/j.ejphar.2005.04.013
Return to citation in text: [1] -
Battaglia, U.; Moody, C. J. J. Nat. Prod. 2010, 73, 1938–1939. doi:10.1021/np100298m
Return to citation in text: [1] -
Baraldi, P. G.; Saponaro, G.; Tabrizi, M. A.; Baraldi, S.; Romagnoli, R.; Moorman, A. R.; Varani, K.; Borea, P. A.; Preti, D. Bioorg. Med. Chem. 2012, 20, 1046–1059. doi:10.1016/j.bmc.2011.11.037
Return to citation in text: [1] -
Guetzoyan, L. J.; Spooner, R. A.; Lord, J. M.; Roberts, L. M.; Clarkson, G. J. Eur. J. Med. Chem. 2010, 45, 275–283. doi:10.1016/j.ejmech.2009.10.007
Return to citation in text: [1] -
Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 1999, 73, 131–176. doi:10.1016/S0065-2725(08)60942-3
Return to citation in text: [1] [2] -
Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 2000, 77, 345–394. doi:10.1016/S0065-2725(00)77009-7
Return to citation in text: [1] [2] -
Rashad, A. E.; Heikal, O. A.; El-Nezhawy, A. O. H.; Abdel-Megeid, F. M. E. Heteroat. Chem. 2005, 16, 226–234. doi:10.1002/hc.20114
Return to citation in text: [1] -
Neustadt, B. R.; Liu, H.; Hao, J.; Greenlee, W. J.; Stamford, A. W.; Foster, C.; Arik, L.; Lachowicz, J.; Zhang, H.; Bertorelli, R.; Fredduzzi, S.; Varty, G.; Cohen-Williams, M.; Ng, K. Bioorg. Med. Chem. Lett. 2009, 19, 967–971. doi:10.1016/j.bmcl.2008.11.075
Return to citation in text: [1] -
Thiel, O. R.; Achmatowicz, M. M.; Reichelt, A.; Larsen, R. D. Angew. Chem. 2010, 122, 8453–8456. doi:10.1002/ange.201001999
Angew. Chem., Int. Ed. 2010, 49, 8395–8398. doi:10.1002/anie.201001999
Return to citation in text: [1] -
Li, C.; Li, Z.; Wang, Q. Synlett 2010, 21, 2179–2183. doi:10.1055/s-0030-1258514
Return to citation in text: [1] [2] -
McCluskey, A.; Keller, P. A.; Morgan, J.; Garner, J. Org. Biomol. Chem. 2003, 1. doi:10.1039/b305458f
Return to citation in text: [1] -
Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Lucacchini, A. Farmaco 1997, 52, 61–66.
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Zhdankin, V. V. J. Org. Chem. 2011, 76, 1185–1197. doi:10.1021/jo1024738
Return to citation in text: [1] -
Salgado, A.; Varela, C.; Collazo, A. M. G.; García, F.; Pevarello, P.; Alkorta, I.; Elguero, J. J. Mol. Struct. 2011, 987, 13–24. doi:10.1016/j.molstruc.2010.11.054
Return to citation in text: [1] -
Snyder, N. L.; Adams, T. P. Miscellaneous Name Reactions. In Name Reactions in Heterocyclic Chemistry II; Li, J. J.; Corey, E. G., Eds.; Wiley & Sons: Hoboken, New Jersey, 2011; pp 554–590. doi:10.1002/9781118092828.ch9
Return to citation in text: [1] -
A single crystal of 6h suitable for X-ray diffraction analysis was obtained by recrystallization from CH2Cl2/n-hexane (1:1). CCDC 959624 contains supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Cikotiene, I.; Jonusis, M.; Jakubkiene, V. Beilstein J. Org. Chem. 2013, 9, 1819–1825. doi:10.3762/bjoc.9.212
Return to citation in text: [1]
| 23. | Cikotiene, I.; Jonusis, M.; Jakubkiene, V. Beilstein J. Org. Chem. 2013, 9, 1819–1825. doi:10.3762/bjoc.9.212 |
| 10. | Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 1999, 73, 131–176. doi:10.1016/S0065-2725(08)60942-3 |
| 11. | Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 2000, 77, 345–394. doi:10.1016/S0065-2725(00)77009-7 |
| 21. | Snyder, N. L.; Adams, T. P. Miscellaneous Name Reactions. In Name Reactions in Heterocyclic Chemistry II; Li, J. J.; Corey, E. G., Eds.; Wiley & Sons: Hoboken, New Jersey, 2011; pp 554–590. doi:10.1002/9781118092828.ch9 |
| 22. | A single crystal of 6h suitable for X-ray diffraction analysis was obtained by recrystallization from CH2Cl2/n-hexane (1:1). CCDC 959624 contains supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 7. | Battaglia, U.; Moody, C. J. J. Nat. Prod. 2010, 73, 1938–1939. doi:10.1021/np100298m |
| 18. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 19. | Zhdankin, V. V. J. Org. Chem. 2011, 76, 1185–1197. doi:10.1021/jo1024738 |
| 5. | Cheng, Y.; Liu, P.; Chen, H.; Zeng, F. J. Cardiovasc. Pharm. 2000, 35, 1–6. doi:10.1097/00005344-200001000-00001 |
| 6. | Kenji, I.; Hisayo, F.; Hida, M.; Awazu, M.; Midori, A. Eur. J. Pharmacol. 2005, 515, 28–33. doi:10.1016/j.ejphar.2005.04.013 |
| 20. | Salgado, A.; Varela, C.; Collazo, A. M. G.; García, F.; Pevarello, P.; Alkorta, I.; Elguero, J. J. Mol. Struct. 2011, 987, 13–24. doi:10.1016/j.molstruc.2010.11.054 |
| 4. | Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.-Y.; Liu, P.; Zhou, J. Org. Lett. 2009, 11, 1999–2002. doi:10.1021/ol900350k |
| 15. | Li, C.; Li, Z.; Wang, Q. Synlett 2010, 21, 2179–2183. doi:10.1055/s-0030-1258514 |
| 2. | Gribble, G. W.; Joule, J. A. Progress in Heterocyclic Chemistry; Elsevier: Oxford, 2007; Vol. 18. |
| 3. | Yet, L. Five-membered ring systems: With more than one N atom in Progress in Heterocyclic Chemistry 2011, Vol. 23, pp 231–266. |
| 16. | McCluskey, A.; Keller, P. A.; Morgan, J.; Garner, J. Org. Biomol. Chem. 2003, 1. doi:10.1039/b305458f |
| 17. | Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Lucacchini, A. Farmaco 1997, 52, 61–66. |
| 12. | Rashad, A. E.; Heikal, O. A.; El-Nezhawy, A. O. H.; Abdel-Megeid, F. M. E. Heteroat. Chem. 2005, 16, 226–234. doi:10.1002/hc.20114 |
| 14. |
Thiel, O. R.; Achmatowicz, M. M.; Reichelt, A.; Larsen, R. D. Angew. Chem. 2010, 122, 8453–8456. doi:10.1002/ange.201001999
Angew. Chem., Int. Ed. 2010, 49, 8395–8398. doi:10.1002/anie.201001999 |
| 10. | Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 1999, 73, 131–176. doi:10.1016/S0065-2725(08)60942-3 |
| 11. | Shaban, M. A. E.; Morgaan, A. E. A. Adv. Heterocycl. Chem. 2000, 77, 345–394. doi:10.1016/S0065-2725(00)77009-7 |
| 15. | Li, C.; Li, Z.; Wang, Q. Synlett 2010, 21, 2179–2183. doi:10.1055/s-0030-1258514 |
| 9. | Guetzoyan, L. J.; Spooner, R. A.; Lord, J. M.; Roberts, L. M.; Clarkson, G. J. Eur. J. Med. Chem. 2010, 45, 275–283. doi:10.1016/j.ejmech.2009.10.007 |
| 8. | Baraldi, P. G.; Saponaro, G.; Tabrizi, M. A.; Baraldi, S.; Romagnoli, R.; Moorman, A. R.; Varani, K.; Borea, P. A.; Preti, D. Bioorg. Med. Chem. 2012, 20, 1046–1059. doi:10.1016/j.bmc.2011.11.037 |
| 13. | Neustadt, B. R.; Liu, H.; Hao, J.; Greenlee, W. J.; Stamford, A. W.; Foster, C.; Arik, L.; Lachowicz, J.; Zhang, H.; Bertorelli, R.; Fredduzzi, S.; Varty, G.; Cohen-Williams, M.; Ng, K. Bioorg. Med. Chem. Lett. 2009, 19, 967–971. doi:10.1016/j.bmcl.2008.11.075 |
© 2013 Tang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)