Abstract
β-Lactams were diastereoselectively formed by the reaction of SF5-containing aldimines, or an SF5-containing ketimine, with benzyloxyketene in a conrotatory ring closure process. Imine formation and cyclization were possible in spite of the acidification of protons on the carbon bound to SF5. The reactions of the aldimines demonstrated very good 1,2-lk diastereoselectivity, however lack of stereochemical control of the C–N ketimine geometry was reflected in the stereochemistry of the product β-lactam. Cyclization of imines with a stereogenic center bearing SF5 was reflected in the 1,2-lk,lk selectivity of the β-lactam.

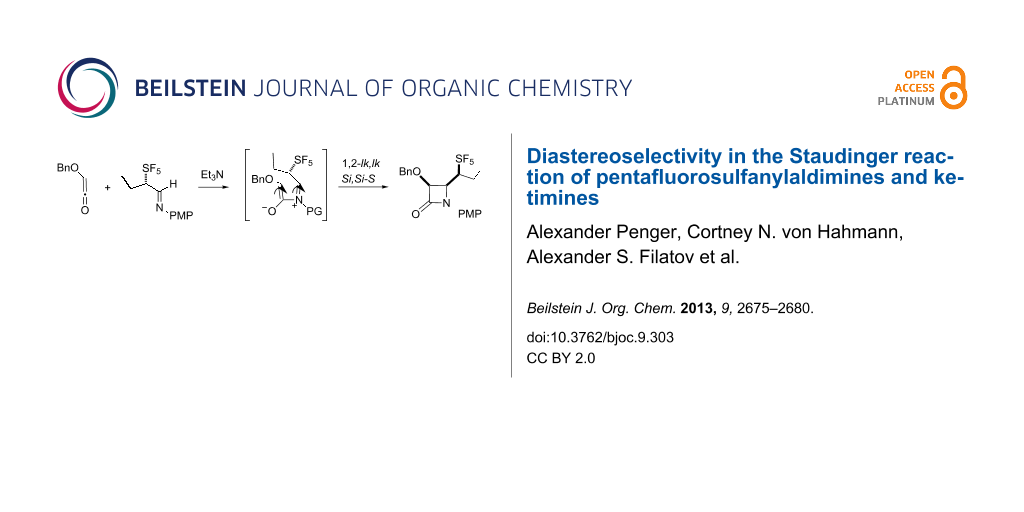
Graphical Abstract
Introduction
The pentafluorosulfanyl (SF5) group is one of the few truly new functional groups to be introduced to synthetic organic chemistry in the last 100 years [1-6]. With pseudooctahedral geometry around sulfur, the SF5 group is a unique substituent for the medicinal or pharmaceutical chemist. While the electron withdrawing properties of the SF5 and CF3 groups are similar [7,8], the electronegativity of the SF5 group has been suggested to be as high as 3.65 in contrast to 3.36 for CF3 [9]. The inductive σI and σR values for SF5, 0.55 and 0.11 [10], contrast with σI and σR values for CF3 of 0.39 and 0.12 [11,12] illustrating the increased inductive effect of the SF5 group relative to the CF3 group. This effect can also be seen in the calculated dipole moments of 1,1,1-trifluoroethane and pentafluorosulfanylmethane of 2.589 and 3.556 Debye respectively [7,8]. When these electronic effects are combined with an occupied volume only slightly less than that of a tert-butyl group [3,13], the SF5 group can have unanticipated influences on the structure such as anchoring side chain and neighboring hydroxy group conformations [14,15].
Given the unique potential of the SF5 group, the rarity of its application in medicinal or agrochemical materials may be surprising. However, synthesis of aliphatic SF5-containing building blocks (SF5-substituted aromatic compounds are more accessible) [1,6], is challenging and often beset by confusing reactivity, largely because of the very properties that make SF5 an attractive substituent. Studies of aliphatic compounds have lagged as a consequence, a fact that is compounded by the lack of commercially available aliphatic SF5-containing building blocks. Although the number of known aliphatic pentafluorosulfanylated compounds is constantly increasing [2], this expansion has not generally been accompanied by applications of these materials. Among aliphatic SF5-substituted compounds α-pentafluorosulfanylated aliphatic carbonyl compounds [16], readily prepared from the corresponding enol acetates or enol ethers, are especially valuable as starting materials [17-22]. In these compounds the profound steric influence of the SF5 group [14,15] is accompanied by a dramatic increase in the acidity of the adjacent α-proton, a phenomenon underlying the abstraction of protons by methyllithium and a number of different ylides [16]. The reactions of α-pentafluorosulfanyl carbonyl compounds are governed by a combination of the substantial dipole moment and unique steric effects of the octahedral SF5 group. Aliphatic SF5-containing derivatives of biologically active compounds are not well-known. One exception is the inclusion of an ω-SF5-substituted amino acid incorporated at the crucial first and fourth positions of a heptapeptide [23]. When introduced at these positions, the SF5-substituted amino acid had a strong propensity to direct α-helix formation of even this short heptapeptide presumably by minimization of unfavourable hydrophobic interactions.
Results and Discussion
The use of fluoroalkylimines to form β-lactams has proven especially useful in synthesis [24-26] especially in the Ojima β-lactam synthon method [27-29] used to prepare docetaxel analogs [26]. The general utility of the familiar Staudinger reaction of imines to transform readily accessible aldehydes to β-lactams has been well reviewed [30-33], yet in spite of this familiarity, the mechanism of this process remains a topic of interest [34-36].
Previously, it has been shown that fluorinated imines can undergo a manifold of reactions that are difficult to access with fluorinated aldehydes or ketones [37-40]. While there are many reports of the utility of trifluoroacetaldimines [41-44], there is only a single report of the preparation of N-ethyl 3,3,3-trifluoropropanaldimine [45], the trifluoromethyl analog of pentafluorosulfanylacetaldehyde (1a). The imine (3,3,3-trifluoropropanaldimine), in combination with the isomeric trifluoropropenamine, was not formed by the condensation of an amine with the aldehyde but rather by addition of an amine to 3,3,3-trifluoropropyne. The imine was described as “extremely unstable” [45] with no selective reactions reported. In light of this precedence the reaction of α-SF5-substituted aldehydes and ketones with amines was particularly intriguing.
α-SF5-Substituted aldehydes and ketones
In this work the SF5-bearing aldehyde 1 was prepared by the addition of SF5Cl to the enol ether 2 instead of the previously described additions to enol acetates [16] (Scheme 1). In earlier studies, it was found that the yield of SF5Cl addition to enol acetates was highly dependent upon the purity of the enol acetate substrate, compounds surprisingly difficult to purify. Since vinyl acetate is the only enol acetate readily accessible for this reaction, the commercial availability of high purity propenyl and butenyl ethers 2b and 2c rendered these starting materials highly attractive for the formation of SF5-bearing aldehydes.
![[1860-5397-9-303-i1]](/bjoc/content/inline/1860-5397-9-303-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2-pentafluorosulfanylaldehydes by addition of SF5Cl to enol ethers.
Scheme 1: Synthesis of 2-pentafluorosulfanylaldehydes by addition of SF5Cl to enol ethers.
After addition of SF5Cl to 2, intermediate 3 was typically formed as a 9:1 mixture of diastereomers. Hydrolysis of 3 was easily followed by 19F NMR, e.g., the resonance for 3b appeared approximately 7 ppm upfield from that of the aldehyde 1b. The JH,F coupling constant of 5.0 Hz contrasts with the JFeq,Fax value of 144 Hz.
The ready availability of the α-pentafluorosulfanyl carbonyl compounds facilitated an effort to dramatically expand the utility of these intriguing materials by synthesis and characterization of the corresponding pentafluorosulfanylated β-lactams.
α-SF5-Substituted aldimines and ketimines
The aldehydes 1 were readily converted to the corresponding imine 5 in dichloromethane using anhydrous magnesium sulfate as a dehydrating agent (Scheme 2). Other desiccants, especially inherently basic materials, such as potassium carbonate lead to little imine formation. While the crude imine solution likely contained unreacted amine, attempted separation by silica gel chromatography led to extensive decomposition. The product imine consisted of a single stereoisomer as determined by 19F NMR, tentatively assigned as the E-isomer. Similar to the formation of 1, it was easy to follow formation of the imine by 19F NMR with the resonance attributed to the equatorial fluorines of the imine 5a (δ 68.8 ppm) appearing upfield of those assigned to the aldehyde 1a (δ 72.4 ppm). In the case of imines 5b–d, the solution also contained between 15–20% of the putative enamine 6b–d, where, for example, the equatorial fluorine resonances of 6b appeared at δ 65.5 ppm in contrast to those of 5b that appeared at δ 59.5 ppm. The tendency for the enamine resonances to appear downfield of imine resonances was confirmed by the preparation of the morpholine enamine of 1a for which the equatorial fluorine resonance is also shifted downfield [46]. As mentioned above, the preparation of N-ethyl 3,3,3-trifluoropropanaldimine, the Schiff base of 3,3,3-trifluoropropanal, was accompanied by enamine formation, likely as a consequence of the acidity of the proton α to the trifluoromethyl group [45,47]. Not surprisingly, 5 was relatively reactive. After careful filtration of the desiccant, the dichloromethane solution of the imine was used in further reactions without purification or separation of unreacted amine or the enamine side product.
![[1860-5397-9-303-i2]](/bjoc/content/inline/1860-5397-9-303-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of pentafluorosulfanylaldimines with benzyloxyketene.
Scheme 2: Reaction of pentafluorosulfanylaldimines with benzyloxyketene.
Ketene imine cycloaddition reactions of α-SF5-substituted aldimines and ketimines
Dropwise addition of the crude dichloromethane solution of 5 to benzyloxyacetyl chloride and triethylamine in dichloromethane was completed at 0 °C. The solution was then allowed to warm to room temperature with stirring overnight. Not surprisingly, the use of the crude solution of the imine led to the formation of a complex mixture where the desired product β-lactam 7 was a minor constituent. However, the yields of 7 are of product purified by silica gel chromatography and crystallization. The only other fluorinated products observed prior to purification were unreacted imine 5 and the tentatively designated N-acylenamine 8. In the case of 7b–d the de of the 1,2-lk to 1,2-ul products was 76%, 84% and 50% respectively. The relative proportion of the product mixture that was comprised of enamine 8b–d was 10%, 4% and 21% respectively.
In an effort to improve the reaction, the addition of triethylamine to a solution of the acid chloride and imine 5 resulted only in decomposition. Excess amine 4 that was present was acylated by benzyloxyacetyl chloride to form the corresponding amide.
The utility of the ketene–imine cyclization was not limited to aldimines. The addition of SF5Br to the enol acetate of ethyl pyruvate 9 formed ethyl pentafluorosulfanylpyruvate 11 (Scheme 3). The ketimine 12, prepared via amine condensation with 11, was reacted as described for the aldimines 5 to form the desired β-lactam 7e.
![[1860-5397-9-303-i3]](/bjoc/content/inline/1860-5397-9-303-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of ethyl pentafluorosulfanylpyruvate and formation of the corresponding β-lactam.
Scheme 3: Preparation of ethyl pentafluorosulfanylpyruvate and formation of the corresponding β-lactam.
Formation of 7e was accompanied by significantly greater decomposition of 12 and hence 7e was formed in lower overall yields. In contrast to the stereoselective formation of 7b–d, the diastereoselectivity of the Staudinger reactions as determined by 19F NMR was dramatically reduced for 7e to a de of 14%, a value consistent with the Z/E ratio for 12 of 0.7. While the isolated yields of purified 7b–e are especially modest, the reaction conditions have not been optimized. But the significance of these findings lies not only in the difference in reactivity in comparison with trifluoropropanaldimine but also in the relative diastereoselectivity of the ketene–imine cycloaddition reaction in comparison with the other reactions of SF5-bearing aldehydes [16].
Structural characterization of SF5-containing β-lactams
Isolated as a single diastereomer, the relative stereochemistry of 7a, the product of the Staudinger reaction of 5a, is shown in Figure 1. The cis relative stereochemistry of β-lactam is consistent with 1,2-lk conrotatory ring closure of the E-imine 5a as would be predicted for a reaction with the Bose–Evans ketene formed from benzyloxyacetyl chloride [30]. The low yield of β-lactam product is better understood when the reactivity of the intermediate pentafluorosulfanylated imine and the subsequently formed iminium ion are considered. The SF5 group increases the acidity of the α-proton of the imine 5 and of the iminium ion intermediate B formed on the initial nucleophilic attack of the imine on the ketene as illustrated in Scheme 4. The ring closure step requires bond formation between the iminium ion carbon and the enolate carbon B to be particularly facile for the stereoselectivity of the process to be preserved. The ring closure process must compete successfully with loss of the acidic proton from B to form 8 [16]. Another indication of the rapidity of ring closure is the failure to detect the 1,2-ul product. The absence of 1,2-ul product is consistent with retardation the rate of E/Z imine isomerization by the electron withdrawing pentafluorosulfanyl group [35]. In the reaction of 12, the E/Z ratio of the imine was reflected very well in the 1,2-diastereomeric excess of the product β-lactam 7e .
![[1860-5397-9-303-1]](/bjoc/content/figures/1860-5397-9-303-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The 1,2-lk stereochemistry of 7a as determined by single crystal X-ray diffraction. Thermal ellipsoids are set at 50% probability.
Figure 1: The 1,2-lk stereochemistry of 7a as determined by single crystal X-ray diffraction. Thermal ellipso...
![[1860-5397-9-303-i4]](/bjoc/content/inline/1860-5397-9-303-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Influence of the SF5 group on the initial attack of the ketene on the imine nitrogen (A) and on the sense of conrotatory ring closure (B).
Scheme 4: Influence of the SF5 group on the initial attack of the ketene on the imine nitrogen (A) and on the...
The ketene–imine condensation of 7c is influenced by the presence of the pentafluorosulfanyl group at a stereogenic center. The 1,2-lk,lk (Si,Si-S) (or (Re,Re-R)) stereochemistry of 7c (Figure 2) suggests the profound dipole associated with the introduction of the SF5 group may influence the diastereoselectivity of ring closure. The initial approach of the ketene (A in Scheme 4) appears to be influenced by avoidance of unfavorable interaction of the ketene with the sterically demanding SF5 group. Previously it was found in single crystal X-ray diffraction studies that the pentafluorosulfanyl group [48] is predictably orthogonal to a carbonyl group as shown in A. Cornforth control of the ring closure step of the zwitterion (B in Scheme 4) where the SF5 would be antiperiplanar to the iminium ion would lead to the observed diastereoselectivity (Scheme 1). This finding is consistent with dipolar effects being most important in reactions with highly charged transition states such as B.
![[1860-5397-9-303-2]](/bjoc/content/figures/1860-5397-9-303-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The stereochemistry of 7c, 1,2-lk,lk (Si, Si-S), as determined by single crystal X-ray diffraction studies. Thermal ellipsoids are set at 50% probability. (a) Complete structure of 7c. (b) PMP protecting group hidden for clarity.
Figure 2: The stereochemistry of 7c, 1,2-lk,lk (Si, Si-S), as determined by single crystal X-ray diffraction ...
In both structures, consistent with the opposing dipole geometry of the Cornforth transition state, the N–C–C–S torsional angles remain near 170° (169° and 167° for 7a and 7c respectively).
The 1,2-lk,ul ring closure product may be formed in the reaction of 5c with benzyloxyketene and simply remain undetected, but it is clear that the control of diastereofacial selectivity in the formation of the principal β-lactam is strongly under the control of the SF5 group.
Conclusion
Low molecular weight pentafluorosulfanylated aldehydes 1 were prepared by addition of SF5Cl to enol ethers and the subsequent acidic hydrolysis of 3. Formation of Schiff base 5 is problematic but, in contrast to the reactions of the analogous trifluoromethyl compounds, does successfully proceed. Even with a manifold of possible side reactions, β-lactam formation by the ketene–imine cycloaddition reaction of 5 occurs, albeit in very modest yields. The 1,2-lk stereochemistry of the β-lactam 7 was consistent with rapid cyclization and a failure of the imines 5 to isomerize. The presence of a pentafluorosulfanylated stereogenic carbon as in 5c, apparently also influences the 2,3-lk stereochemistry. Optimization of β-lactam synthesis will require a better understanding of the nature of the competing, undesirable reactions and enable utilization of this unique construct in further synthetic transformations. The product β-lactams are a useful entrée to the diastereoselective synthesis of pentafluorosulfanyl β-amino acids and suggest a path to the preparation of more extensively functionalized SF5-containing β-lactams.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and spectroscopic data for 1a–e, 10, 5a–d, 7a–e and 11. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: X-ray crystallographic data for 7a and 7c, CCDC 937908 and 937909. | ||
| Format: CIF | Size: 32.3 KB | Download |
References
-
Welch, J. T. Applications of pentafluorosulfanyl substitution in life sciences research. In Fluorine in Pharmaceutical and Medicinal Chemistry; Gouverneur, V.; Müller, K., Eds.; Imperial College Press: London, 2012; pp 175–207.
Return to citation in text: [1] [2] -
Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030
Return to citation in text: [1] [2] -
Lentz, D.; Seppelt, K. The –SF5, –SeF5, and –TeF5 groups in organic chemistry. In Chemistry of Hypervalent Compounds; Akiba, K.-y., Ed.; Wiley-VCH: New York, 1999; pp 295–323.
Return to citation in text: [1] [2] -
Winter, R. W.; Dodean, R. A.; Gard, G. L. SF5 synthons: Pathways to organic derivatives of SF6. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; American Chemical Society: Washington, D. C., 2005; pp 87–118. doi:10.1021/bk-2005-0911.ch004
Return to citation in text: [1] -
Gard, G. L. Chim. Oggi 2009, 27, 10–13.
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry. Synthesis, Reactivity and Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] [2] -
Brant, P.; Berry, A. D.; DeMarco, R. A.; Carter, F. L.; Fox, W. B.; Hashmall, J. A. J. Electron Spectrosc. Relat. Phenom. 1981, 22, 119–129. doi:10.1016/0368-2048(81)80021-7
Return to citation in text: [1] [2] -
True, J. E.; Thomas, T. D.; Winter, R. W.; Gard, G. L. Inorg. Chem. 2003, 42, 4437–4441. doi:10.1021/ic0343298
Return to citation in text: [1] [2] -
Sæthre, L. J.; Berrah, N.; Bozek, J. D.; Børve, K. J.; Carroll, T. X.; Kukk, E.; Gard, G. L.; Winter, R.; Thomas, T. D. J. Am. Chem. Soc. 2001, 123, 10729–10737. doi:10.1021/ja016395j
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007
Return to citation in text: [1] -
Taft, R. W., Jr.; Lewis, I. C. J. Am. Chem. Soc. 1959, 81, 5343–5352. doi:10.1021/ja01529a025
Return to citation in text: [1] -
Taft, R. W., Jr. J. Phys. Chem. 1960, 64, 1805–1815. doi:10.1021/j100841a003
Return to citation in text: [1] -
Anthony, M. Aust. N. Z. J. Med. 1984, 14, 888–895. doi:10.1111/j.1445-5994.1984.tb03802.x
Return to citation in text: [1] -
Savoie, P. R.; Higashiya, S.; Lin, J.-H.; Wagle, D. V.; Welch, J. T. J. Fluorine Chem. 2012, 143, 281–286. doi:10.1016/j.jfluchem.2012.06.027
Return to citation in text: [1] [2] -
Savoie, P. R.; Welch, J. M.; Higashiya, S.; Welch, J. T. J. Fluorine Chem. 2013, 148, 1–5. doi:10.1016/j.jfluchem.2013.01.013
Return to citation in text: [1] [2] -
Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763
Return to citation in text: [1] [2] [3] [4] [5] -
Winter, R.; Gard, G. L. J. Fluorine Chem. 1994, 66, 109–116. doi:10.1016/0022-1139(93)03005-7
Return to citation in text: [1] -
Ray, N. H. J. Chem. Soc. 1963, 1440–1441. doi:10.1039/JR9630001440
Return to citation in text: [1] -
Kleemann, G.; Seppelt, K. Chem. Ber. 1979, 112, 1140–1146. doi:10.1002/cber.19791120409
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003
Return to citation in text: [1] -
Coffman, D. D.; Tullock, C. W. Carbonylic compounds containing the SF5 function. U.S. Patent US3,102,903, Sept 3, 1963.
Return to citation in text: [1] -
Winter, R.; Willett, R. D.; Gard, G. L. Inorg. Chem. 1989, 28, 2499–2501. doi:10.1021/ic00311a054
Return to citation in text: [1] -
Lim, D. S.; Lin, J.-H.; Welch, J. T. Eur. J. Org. Chem. 2012, 3946–3954. doi:10.1002/ejoc.201200327
Return to citation in text: [1] -
Abouabdellah, A.; Bégué, J.-P.; Bonnet-Delpon, D.; Thanh Nga, T. T. J. Org. Chem. 1997, 62, 8826–8833. doi:10.1021/jo971381a
Return to citation in text: [1] -
Petrik, V.; Röschenthaler, G.-V.; Cahard, D. Tetrahedron 2011, 67, 3254–3259. doi:10.1016/j.tet.2011.03.001
Return to citation in text: [1] -
Pepe, A.; Kuznetsova, L.; Sun, L.; Ojima, I. Fluoro-taxoid anticancer agents. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; John Wiley & Sons Ltd.: Chichester, 2009; pp 117–139. doi:10.1002/9781444312096.ch5
Return to citation in text: [1] [2] -
Ojima, I. β-Lactam synthon method: enantiomerically pure β-lactams as synthetic intermediates. In Organic Chemistry of β-lactams; Georg, G. I., Ed.; VCH: New York, NY, 1993; pp 197–255.
Return to citation in text: [1] -
Kamath, A.; Ojima, I. Tetrahedron 2012, 68, 10640–10664. doi:10.1016/j.tet.2012.07.090
Return to citation in text: [1] -
Ojima, I.; Zuniga, E. S.; Seitz, J. D. Top. Heterocycl. Chem. 2013, 30, 1–63. doi:10.1007/7081_2012_86
Return to citation in text: [1] -
Georg, G. I.; Ravikumar, V. T. Stereocontrolled ketene-imine cycloaddition reactions. In Organic Chemistry of β-lactams; Georg, G. I., Ed.; VCH: New York, NY, 1993; pp 295–368.
Return to citation in text: [1] [2] -
Palomo, C.; Aizpurua, J. M.; Ganboa, I.; Oiarbide, M. Eur. J. Org. Chem. 1999, 3223–3235. doi:10.1002/(SICI)1099-0690(199912)1999:12<3223::AID-EJOC3223>3.0.CO;2-1
Return to citation in text: [1] -
Palomo, C.; Aizpurua, J. M.; Ganboa, I.; Oiarbide, M. Curr. Med. Chem. 2004, 11, 1837–1872. doi:10.2174/0929867043364900
Return to citation in text: [1] -
Thompson, S.; Coyne, A. G.; Knipe, P. C.; Smith, M. D. Chem. Soc. Rev. 2011, 40, 4217–4231. doi:10.1039/c1cs15022g
Return to citation in text: [1] -
Cossio, F. P.; Arrieta, A.; Sierra, M. A. Acc. Chem. Res. 2008, 41, 925–936. doi:10.1021/ar800033j
Return to citation in text: [1] -
Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060–6069. doi:10.1021/ja056711k
Return to citation in text: [1] [2] -
Xu, J. Tetrahedron 2012, 68, 10696–10747. doi:10.1016/j.tet.2012.04.007
Return to citation in text: [1] -
Fustero, S.; Jiménez, D.; Sanz-Cervera, J. F.; Sánchez-Roselló, M.; Esteban, E.; Simón-Fuentes, A. Org. Lett. 2005, 7, 3433–3436. doi:10.1021/ol050791f
Return to citation in text: [1] -
Zanda, M.; Bravo, P.; Volonterio, A. Stereoselective synthesis of β-fluoroalkyl β-amino alcohol units. In Asymmetric Fluoroorganic Chemistry; Ramachandran, P. V., Ed.; American Chemical Society: Washington, D.C., 2000; Vol. 746, pp 127–141. doi:10.1021/bk-2000-0746.ch010
Return to citation in text: [1] -
Welch, J. T.; De Corte, B.; De Kimpe, N. J. Org. Chem. 1990, 55, 4981–4983. doi:10.1021/jo00304a002
Return to citation in text: [1] -
Welch, J. T.; Seper, K. W. J. Org. Chem. 1988, 53, 2991–2999. doi:10.1021/jo00248a017
Return to citation in text: [1] -
Carroccia, L.; Fioravanti, S.; Pellacani, L.; Tardella, P. A. Synthesis 2010, 4096–4100. doi:10.1055/s-0030-1258280
Return to citation in text: [1] -
Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a
Return to citation in text: [1] -
Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2005, 1258–1265. doi:10.1002/ejoc.200400719
Return to citation in text: [1] -
Crousse, B.; Narizuka, S.; Bonnet-Delpon, D.; Bégué, J.-P. Synlett 2001, 679–681. doi:10.1055/s-2001-13364
Return to citation in text: [1] -
Stepanova, N. P.; Lebedev, V. B.; Orlova, N. A.; Turbanova, E. S.; Petrov, A. A. Zh. Org. Khim. 1988, 24, 692–699.
Return to citation in text: [1] [2] [3] -
Welch, J. T.; Penger, A. unpublished results.
Return to citation in text: [1] -
Yamazaki, T.; Kobayashi, R.; Kitazume, T.; Kubota, T. J. Org. Chem. 2006, 71, 2499–2502. doi:10.1021/jo052434o
Return to citation in text: [1] -
Welch, J. T.; Zhong, L.; Filatov, A. S. unpublished results.
Return to citation in text: [1]
| 16. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 30. | Georg, G. I.; Ravikumar, V. T. Stereocontrolled ketene-imine cycloaddition reactions. In Organic Chemistry of β-lactams; Georg, G. I., Ed.; VCH: New York, NY, 1993; pp 295–368. |
| 16. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 1. | Welch, J. T. Applications of pentafluorosulfanyl substitution in life sciences research. In Fluorine in Pharmaceutical and Medicinal Chemistry; Gouverneur, V.; Müller, K., Eds.; Imperial College Press: London, 2012; pp 175–207. |
| 2. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 3. | Lentz, D.; Seppelt, K. The –SF5, –SeF5, and –TeF5 groups in organic chemistry. In Chemistry of Hypervalent Compounds; Akiba, K.-y., Ed.; Wiley-VCH: New York, 1999; pp 295–323. |
| 4. | Winter, R. W.; Dodean, R. A.; Gard, G. L. SF5 synthons: Pathways to organic derivatives of SF6. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; American Chemical Society: Washington, D. C., 2005; pp 87–118. doi:10.1021/bk-2005-0911.ch004 |
| 5. | Gard, G. L. Chim. Oggi 2009, 27, 10–13. |
| 6. | Kirsch, P. Modern Fluoroorganic Chemistry. Synthesis, Reactivity and Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 11. | Taft, R. W., Jr.; Lewis, I. C. J. Am. Chem. Soc. 1959, 81, 5343–5352. doi:10.1021/ja01529a025 |
| 12. | Taft, R. W., Jr. J. Phys. Chem. 1960, 64, 1805–1815. doi:10.1021/j100841a003 |
| 23. | Lim, D. S.; Lin, J.-H.; Welch, J. T. Eur. J. Org. Chem. 2012, 3946–3954. doi:10.1002/ejoc.201200327 |
| 10. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007 |
| 24. | Abouabdellah, A.; Bégué, J.-P.; Bonnet-Delpon, D.; Thanh Nga, T. T. J. Org. Chem. 1997, 62, 8826–8833. doi:10.1021/jo971381a |
| 25. | Petrik, V.; Röschenthaler, G.-V.; Cahard, D. Tetrahedron 2011, 67, 3254–3259. doi:10.1016/j.tet.2011.03.001 |
| 26. | Pepe, A.; Kuznetsova, L.; Sun, L.; Ojima, I. Fluoro-taxoid anticancer agents. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; John Wiley & Sons Ltd.: Chichester, 2009; pp 117–139. doi:10.1002/9781444312096.ch5 |
| 9. | Sæthre, L. J.; Berrah, N.; Bozek, J. D.; Børve, K. J.; Carroll, T. X.; Kukk, E.; Gard, G. L.; Winter, R.; Thomas, T. D. J. Am. Chem. Soc. 2001, 123, 10729–10737. doi:10.1021/ja016395j |
| 14. | Savoie, P. R.; Higashiya, S.; Lin, J.-H.; Wagle, D. V.; Welch, J. T. J. Fluorine Chem. 2012, 143, 281–286. doi:10.1016/j.jfluchem.2012.06.027 |
| 15. | Savoie, P. R.; Welch, J. M.; Higashiya, S.; Welch, J. T. J. Fluorine Chem. 2013, 148, 1–5. doi:10.1016/j.jfluchem.2013.01.013 |
| 7. | Brant, P.; Berry, A. D.; DeMarco, R. A.; Carter, F. L.; Fox, W. B.; Hashmall, J. A. J. Electron Spectrosc. Relat. Phenom. 1981, 22, 119–129. doi:10.1016/0368-2048(81)80021-7 |
| 8. | True, J. E.; Thomas, T. D.; Winter, R. W.; Gard, G. L. Inorg. Chem. 2003, 42, 4437–4441. doi:10.1021/ic0343298 |
| 16. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 1. | Welch, J. T. Applications of pentafluorosulfanyl substitution in life sciences research. In Fluorine in Pharmaceutical and Medicinal Chemistry; Gouverneur, V.; Müller, K., Eds.; Imperial College Press: London, 2012; pp 175–207. |
| 6. | Kirsch, P. Modern Fluoroorganic Chemistry. Synthesis, Reactivity and Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 16. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 14. | Savoie, P. R.; Higashiya, S.; Lin, J.-H.; Wagle, D. V.; Welch, J. T. J. Fluorine Chem. 2012, 143, 281–286. doi:10.1016/j.jfluchem.2012.06.027 |
| 15. | Savoie, P. R.; Welch, J. M.; Higashiya, S.; Welch, J. T. J. Fluorine Chem. 2013, 148, 1–5. doi:10.1016/j.jfluchem.2013.01.013 |
| 17. | Winter, R.; Gard, G. L. J. Fluorine Chem. 1994, 66, 109–116. doi:10.1016/0022-1139(93)03005-7 |
| 18. | Ray, N. H. J. Chem. Soc. 1963, 1440–1441. doi:10.1039/JR9630001440 |
| 19. | Kleemann, G.; Seppelt, K. Chem. Ber. 1979, 112, 1140–1146. doi:10.1002/cber.19791120409 |
| 20. | Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003 |
| 21. | Coffman, D. D.; Tullock, C. W. Carbonylic compounds containing the SF5 function. U.S. Patent US3,102,903, Sept 3, 1963. |
| 22. | Winter, R.; Willett, R. D.; Gard, G. L. Inorg. Chem. 1989, 28, 2499–2501. doi:10.1021/ic00311a054 |
| 3. | Lentz, D.; Seppelt, K. The –SF5, –SeF5, and –TeF5 groups in organic chemistry. In Chemistry of Hypervalent Compounds; Akiba, K.-y., Ed.; Wiley-VCH: New York, 1999; pp 295–323. |
| 13. | Anthony, M. Aust. N. Z. J. Med. 1984, 14, 888–895. doi:10.1111/j.1445-5994.1984.tb03802.x |
| 35. | Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060–6069. doi:10.1021/ja056711k |
| 7. | Brant, P.; Berry, A. D.; DeMarco, R. A.; Carter, F. L.; Fox, W. B.; Hashmall, J. A. J. Electron Spectrosc. Relat. Phenom. 1981, 22, 119–129. doi:10.1016/0368-2048(81)80021-7 |
| 8. | True, J. E.; Thomas, T. D.; Winter, R. W.; Gard, G. L. Inorg. Chem. 2003, 42, 4437–4441. doi:10.1021/ic0343298 |
| 2. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 30. | Georg, G. I.; Ravikumar, V. T. Stereocontrolled ketene-imine cycloaddition reactions. In Organic Chemistry of β-lactams; Georg, G. I., Ed.; VCH: New York, NY, 1993; pp 295–368. |
| 31. | Palomo, C.; Aizpurua, J. M.; Ganboa, I.; Oiarbide, M. Eur. J. Org. Chem. 1999, 3223–3235. doi:10.1002/(SICI)1099-0690(199912)1999:12<3223::AID-EJOC3223>3.0.CO;2-1 |
| 32. | Palomo, C.; Aizpurua, J. M.; Ganboa, I.; Oiarbide, M. Curr. Med. Chem. 2004, 11, 1837–1872. doi:10.2174/0929867043364900 |
| 33. | Thompson, S.; Coyne, A. G.; Knipe, P. C.; Smith, M. D. Chem. Soc. Rev. 2011, 40, 4217–4231. doi:10.1039/c1cs15022g |
| 27. | Ojima, I. β-Lactam synthon method: enantiomerically pure β-lactams as synthetic intermediates. In Organic Chemistry of β-lactams; Georg, G. I., Ed.; VCH: New York, NY, 1993; pp 197–255. |
| 28. | Kamath, A.; Ojima, I. Tetrahedron 2012, 68, 10640–10664. doi:10.1016/j.tet.2012.07.090 |
| 29. | Ojima, I.; Zuniga, E. S.; Seitz, J. D. Top. Heterocycl. Chem. 2013, 30, 1–63. doi:10.1007/7081_2012_86 |
| 26. | Pepe, A.; Kuznetsova, L.; Sun, L.; Ojima, I. Fluoro-taxoid anticancer agents. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; John Wiley & Sons Ltd.: Chichester, 2009; pp 117–139. doi:10.1002/9781444312096.ch5 |
| 45. | Stepanova, N. P.; Lebedev, V. B.; Orlova, N. A.; Turbanova, E. S.; Petrov, A. A. Zh. Org. Khim. 1988, 24, 692–699. |
| 47. | Yamazaki, T.; Kobayashi, R.; Kitazume, T.; Kubota, T. J. Org. Chem. 2006, 71, 2499–2502. doi:10.1021/jo052434o |
| 45. | Stepanova, N. P.; Lebedev, V. B.; Orlova, N. A.; Turbanova, E. S.; Petrov, A. A. Zh. Org. Khim. 1988, 24, 692–699. |
| 16. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 41. | Carroccia, L.; Fioravanti, S.; Pellacani, L.; Tardella, P. A. Synthesis 2010, 4096–4100. doi:10.1055/s-0030-1258280 |
| 42. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 43. | Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2005, 1258–1265. doi:10.1002/ejoc.200400719 |
| 44. | Crousse, B.; Narizuka, S.; Bonnet-Delpon, D.; Bégué, J.-P. Synlett 2001, 679–681. doi:10.1055/s-2001-13364 |
| 45. | Stepanova, N. P.; Lebedev, V. B.; Orlova, N. A.; Turbanova, E. S.; Petrov, A. A. Zh. Org. Khim. 1988, 24, 692–699. |
| 34. | Cossio, F. P.; Arrieta, A.; Sierra, M. A. Acc. Chem. Res. 2008, 41, 925–936. doi:10.1021/ar800033j |
| 35. | Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060–6069. doi:10.1021/ja056711k |
| 36. | Xu, J. Tetrahedron 2012, 68, 10696–10747. doi:10.1016/j.tet.2012.04.007 |
| 37. | Fustero, S.; Jiménez, D.; Sanz-Cervera, J. F.; Sánchez-Roselló, M.; Esteban, E.; Simón-Fuentes, A. Org. Lett. 2005, 7, 3433–3436. doi:10.1021/ol050791f |
| 38. | Zanda, M.; Bravo, P.; Volonterio, A. Stereoselective synthesis of β-fluoroalkyl β-amino alcohol units. In Asymmetric Fluoroorganic Chemistry; Ramachandran, P. V., Ed.; American Chemical Society: Washington, D.C., 2000; Vol. 746, pp 127–141. doi:10.1021/bk-2000-0746.ch010 |
| 39. | Welch, J. T.; De Corte, B.; De Kimpe, N. J. Org. Chem. 1990, 55, 4981–4983. doi:10.1021/jo00304a002 |
| 40. | Welch, J. T.; Seper, K. W. J. Org. Chem. 1988, 53, 2991–2999. doi:10.1021/jo00248a017 |
© 2013 Penger et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)