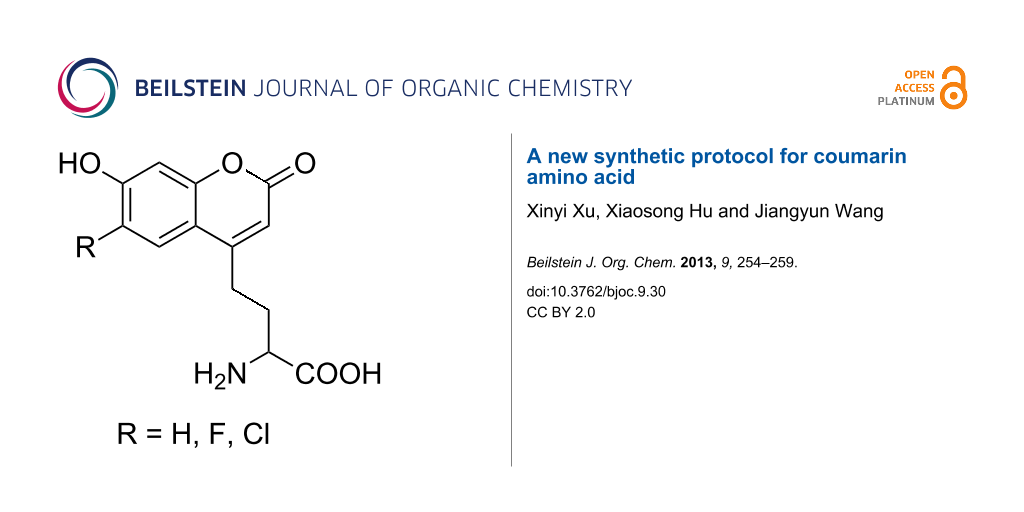
Abstract
The hydrochloride of the racemic amino acid (2-(7-hydroxycoumarin-4-yl)ethyl)glycine, which can serve as a fluorescent probe in proteins, and two halogen derivatives of it, were synthesized by using a new synthetic protocol in five steps. It is less costly and relatively easy to prepare this kind of fluorescent amino acid with the new synthetic method. Furthermore, it can be applied to synthesize other derivatives of the coumarin amino acid with some specific properties.

Graphical Abstract
Introduction
Since α-L-(2-(7-hydroxycoumarin-4-yl)ethyl)glycine (1a, Figure 1), a fluorescent non-natural amino acid, was genetically incorporated at a defined site in proteins in living organisms for the first time by Schultz and co-workers [1] there have been more and more applications related to it [2-8]. Compound 1a is of great interest to scientists because the 7-hydroxycoumarin moiety has a high fluorescence quantum yield and a large Stokes shift. Its excellent fluorescence properties make it a great candidate to substitute green fluorescent protein (GFP) in the application of fluorescent labeling of living cells. Compared with GFP, compound 1a is small, and can be incorporated at any defined site in proteins; whereas GFP is large, which will cause significant perturbation, and can only be fused to the C- or N-terminus of the target protein [9]. The coumarin amino acid 1a (Figure 1) is sensitive to both pH and solvent polarity, which makes it a good probe to investigate protein functionalities and biological processes related to them. The following examples are several applications of it. Shan and co-workers used compound 1a to form a FRET pair with the dye BODIPY-Fl to study the dynamics of protein–protein interactions [2]. Wang and co-workers genetically incorporated 1a to a position near to amino acids, which can be phosphorylated to investigate how phosphorylation affects the fluorescence properties of 1a, and the variation in the fluorescence intensity can be used to probe the phosphorylation status of certain amino acids [3]. Chapman and co-workers studied the FtsZ protein with genetically incorporated 1a [4]. The fluorescence of 1a was utilized to study the assembling of FtsZ in vivo, especially how the Z-ring is formed by FtsZ. This cannot be achieved by using GFP labeling technique since GFP is relatively insensitive to the pH and the solvent polarity of the solution. Many more examples of the usage of 1a have also been reported [5-8]. Due to the high importance of 1a, there has been a great need for a highly efficient and practical protocol for its synthesis.
![[1860-5397-9-30-1]](/bjoc/content/figures/1860-5397-9-30-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The chemical structure of a series of fluorescent amino acids. (1a) α-(2-(7-hydroxycoumarin-4-yl)ethyl)glycine; (1b) α-(2-(6-fluoro-7-hydroxycoumarin-4-yl)ethyl)glycine; (1c) α-(2-(6-chloro-7-hydroxycoumarin-4-yl)ethyl)glycine.
Figure 1: The chemical structure of a series of fluorescent amino acids. (1a) α-(2-(7-hydroxycoumarin-4-yl)et...
There are two major different approaches available in the literature to synthesize compound 1a. Approach 1 was reported by Schultz and co-workers, and it was the first synthetic protocol to provide 1a as an enantiomerically pure amino acid in L-configuration [1]. In this approach, N-α-Cbz-L-glutamic acid α-benzyl ester was first converted into the side-chain β-keto ester and then it was reacted with resorcinol in methanesulfonic acid to afford 1a. The biggest shortcoming of this approach is that the purification of the final product requires a costly preparative reversed-phase HPLC system. Other drawbacks of this approach include that the reactant Z-Glu-OBzl is expensive, and it is difficult to characterize some of the intermediates formed. Approach 2 was designed by Braun and Dittrich to provide an alternative path for the synthesis of 1a [10]. It started from a coumarinylacetic acid, which was then reduced to an alcohol by borane-dimethyl sulfide. After the phenolic hydroxy group was protected with a tert-butyl(dimethyl)silyl group, the alcohol was converted into a bromide and was used to alkylate a glycine enolate synthon to afford an imine. All the protecting groups were then removed and racemic amino acid 1a was afforded. This approach prevents the tedious and costly HPLC purification step used in approach 1. However, it can only provide compound 1a as a racemic mixture, and some reagents used are not readily available. Due to the great importance of coumarin amino acid, we designed a new synthetic protocol for compound 1a and two further halogen derivatives (1b and 1c). This new approach avoids some of the problems discussed above. It has a good overall yield and only requires reagents that are relatively cheap or easy to prepare. Compounds 1b and 1c have different sensitivities to pH and solvent polarity, and can serve as new fluorescent probes in a variety of applications.
Results and Discussion
Scheme 1 gives an outline of the new protocol used to synthesize compound 1. First, the coumarin ring with a 4-chloromethyl group (compound 3) was formed through Pechmann condensation [11] from ethyl 4-chloroacetoacetate and resorcinol or its 6-halogenated derivatives (3a, 77.4% yield; 3b, 78.0%; 3c, 70.0%). By suspending compound 3 in triethyl phosphate with a catalytic amount of sodium iodide and heating the mixture under reflux, phosphonate 4 was prepared and used directly in the next step without purification. Compound 4 was first treated with sodium hydride or other bases and then reacted with formaldehyde to form terminal alkene 5 through a Horner–Wadsworth–Emmons reaction [12] (two-step overall yields are 27%, 53% and 55% for 3a to 5a, 3b to 5b and 3c to 5c, respectively, if sodium hydride was used). In the presence of potassium tert-butoxide and a catalytic amount of tetrabutylammonium bromide, alkene 5 reacted with diethyl acetamidomalonate (DEAM) [13] to form malonate 6 in high yield (6a, 73.1% yield; 6b, 86.0%; 6c, 86.0%). It was then heated under reflux in aqueous HCl solution (12 M) to completely remove the protecting groups to afford the racemic coumarin amino acid 1 (1a, 95% yield; 1b, 83%; 1c, 84.3%).
![[1860-5397-9-30-i1]](/bjoc/content/inline/1860-5397-9-30-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route to fluorescent amino acids 1a, 1b and 1c.
Scheme 1: Synthetic route to fluorescent amino acids 1a, 1b and 1c.
This is a short and practical approach for the synthesis of coumarin amino acid 1. The Pechmann condensation was chosen to assemble the coumarin ring, since the yield is high and derivatives with substituents at the 5-, 6- or 8-position can be prepared [14-16]. In this paper, we report the synthesis of 6-fluoro and 6-chloro derivatives of compound 1a through Pechmann condensation. The functional group introduced at these positions will further improve the fluorescent property of the coumarin amino acids, or add new chemical handles to the coumarin ring for some specific investigations. The Horner–Wadsworth–Emmons reaction was applied to install the terminal alkene at the 4-position. Compared with the Wittig reaction, the Horner–Wadsworth–Emmons reaction has a significant advantage: The resulting phosphate byproduct can be readily separated, whereas the byproduct triphenylphosphine oxide generated in the Wittig reaction is difficult to remove [17]. The effect of the base used in the Horner–Wadsworth–Emmons reaction on the reaction yield was investigated (Table 1). Three different bases including sodium hydride, 1,8-diazabicycloundec-7-ene (DBU) and potassium tert-butoxide (KOt-Bu) were used, and the reactions were carried out at 25 °C for 5 h. DBU gave the best yield in the synthesis of 5a, 5b or 5c, whereas there was no reaction at all when KOt-Bu was used. This indicates that KOt-Bu is not basic enough to deprotonate the α-hydrogen of phosphonate 4 at 25 °C. However, when the reaction temperature was raised to 55 °C, the deprotonation happened and compound 5 was afforded with good yield (Table 2). This indicates that the reaction temperature has a significant effect on the Horner–Wadsworth–Emmons reaction. Since the coumarin ring could be opened under basic conditions, especially at high temperature [18], we need to choose proper base and reaction temperature to get the optimized yield of terminal alkene 5. Based on the experimental results, DBU is the best base among the three, and the reaction can be carried out at room temperature to obtain a relatively good yield. Alkylation of DEAM is widely used to synthesize α-amino acids [13], which was also applied here to prepare the coumarin amino acids. Through Michael addition reaction of DEAM to terminal alkene 5, malonate 6 was formed in high yield, and it was followed by the total hydrolysis of compound 6 with concentrated aqueous HCl solution to afford the coumarin amino acid 1. We found that the hydrolysis needed a high concentration of aqueous HCl solution (12 M) and a relatively long reaction time (10 h) to reach completion.
Table 1: Effect of different bases on the yield of compounds 5a, 5b and 5ca.
| Compound | NaH | DBU | KOt-Bu |
|---|---|---|---|
| 5a | 27% | 45% | No reaction |
| 5b | 51% | 59% | No reaction |
| 5c | 45% | 55% | No reaction |
aReaction temperature: 25 °C; reaction time: 5 h; solvent: THF; equiv of compound 4/equiv of base = 1/5.
Table 2: Effect of temperature on the yield of compounds 5a, 5b and 5ca.
| Compound | 25 °C | 55 °C |
|---|---|---|
| 5a | No reaction | 52% |
| 5b | No reaction | 47% |
| 5c | No reaction | 48% |
aBase: KOt-Bu; reaction time: 5 h; solvent: THF; equiv of compound 4/equiv of base = 1/5.
Though this new protocol also affords the coumarin non-natural amino acids in racemic form, similar to approach 2, it has some advantages that make it practical for the preparation of these fluorescent amino acids. First, the overall yields of the products are good compared with the two approaches reported, and all the reagents are commercially available and relatively cheap. Second, a tedious HPLC purification step is not necessary and only flash chromatography is required. Last, but most important, derivatives with a substituent at the 6-position can be prepared, which greatly expands the usage of this synthetic protocol. Coumarin non-natural amino acids with a substituent at the 5- or 8-position can also be prepared and a respective study is ongoing in our laboratory.
The L-enantiomer was proved to be able to be incorporated into a protein exclusively from the racemic mixture. In the presence of the synthetase CouRS and MjtRNATyrCUA, in E. coli [1], an amber codon was substituted for Ile-38 in thioredoxin-1 (TRX-TAG38), and protein expression was carried out in the LB medium with the addition of 1 mM of racemic coumarin amino acid 1a. The protein expression was also carried out without 1a as a negative control. The synthetase/tRNA pair could uniquely specify 1a in response to the TAG codon, which made the expression of TRX-TAG38 possible. The protein was then purified and analyzed by the SDS-PAGE gel (Figure 2). It showed that the full length protein (12.9 kDa) was only expressed in the presence of 1a; even the final product was racemic. Since thioredoxin has no intrinsic fluorescence, the fluorescent band corresponding to mutant thioredoxin in the right panel of Figure 2 must come from incorporated 1a. The L-enantiomer is assumed to be accepted exclusively from the racemic mixture since there is not any report indicating that D-amino acids exist in proteins. A related report proved that in the incorporation of a non-natural amino acid into a protein, the L-enantiomer is accepted exclusively from the racemic mixture [19]. Nevertheless, crystallization of the TAG38 mutant thioredoxin is under way to obtain the X-ray crystal structure of it, which will give direct proof of the exclusive incorporation of the L-enantiomer of 1a.
![[1860-5397-9-30-2]](/bjoc/content/figures/1860-5397-9-30-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Coomassie-stained SDS-PAGE (left) of TAG38 mutant thioredoxin (indicated by the black arrow) expression in the presence and absence of 1 mM 1a. The right panel shows the fluorescence image of wild-type and TAG38 mutant thioredoxin.
Figure 2: Coomassie-stained SDS-PAGE (left) of TAG38 mutant thioredoxin (indicated by the black arrow) expres...
6-Halogenated derivatives were also synthesized with this new protocol; these derivatives should have similar extinction coefficients and quantum yields as compound 1a, according to the literature [20,21]. However, their pKa values are significantly different from those of 1a, as shown in Table 3. The pKa values are calculated from the absorbance at 360 nm at different pH values illustrated in Figure 3, by using the Henderson–Hasselbalch equation. The halogenation of 1a at the 6-position decreases the pKa value, which makes compounds 1b and 1c good substitutes for 1a in fluorescent labeling and other investigations in biological systems. Screening for the synthetase/tRNA pair for 1b and 1c is under way. Fluorescent emission spectra of 1b and 1c at different pH values were both acquired (Figure 4). The fluorescence intensities of these two compounds are greatly enhanced upon increasing pH value of the solutions. This fluorescence property can be used to monitor the pH value of some acidic organelles, which may be difficult or even impossible for other investigating technologies. For the 6-fluorinated compound 1b, its 19F NMR is another important property suitable for probing the biological system. Coupling the fluorescent emission spectrum with its 19F NMR spectrum, it will provide a very powerful means in biological investigations and analysis with small molecules.
![[1860-5397-9-30-3]](/bjoc/content/figures/1860-5397-9-30-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Absorbance of compounds 1b and 1c at 360 nm as a function of pH value. (A) Absorption spectrum of 50 μM compound 1b in 200 mM sodium phosphate buffer (pH 5.8–8.0), 200 mM sodium acetate buffer (pH 3.7–5.6) or 50 mM Tris-HCl buffer (pH 8.2–8.9). (B) Absorption spectrum of 25 μM compound 1c in 200 mM sodium phosphate buffer (pH 5.8–8.0) or sodium acetate buffer (pH 3.7–5.6).
Figure 3: Absorbance of compounds 1b and 1c at 360 nm as a function of pH value. (A) Absorption spectrum of 5...
![[1860-5397-9-30-4]](/bjoc/content/figures/1860-5397-9-30-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Effect of the pH value of the solution on the fluorescence emission spectra of compounds 1b and 1c. (A) Fluorescence emission spectrum of compound 1b. (B) Fluorescence emission spectrum of compound 1c.
Figure 4: Effect of the pH value of the solution on the fluorescence emission spectra of compounds 1b and 1c....
Conclusion
In summary, a new protocol for the synthesis of several fluorescent coumarin non-natural amino acids in good yields was designed. This protocol only requires relatively cheap reagents and five reaction steps in total. The separation and purification processes are much easier and an HPLC purification step is unnecessary. Thus, it is more economical and less tedious compared with previously reported protocols. 6-Halogenated coumarin non-natural amino acids have different fluorescence properties and other functionalities, which makes them good probes for various biological studies. That a series of related coumarin non-natural amino acids can be prepared with this synthetic protocol is its most important advantages. Other derivatives with a 5- and 8-substituent are being synthesized and their fluorescence properties will be studied. The drawback of this protocol is that we cannot obtain the pure L-enantiomer, and only a racemic mixture was synthesized. However we proved that the L-enantiomer can be incorporated into protein exclusively, which indicated that the racemic product is good enough for biological studies. Nevertheless, pursuit of a synthetic protocol affording the pure L-enantiomer is still our next goal.
By the chemical synthesis and genetic encoding of the fluorescent non-natural amino acids, fluorescent groups can easily be incorporated at defined sites of proteins directly in living organisms. The fluorescent group wouldn’t cause significant perturbations on proteins due to its small size, thus, it greatly extends the scope of fluorescence imaging techniques. Compound 1a has a pKa of 7.8 and only its conjugate base is fluorescent. This limits its usage in the fluorescence imaging in vivo, such as in the study of receptor-mediated endocytosis. The protein involved in the endocytosis needs to be translocated from cytoplasm, which has a pH around 7, to the endosomes and lysosomes, which have pH values around 5 [22]. Since the fluorescence of compound 1a is relatively weak at acidic conditions, compound 1b and 1c with lower pKa are important and thus synthesized. They will be more fluorescent in an acidic environment, which makes them better probes for endocytosis than compound 1a. With the new synthetic protocol, compound 1b and 1c now can be prepared in a straightforward manner. The remaining unsolved problem in our work is on the molecular biology side, the focus of which is the screening of the specific aminoacyl-tRNA synthetases capable of recognizing 1b or 1c. Once we get the synthetases, compound 1b and 1c will be very useful probes of organellar pH and pH-dependent cellular processes. More coumarin amino acids with specific properties can also be prepared by using our new synthetic protocol, which makes it an important one in this research area.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 664.5 KB | Download |
Acknowledgements
We gratefully acknowledge the National Science Foundation of China (21102172 to X.H. and 90913022 to J.W.) and the Major State Basic Research Program of China (2010CB912301 and 2009CB825505 to J.W.) for financial support. X.H. also gratefully thank the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, the Fundamental Research Funds for the Central Universities (Wuhan University of Technology: 133114001)”, and “the Self-determined and Innovative Research Funds of Wuhan University of Technology” for financial support.
References
-
Wang, J.; Xie, J.; Schultz, P. G. J. Am. Chem. Soc. 2006, 128, 8738–8739. doi:10.1021/ja062666k
Return to citation in text: [1] [2] [3] -
Saraogi, I.; Zhang, D.; Chandrasekaran, S.; Shan, S.-o. J. Am. Chem. Soc. 2011, 133, 14936–14939. doi:10.1021/ja206626g
Return to citation in text: [1] [2] -
Lacey, V. K.; Parrish, A. R.; Han, S.; Shen, Z.; Briggs, S. P.; Ma, Y.; Wang, L. Angew. Chem., Int. Ed. 2011, 50, 8692–8696. doi:10.1002/anie.201102923
Return to citation in text: [1] [2] -
Charbon, G.; Brustad, E.; Scott, K. A.; Wang, J.; Løbner-Olesen, A.; Schultz, P. G.; Jacobs-Wagner, C.; Chapman, E. ChemBioChem 2011, 12, 1818–1821. doi:10.1002/cbic.201100282
Return to citation in text: [1] [2] -
Ugwumba, I. N.; Ozawa, K.; Xu, Z.-Q.; Ely, F.; Foo, J.-L.; Herlt, A. J.; Coppin, C.; Brown, S.; Taylor, M. C.; Ollis, D. L.; Mander, L. N.; Schenk, G.; Dixon, N. E.; Otting, G.; Oakeshott, J. G.; Jackson, C. J. J. Am. Chem. Soc. 2011, 133, 326–333. doi:10.1021/ja106416g
Return to citation in text: [1] [2] -
Li, S. ChemBioChem 2011, 12, 2729–2731. doi:10.1002/cbic.201100554
Return to citation in text: [1] [2] -
Charbon, G.; Wang, J.; Brustad, E.; Schultz, P. G.; Horwich, A. L.; Jacobs-Wagner, C.; Chapman, E. Bioorg. Med. Chem. Lett. 2011, 21, 6067–6070. doi:10.1016/j.bmcl.2011.08.057
Return to citation in text: [1] [2] -
Ho, D.; Lugo, M. R.; Lomize, A. L.; Pogozheva, I. D.; Singh, S. P.; Schwan, A. L.; Merrill, A. R. Biochemistry 2011, 50, 4830–4842. doi:10.1021/bi101934e
Return to citation in text: [1] [2] -
Tsien, R. Y. Annu. Rev. Biochem. 1998, 67, 509–544. doi:10.1146/annurev.biochem.67.1.509
Return to citation in text: [1] -
Braun, M.; Dittrich, T. Beilstein J. Org. Chem. 2010, 6, No. 69. doi:10.3762/bjoc.6.69
Return to citation in text: [1] -
von Pechmann, H.; Duisberg, C. Ber. Dtsch. Chem. Ges. 1883, 16, 2119–2128.
Return to citation in text: [1] -
Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042
Return to citation in text: [1] -
Haudegond, J. P.; Chauvin, Y.; Commereuc, D. J. Org. Chem. 1979, 44, 3063–3065. doi:10.1021/jo01331a020
Return to citation in text: [1] [2] -
Trost, B. M.; Toste, F. D.; Greenman, K. J. Am. Chem. Soc. 2003, 125, 4518–4526. doi:10.1021/ja0286573
Return to citation in text: [1] -
Herath, H. M. T. B.; Müller, K.; Diyabalanage, H. V. K. J. Heterocycl. Chem. 2004, 41, 23–28. doi:10.1002/jhet.5570410104
Return to citation in text: [1] -
Gündüz, C.; Salan, Ü.; Bulut, M. J. Heterocycl. Chem. 2009, 46, 567–570. doi:10.1002/jhet.90
Return to citation in text: [1] -
Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. doi:10.1021/cr60287a005
Return to citation in text: [1] -
Ramesh, D.; Srinivasan, M. Curr. Sci. 1984, 53, 369–371.
Return to citation in text: [1] -
Cellitti, S. E.; Jones, D. H.; Lagpacan, L.; Hao, X.; Zhang, Q.; Hu, H.; Brittain, S. M.; Brinker, A.; Caldwell, J.; Bursulaya, B.; Spraggon, G.; Brock, A.; Ryu, Y.; Uno, T.; Schultz, P. G.; Geierstanger, B. H. J. Am. Chem. Soc. 2008, 130, 9268–9281. doi:10.1021/ja801602q
Return to citation in text: [1] -
Sun, W.-C.; Gee, K. R.; Haugland, R. P. Bioorg. Med. Chem. Lett. 1998, 8, 3107–3110. doi:10.1016/S0960-894X(98)00578-2
Return to citation in text: [1] -
Brun, M.-P.; Bischoff, L.; Garbay, C. Angew. Chem., Int. Ed. 2004, 43, 3432–3436. doi:10.1002/anie.200454116
Return to citation in text: [1] -
Conner, S. D.; Schmid, S. L. Nature 2003, 422, 37–44. doi:10.1038/nature01451
Return to citation in text: [1]
| 20. | Sun, W.-C.; Gee, K. R.; Haugland, R. P. Bioorg. Med. Chem. Lett. 1998, 8, 3107–3110. doi:10.1016/S0960-894X(98)00578-2 |
| 21. | Brun, M.-P.; Bischoff, L.; Garbay, C. Angew. Chem., Int. Ed. 2004, 43, 3432–3436. doi:10.1002/anie.200454116 |
| 1. | Wang, J.; Xie, J.; Schultz, P. G. J. Am. Chem. Soc. 2006, 128, 8738–8739. doi:10.1021/ja062666k |
| 19. | Cellitti, S. E.; Jones, D. H.; Lagpacan, L.; Hao, X.; Zhang, Q.; Hu, H.; Brittain, S. M.; Brinker, A.; Caldwell, J.; Bursulaya, B.; Spraggon, G.; Brock, A.; Ryu, Y.; Uno, T.; Schultz, P. G.; Geierstanger, B. H. J. Am. Chem. Soc. 2008, 130, 9268–9281. doi:10.1021/ja801602q |
| 1. | Wang, J.; Xie, J.; Schultz, P. G. J. Am. Chem. Soc. 2006, 128, 8738–8739. doi:10.1021/ja062666k |
| 3. | Lacey, V. K.; Parrish, A. R.; Han, S.; Shen, Z.; Briggs, S. P.; Ma, Y.; Wang, L. Angew. Chem., Int. Ed. 2011, 50, 8692–8696. doi:10.1002/anie.201102923 |
| 2. | Saraogi, I.; Zhang, D.; Chandrasekaran, S.; Shan, S.-o. J. Am. Chem. Soc. 2011, 133, 14936–14939. doi:10.1021/ja206626g |
| 13. | Haudegond, J. P.; Chauvin, Y.; Commereuc, D. J. Org. Chem. 1979, 44, 3063–3065. doi:10.1021/jo01331a020 |
| 9. | Tsien, R. Y. Annu. Rev. Biochem. 1998, 67, 509–544. doi:10.1146/annurev.biochem.67.1.509 |
| 14. | Trost, B. M.; Toste, F. D.; Greenman, K. J. Am. Chem. Soc. 2003, 125, 4518–4526. doi:10.1021/ja0286573 |
| 15. | Herath, H. M. T. B.; Müller, K.; Diyabalanage, H. V. K. J. Heterocycl. Chem. 2004, 41, 23–28. doi:10.1002/jhet.5570410104 |
| 16. | Gündüz, C.; Salan, Ü.; Bulut, M. J. Heterocycl. Chem. 2009, 46, 567–570. doi:10.1002/jhet.90 |
| 2. | Saraogi, I.; Zhang, D.; Chandrasekaran, S.; Shan, S.-o. J. Am. Chem. Soc. 2011, 133, 14936–14939. doi:10.1021/ja206626g |
| 3. | Lacey, V. K.; Parrish, A. R.; Han, S.; Shen, Z.; Briggs, S. P.; Ma, Y.; Wang, L. Angew. Chem., Int. Ed. 2011, 50, 8692–8696. doi:10.1002/anie.201102923 |
| 4. | Charbon, G.; Brustad, E.; Scott, K. A.; Wang, J.; Løbner-Olesen, A.; Schultz, P. G.; Jacobs-Wagner, C.; Chapman, E. ChemBioChem 2011, 12, 1818–1821. doi:10.1002/cbic.201100282 |
| 5. | Ugwumba, I. N.; Ozawa, K.; Xu, Z.-Q.; Ely, F.; Foo, J.-L.; Herlt, A. J.; Coppin, C.; Brown, S.; Taylor, M. C.; Ollis, D. L.; Mander, L. N.; Schenk, G.; Dixon, N. E.; Otting, G.; Oakeshott, J. G.; Jackson, C. J. J. Am. Chem. Soc. 2011, 133, 326–333. doi:10.1021/ja106416g |
| 6. | Li, S. ChemBioChem 2011, 12, 2729–2731. doi:10.1002/cbic.201100554 |
| 7. | Charbon, G.; Wang, J.; Brustad, E.; Schultz, P. G.; Horwich, A. L.; Jacobs-Wagner, C.; Chapman, E. Bioorg. Med. Chem. Lett. 2011, 21, 6067–6070. doi:10.1016/j.bmcl.2011.08.057 |
| 8. | Ho, D.; Lugo, M. R.; Lomize, A. L.; Pogozheva, I. D.; Singh, S. P.; Schwan, A. L.; Merrill, A. R. Biochemistry 2011, 50, 4830–4842. doi:10.1021/bi101934e |
| 10. | Braun, M.; Dittrich, T. Beilstein J. Org. Chem. 2010, 6, No. 69. doi:10.3762/bjoc.6.69 |
| 12. | Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042 |
| 1. | Wang, J.; Xie, J.; Schultz, P. G. J. Am. Chem. Soc. 2006, 128, 8738–8739. doi:10.1021/ja062666k |
| 13. | Haudegond, J. P.; Chauvin, Y.; Commereuc, D. J. Org. Chem. 1979, 44, 3063–3065. doi:10.1021/jo01331a020 |
| 5. | Ugwumba, I. N.; Ozawa, K.; Xu, Z.-Q.; Ely, F.; Foo, J.-L.; Herlt, A. J.; Coppin, C.; Brown, S.; Taylor, M. C.; Ollis, D. L.; Mander, L. N.; Schenk, G.; Dixon, N. E.; Otting, G.; Oakeshott, J. G.; Jackson, C. J. J. Am. Chem. Soc. 2011, 133, 326–333. doi:10.1021/ja106416g |
| 6. | Li, S. ChemBioChem 2011, 12, 2729–2731. doi:10.1002/cbic.201100554 |
| 7. | Charbon, G.; Wang, J.; Brustad, E.; Schultz, P. G.; Horwich, A. L.; Jacobs-Wagner, C.; Chapman, E. Bioorg. Med. Chem. Lett. 2011, 21, 6067–6070. doi:10.1016/j.bmcl.2011.08.057 |
| 8. | Ho, D.; Lugo, M. R.; Lomize, A. L.; Pogozheva, I. D.; Singh, S. P.; Schwan, A. L.; Merrill, A. R. Biochemistry 2011, 50, 4830–4842. doi:10.1021/bi101934e |
| 22. | Conner, S. D.; Schmid, S. L. Nature 2003, 422, 37–44. doi:10.1038/nature01451 |
| 4. | Charbon, G.; Brustad, E.; Scott, K. A.; Wang, J.; Løbner-Olesen, A.; Schultz, P. G.; Jacobs-Wagner, C.; Chapman, E. ChemBioChem 2011, 12, 1818–1821. doi:10.1002/cbic.201100282 |
© 2013 Xu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)