Abstract
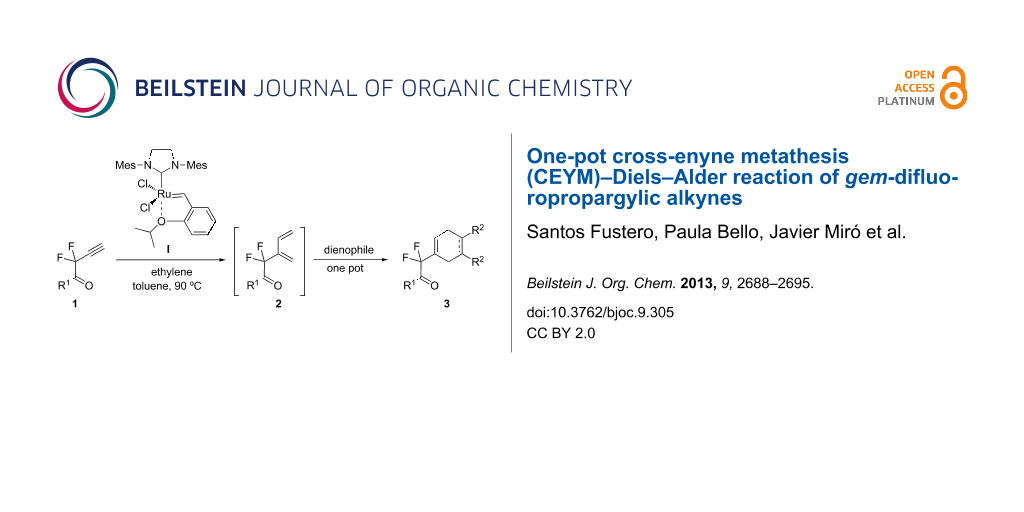
Propargylic difluorides 1 were used as starting substrates in a combination of cross-enyne metathesis and Diels–Alder reactions. Thus, the reaction of 1 with ethylene in the presence of 2nd generation Hoveyda–Grubbs catalyst generates a diene moiety which in situ reacts with a wide variety of dienophiles giving rise to a small family of new fluorinated carbo- and heterocyclic derivatives in moderate to good yields. This is a complementary protocol to the one previously described by our research group, which involved the use of 1,7-octadiene as an internal source of ethylene.

Graphical Abstract
Introduction
In recent years the number of applications of olefin metathesis as a mild and competitive synthetic method for the creation of carbon–carbon bonds has exponentially increased, due to the availability of well-defined catalysts [1-3]. Particularly, enyne metathesis (EYM) is a powerful synthetic tool for generating 1,3-dienes by redistributing unsaturated functionalities between an alkene and an alkyne moiety via vinylalkylidene intermediates [4-6]. This is an atom economical process since it is an addition reaction and non-olefin byproducts are formed. Furthermore, a sequential use of EYM and Diels–Alder reactions generates highly functionalized carbo- and heterocyclic frameworks [7-11].
The intramolecular version of this process, the ring closing enyne metathesis (RCEYM) reaction, has found wide application, and several examples can be found in the literature [12,13]. However, the intermolecular version, i.e. the cross-enyne metathesis (CEYM) reaction, has been much less exploited probably due to its inherent problems of selectivity, which results in the formation of a mixture of E- and Z-isomers [14]. The discovery of the beneficial effect of ethylene has changed this tendency allowing the straightforward preparation of 1,3-dienes [15,16]. Thus, during the ethylene gas promoted CEYM reaction, ethylene does not incorporate into the product but intercepts the secondary metathesis pathways avoiding the formation of secondary products during the process [17,18].
Among organic fluorine compounds, propargylic fluorides constitute a relevant class of fluorinated building blocks. The transformational diversity of the alkynyl group converts them into versatile synthetic intermediates. Additionally, propargylic fluorides are prevalent motifs in life sciences, such as medicinal chemistry or crop protection. In this context, the preparation of monofluorinated propargylic compounds has been the subject of intense research, and efficient methodologies to access these derivatives have been devised [19,20]. However, the analogues bearing the gem-difluoro moiety next to unsaturated bonds have not received comparable attention, probably due to the limited availability of the starting materials. In this context, the recent introduction of difluoropropargyl bromide as fluorinated building block gave access to a wide variety of gem-difluoro-containing alkyne derivatives [21,22]. Recently, we have employed these fluorinated triple bond scaffolds in several types of cyclization reactions for the preparation of different difluoropropargylamides and ketones, having been subjected to intramolecular hydroaminations [23], cascade RCEYM–Diels–Alder reactions [24], [2 + 2 + 2] cycloadditions and gold-mediated dimerization reactions [25].
Although the CEYM reaction has been found to be very fruitful for the preparation of 1,3-dienes, this protocol has remained almost unexplored for propargyl fluorides [26]. In this context, we have recently established a tandem multicomponent protocol CEYM–Diels–Alder reaction of several difluoropropargylic derivatives [27,28] mediated by 1,7-octadiene as an internal source of ethylene [29]. Following our ongoing interest in the use of these fluorinated building blocks, we decided to evaluate the CEYM reaction of several difluoropropargylamides and ketones in combination with a Diels–Alder reaction under Mori´s conditions, in order to compare this protocol with the aforementioned one (Scheme 1).
![[1860-5397-9-305-i1]](/bjoc/content/inline/1860-5397-9-305-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Sequential CEYM–Diels–Alder reaction.
Scheme 1: Sequential CEYM–Diels–Alder reaction.
Results and Discussion
In order to prove the efficiency of ethylene-mediated cross-enyne metathesis on our fluorinated alkynes, substrate 1a was chosen as a model substrate. As expected, when a toluene solution of alkyne 1a and 5 mol % of 2nd generation Hoveyda–Grubbs catalyst I was heated under ethylene atmosphere (1 atm) for 2 h, the clean formation of diene 2a was observed. This newly formed diene was isolated in 70% yield and it reacted smoothly with 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione as dienophile at room temperature to afford, after chromatographic purification, the corresponding Diels–Alder adduct 3a in 60% yield (Scheme 2). It was interesting to find that this sequence can be performed as a one-pot procedure. Thus, when the formation of the diene intermediate 2a was completed (determined by TLC), the dienophile was added to the reaction mixture and was allowed to react for two additional hours. Flash chromatography of the crude product afforded the desired tandem derivative 3a in 60% overall yield (Scheme 2).
![[1860-5397-9-305-i2]](/bjoc/content/inline/1860-5397-9-305-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: One-pot CM–Diels–Alder reaction with fluorinated alkyne 1a.
Scheme 2: One-pot CM–Diels–Alder reaction with fluorinated alkyne 1a.
Next, the one-pot protocol was extended to other starting difluoropropargylic alkynes and dienophiles, affording a new family of carbo- and heterocyclic derivatives in moderate to good yields (Table 1).
Table 1: Preparation of compounds 3 by one-pot CEYM–Diels–Alder reaction of substrates 1 (method A).
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-305-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | 1 | R1 | dienophile | product |
% yield
method Aa (time, h) |
% yield
method Bb (time, h) |
|---|---|---|---|---|---|---|
| 1 | 1a | Bn–NH |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-305-i4.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-305-i5.svg?max-width=637&scale=1.0)
3a |
60 (2 h)c | – |
| 2 | 1a | Bn–NH |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-305-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-305-i7.svg?max-width=637&scale=1.0)
3b |
55 (8 h) | 70 (20 h) |
| 3 | 1a | Bn–NH |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-305-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-305-i9.svg?max-width=637&scale=1.0)
3c |
51 (6 h) | 55 (6 h) |
| 4 | 1a | Bn–NH |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-305-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-305-i11.svg?max-width=637&scale=1.0)
3d |
50 (4 h) | 63 (24 h) |
| 5 | 1a | Bn–NH |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-305-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-305-i13.svg?max-width=637&scale=1.0)
3e |
47 (24 h) | 50 (24 h) |
| 6 | 1a | Bn–NH |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-305-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-305-i15.svg?max-width=637&scale=1.0)
3f |
85 (120 h)c | – |
| 7 | 1b | (R)-Ph(Me)–CHNH |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-305-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-305-i17.svg?max-width=637&scale=1.0)
3g + ![[Graphic 16]](/bjoc/content/inline/1860-5397-9-305-i18.svg?max-width=637&scale=1.0)
3g’ |
87 (4 h)d | 73 (24 h)d |
| 8 | 1b | (R)-Ph(Me)–CHNH |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-305-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-305-i20.svg?max-width=637&scale=1.0)
3h + ![[Graphic 19]](/bjoc/content/inline/1860-5397-9-305-i21.svg?max-width=637&scale=1.0)
3h’ |
74 (2 h)d | 70 (10 h)d |
| 9 | 1b | (R)-Ph(Me)–CHNH |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-305-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-305-i23.svg?max-width=637&scale=1.0)
3i + ![[Graphic 22]](/bjoc/content/inline/1860-5397-9-305-i24.svg?max-width=637&scale=1.0)
3i’ |
74 (20 h)c,d | – |
| 10 | 1b | (R)-Ph(Me)–CHNH |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-305-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-305-i26.svg?max-width=637&scale=1.0)
3j |
63 (10 h) | 60 (24 h) |
| 11 | 1c | PropylNH |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-305-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-305-i28.svg?max-width=637&scale=1.0)
3k |
40 (5 h) | – |
| 12 | 1d | Ph |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-305-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-305-i30.svg?max-width=637&scale=1.0)
3l |
48 (4 h) | 55 (15 h) |
| 13 | 1d | Ph |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-305-i31.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-305-i32.svg?max-width=637&scale=1.0)
3m |
28 (8 h) | – |
| 14 | 1d | Ph |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-305-i33.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-305-i34.svg?max-width=637&scale=1.0)
3n |
58 (12 h) | 70 (17 h) |
| 15 | 1e | Cy |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-305-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-305-i36.svg?max-width=637&scale=1.0)
3o |
54 (12 h) | – |
aMethod A: One-pot protocol with Mori´s conditions. The formation of the corresponding diene 2 with ethylene was complete after 2 h at 90 °C for all substrates. bMethod B: Tandem multicomponent protocol mediated by 1,7-octadiene [29]. cWhen maleic anhydride or 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione were used as dienophiles, the Diels–Alder reaction was performed at rt. With maleic anhydride final products were isolated as the corresponding diacid derivatives. dIn all cases, adducts were obtained as an inseparable 1:1 mixture of diastereoisomers.
A wide variety of nucleophiles is compatible with the one-pot protocol (Table 1, method A, entries 1–6). Ethyl fumarate gave the desired product 3b in 55% yield (Table 1, method A, entry 2). It is interesting to point out that diethyl acetylenedicarboxylate (DEAD) afforded adduct 3c in 51% yield, and no aromatization was observed during the process (Table 1, method A, entry 3). When maleic anhydride was used as dienophile, the corresponding diacid 3f, arising from the anhydride ring opening under the reaction conditions, was observed as the major product (Table 1, method A, entries 6 and 9). With chiral starting material 1b, in all cases a 1:1 mixture of diastereoisomers was obtained which could not be separated. This indicates that the chiral information is not close enough to the reacting centre (Table 1, method A, entries 7–10). Finally, fluorinated ketones could also be used as substrates for the sequential process (Table 1, method A, entries 12–15) and again with alkynes as dienophiles, no aromatization of the final products was detected (Table 1, method A, entry 12).
The yields that appear in Table 1 in the last column (method B) represent the reaction performed under the tandem-multicomponent conditions mediated by 1,7-octadiene (Scheme 1). In general, yields are comparable using either methodology, indicating that they are applicable for the synthesis of new carbo- and heterocyclic derivatives bearing a gem-difluoro moiety in an efficient manner. However, at this point it is important to mention that when the 1,7-octadiene protocol was applied using maleic anhydride or 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione as dienophiles (Table 1, method B, entries 1, 6 and 9), a complex mixture was obtained. This is probably due to the fact that under those thermal conditions, it is not possible to use these types of dienophiles since they decompose while being heated. The sequential generation of the dienic intermediate 2 and the Diels–Alder reaction allow performing the second step at rt, avoiding these problems. Thus, although a tandem-multicomponent protocol is more desirable, the use of the one-pot protocol expand the utility and scope of this methodology, since milder conditions can be employed in the cyclization step.
Conclusion
In conclusion, a tandem one-pot enyne-cross metathesis-Diels–Alder reaction of difluoropropargylic alkynes with a variety of dienophiles has been described. The process took place in moderate to good yields, giving rise to a new family of fluorinated carbo- and heterocyclic derivatives in a very simple manner. In comparison with the tandem multicomponent protocol, the one-pot sequence is a complementary methodology, since although comparable yields of the final adducts can be obtained, this methodology is compatible with a greater variety of dienophiles.
Experimental
General experimental methods. Reactions were carried out under argon atmosphere unless otherwise indicated. The solvents were purified prior to use: THF, diethyl ether and toluene were distilled from sodium/benzophenone; dichloromethane and acetonitrile were distilled from calcium hydride. The reactions were monitored with the aid of thin-layer chromatography (TLC) on 0.25 mm precoated silica gel plates. Visualization was carried out with UV light and aqueous ceric ammonium molybdate solution or potassium permanganate stain. Flash column chromatography was performed with the indicated solvents on silica gel 60 (particle size 0.040–0.063 mm). 1H and 13C NMR spectra were recorded on 300 or 400 MHz spectrometers. Chemical shifts are given in ppm (δ), with reference to the residual proton resonances of the solvents. Coupling constants (J) are given in Hertz (Hz). The letters m, s, d, t, and q stand for multiplet, singlet, doublet, triplet and quartet, respectively. The letters br indicate that the signal is broad. Starting fluorinated amides 1 [23-25] and compounds 3b,c,e,g,h,j,l,n [29] were previously described.
General procedure for the one-pot process. Ethylene was bubbled through a solution of catalyst I (5 mol %) in dry toluene (2.4 mL) for 3 minutes at room temperature in a sealed tube. Substrate 1 (0.12–0.25 mmol) was added next and it was heated at 90 °C for 2 hours. Once the intermediate diene was formed (by TLC), it was cooled to room temperature and the corresponding dienophile was added. The reaction mixture was stirred at temperatures and times given in Table 1. Finally, after removal of solvents, the reaction mixture was purified by flash chromatography in hexanes/ethyl acetate (3:1).
N-Benzyl-2,2-difluoro-3-methylenepent-4-enamide (2a). Following the procedure described above and before adding the dienophile, the crude mixture was subjected to flash chromatography affording 41 mg of 2a (70% yield) as a yellow oil starting from 52 mg of 1a. 1H NMR (CDCl3, 300 MHz) δ 4.52 (d, J = 5.8 Hz, 2H), 5.25 (d, J = 11.3 Hz, 1H), 5.52 (d, J = 17.9 Hz, 1H), 5.64 (d, J = 15.9 Hz, 2H), 6.32 (dd, J1 = 11.3 Hz, J2 = 17.7 Hz, 1H), 6.65 (br s, 1H), 7.26–7.39 (m, 5H); 13C NMR (CDCl3, 300 MHz) δ 43.61, 114.40 (t, 1JCF = 253.2 Hz), 118.17, 119.33 (t, 3JCF = 8.6 Hz), 127.81, 127.93, 128.84, 131.02 (t, 3JCF = 2.3 Hz), 136.75, 138.82 (t, 2JCF = 22.3 Hz), 163.23 (t, 2JCF = 29.9 Hz); 19F NMR (CDCl3, 282 MHz) δ −105.57 (s, 2F); HRMS: [M + 1]+ calcd for C13H14F2NO, 238.1038; found, 238.1042.
N-Benzyl-2-(1,3-dioxo-2-phenyl-2,3,5,8-tetrahydro-1H-[1,2,4]triazolo[1,2a]pyridazin-6-yl)-2,2-difluoroacetamide (3a). Following the general procedure described above, 61 mg of 3a (60% yield) were obtained as a white solid starting from 52 mg of 1a. mp = 148–150 °C; 1H NMR (CDCl3, 300 MHz) δ 4.26–4.29 (m, 2H), 4.34 (s, 2H), 4.5 (d, J = 5.7 Hz, 2H), 6.46 (m, 1H), 6.86 (br s, 1H), 7.16–7.53 (m, 10H); 13C NMR (CDCl3, 300 MHz) δ 41.8 (t, 3JCF = 4 Hz), 43.1, 43.8, 113.3 (t, 1JCF = 253.8 Hz), 123.7 (t, 3JCF = 8.7 Hz), 125.4, 126.8 (t, 2JCF = 25.2 Hz), 127.9, 128.2, 128.4, 129.0, 129.2, 130.8, 136.3, 152.3, 152.4, 162.2 (t, 2JCF = 29.9 Hz); 19F NMR (CDCl3, 282 MHz) δ −106.9 (s, 2F); HRMS: [M]+ calcd for C21H18F2N4O3, 412.1347; found, 412.1344.
Diethyl 4-(2-(benzylamino)-1,1-difluoro-2-oxoethyl)cyclohex-4-ene-1,2-dicarboxylate (3b). Following the general procedure described above, 30 mg (55% yield) of 3b were obtained as a white solid starting from 26 mg of 1a.
Diethyl 4-(2-(benzylamino)-1,1-difluoro-2-oxoethyl)cyclohexa-1,4-diene-1,2-dicarboxylate (3c). Following the general procedure described above, 52 mg (51% yield) of 3c were obtained as a dark brown oil starting from 52 mg of 1a.
N-Benzyl-2-(1,3-dioxo-2-phenyl-2,3,3a,4,7,7a-hexahydro-1H-isoindol-5-yl)-2,2-difluoroacetamide (3d). Following the general procedure described above, 25 mg of 3d (50% yield) were obtained as a dark brown oil starting from 26 mg of 1a. 1H NMR (CDCl3, 300 MHz) δ 2.32–2.47 (m, 2H), 2.82–2.93 (m, 2H), 3.27–3.38 (m, 2H), 4.46 (d, J = 5.7 Hz, 2H), 6.49–6.55 (m, 1H), 6.68 (br s, 1H), 7.27–7.47 (m, 10H); 13C NMR (CDCl3, 300 MHz) δ 22.8, 23.9, 38.7, 39.1, 43.7, 114.2 (t, 1JCF = 251.0 Hz), 126.5, 127.9, 128.0, 128.7, 128.9, 129.1, 129.9 (t, 3JCF = 8.9 Hz), 131.8, 131.9 (t, 2JCF = 24.4 Hz), 136.6, 162.8 (t, 2JCF = 30.1 Hz), 177.7, 178.1; 19F NMR (CDCl3, 282 MHz) δ −106.5 (d, JFF = 258.1 Hz, 1F), −108.7 (d, JFF = 258.4 Hz, 1F); HRMS: [M]+ calcd for C23H20F2N2O3, 410.1442; found, 410.1445.
N-Benzyl-2-(9,10-dioxo-1,4,4a,9,9a,10-hexahydroanthracen-2-yl)-2,2-difluoroacetamide (3e). Following the general procedure described above, 24 mg of 3e (47% yield) were obtained as a dark brown oil starting from 26 mg of 1a.
4-[2-(Benzylamino)-1,1-difluoro-2-oxoethyl]cyclohex-4-ene-1,2-dicarboxylic acid (3f). Following the general procedure described above, 71 mg of 3f (85% yield) were obtained as a dark brown oil starting from 52 mg of 1a. 1H NMR (CDCl3, 300 MHz) δ 2.46–2.52 (m, 2H), 2.68–2.75 (m, 2H), 3.07 (m, 1H), 3.17 (m, 1H), 4.47 (d, J = 5.7 Hz, 2H), 6.21 (s, 1H), 6.80–6.84 (m, J = 5.4 Hz, 1H), 7.25–7.37 (m, 5H), 7.77 (br s, 2H); 13C NMR (CDCl3, 300 MHz) δ 23.1, 25.0, 38.5, 38.9, 43.6, 114.8 (t, 1JCF = 251.1 Hz), 127.8, 127.9, 128.8, 136.6, 163.6 (t, 2JCF = 30.6 Hz), 178.3, 178.5; 19F NMR (CDCl3, 282 MHz) δ −117.0 (d, JFF = 258.9 Hz, 1F), −118.0 (d, JFF = 258.7 Hz, 1F); HRMS: [M + Na]+ calcd for C17H17F2NO5, 376.0972; found, 376.0969.
Diethyl 4-(1,1-difluoro-2-oxo-2-[(R)-1-phenylethylamino)ethyl]cyclohex-4-ene-1,2-dicarboxylate (3g + 3g’). Following the general procedure described above, 41 mg (87% yield) of a inseparable mixture of 3g and 3g’ were obtained as a dark brown oil starting from 25 mg of 1b.
2-(1,3-Dioxo-2-phenyl-2,3,3a,4,7,7a-hexahydro-1H-isoindol-5-yl)-2,2-difluoro-N-[(R)-1-phenylethyl]acetamide (3h + 3h’). Following the general procedure described above, 34 mg (74% yield) of a inseparable mixture of 3h and 3h’ were obtained as a dark brown solid starting from 25 mg of 1b.
4-[1,1-difluoro-2-oxo-2-((R)-1-phenylethylamino)ethyl]cyclohexa-4-ene-1,2-dicarboxylate (3i + 3i’). Following the general procedure described above, 24.3 mg of an inseparable mixture of 3i and 3i’ were obtained as a dark brown oil starting from 25 mg of 1b. 1H NMR (CDCl3, 300 MHz) δ1.53 (dd, J = 6.9 Hz, J = 4.8 Hz, 3H), 2.46–2.51 (m, 2H), 2.67–2.74 (m, 2H), 3.02–3.07 (m, 1H), 3.15–3.17 (m, 1H), 5.08–5.17 (m, 1H), 6.17 (d, J = 13.5 Hz, 1H), 6.63–6.68 (br m, 1H), 7.28–7.38 (m, 5H); 13C NMR (CDCl3, 300 MHz) δ 21.2, 23.4, 25.2, 49.2, 114.7 (t, 1JCF = 255.0 Hz), 126.1, 127.7, 128.7, 141.7, 162.5 (t, 2JCF = 30.7 Hz), 177.6, 177.9; 19F NMR (CDCl3, 282 MHz) δ −106.6 (d, JFF = 258.9 Hz, 1F), −106.8 (d, JFF = 256.4Hz, 1F), −107.8 (d, JFF = 255.6 Hz, 1F), −107.9 (d, JFF = 257.5 Hz, 1F); HRMS: [M + Na]+ calcd for C18H19F2NO5, 390.1129; found, 390.1133.
(R)-Diethyl 4-[1,1-difluoro-2-oxo-2-(1-phenylethylamino)ethyl]cyclohexa-1,4-diene-1,2-dicarboxylate (3j). Following the general procedure described above, 59.5 mg (63% yield) of 3j were obtained as a dark brown oil starting from 50 mg of 1b [29].
Diethyl 4-[1,1-difluoro-2-oxo-2-(propylamino)ethyl]cyclohex-4-ene-1,2-dicarboxylate (3k). Following the general procedure described above, 27 mg of 3k (40% yield) were obtained as a dark brown oil starting from 30 mg of 1c. 1H NMR (CDCl3, 300 MHz) δ 0.93 (t, J = 7.5 Hz, 3H), 1.23 (t, J = 6.9 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H), 1.57 (m, 2H), 2.22–2.37 (m, 2H), 2.50–2.62 (m, 2H), 2.86 (m, 2H), 3.27 (q, J = 6.0 Hz, 2H), 4.13 (q, J = 6.9 Hz, 4H), 6.18–6.20 (br m, 1H); 13C NMR (300 MHz) δ 11.2, 14.0, 22.4, 25.0 (t, 4JCF = 2.3 Hz), 27.3, 40.4, 40.6, 41.2, 60.8, 60.9, 114.7 (t, 1JCF = 250.9 Hz), 127.6 (t, 3JCF = 8.6 Hz), 129.0 (t, 2JCF = 24.1 Hz), 163.2 (t, 2JCF = 30.2 Hz), 173.7, 173,9; 19F NMR (CDCl3, 282 MHz) δ −106.8 (d, JFF = 257.8 Hz, 1F), −107.9 (d, JFF = 258.4 Hz, 1F); HRMS: [M + 1]+ calcd for C17H26F2NO5, 362.1774; found, 362.1779.
Diethyl 4-(1,1-difluoro-2-oxo-2-phenylethyl)cyclohexa-1,4-diene-1,2-dicarboxylate (3l). Following the general procedures described above, 20 mg (48% yield) of 3l were obtained as a dark brown oil starting from 20 mg of 1d.
5-(1,1-Difluoro-2-oxo-2-phenylethyl)-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (3m). Following the general procedure described above, 15 mg of 3m (28% yield) were obtained as a dark brown oil starting from 25 mg of 1d. 1H NMR (CDCl3, 300 MHz) δ 2.26–2.41 (m, 2H), 2.73–2.84 (m, 2H), 3.18–3.30 (m, 2H), 6.33–6.38 (m, 1H), 7.09–7.14 (m, 3H), 7.23–7.35 (m, 5H), 7.47–7.52 (m, 1H), 7.91–7.94 (m, 2H); 13C NMR (CDCl3, 300 MHz) δ 23.2 (t, 3JCF = 2.6 Hz), 23.8, 38.8, 39.0, 116.2 (t, 1JCF = 253.0 Hz), 126.4, 128.6, 128.7, 129.1, 129.7 (t, 3JCF = 8.9 Hz), 130.2 (t, 4JCF = 3.0 Hz), 131.8, 133.2 (t, 2JCF = 23.9 Hz), 134.4, 177.7, 178.2, 188.3 (t, 2JCF = 32.1 Hz); 19F NMR (CDCl3, 282 MHz) δ −112.367 (d, JFF = 284.6 Hz, 1F), −111.2 (d, JFF = 284.4 Hz, 1F); HRMS: [M + 1]+ calcd for C22H18F2NO3, 382.1255; found, 382.1258.
Diethyl 4-[1,1-difluoro-2-oxo-2-(phenylamino)ethyl]cyclohex-4-ene-1,2-dicarboxylate (3n). Following the general procedure described above, 24.5 mg (58% yield) of 3n were obtained as a yellow oil starting from 20 mg of 1d.
Diethyl 4-(2-cyclohexyl-1,1-difluoro-2-oxoethyl)cyclohexa-1,4-diene-1,2-dicarboxylate (3o). Following the general procedure described above, 28 mg of 3o (54% yield) were obtained as a dark oil starting from 25 mg of 1e. 1H NMR (CDCl3, 300 MHz) δ 1.11–1.41 (m, 5H), 1.63–1.84 (m, 5H), 2.30–2.46 (m, 5H), 2.71–2.92 (m, 3H), 3.26–3.36 (m, 2H), 6.37–6.41 (m, 1H), 7.25–7.29 (m, 2H), 7.35–7.49 (m, 3H); 13C NMR (300 MHz) δ 30.0 (t, 4JCF = 2.7 Hz), 23.8, 25.3, 25.4, 28.3, 38.7, 45.1, 115.1 (t, 1JCF = 253.7 Hz), 126.4, 128.6, 129.1, 129.5 (t, 3JCF = 9.1 Hz), 132.2 (t, 2JCF = 24.2 Hz), 177.7, 178.2, 202.7 (t, 2JCF = 31.7 Hz); 19F NMR (CDCl3, 282 MHz) δ −109.9 (d, JFF = 276.7 Hz, 1F), −106.0 (d, JFF = 276.7 Hz, 1F); HRMS: [M]+ calcd for C22H23F2NO3, 387.1646; found, 387.1656.
References
-
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746. doi:10.1021/cr9002424
Return to citation in text: [1] -
Lozano-Vila, A. M.; Monsaert, S.; Bajek, A.; Verpoort, F. Chem. Rev. 2010, 110, 4865. doi:10.1021/cr900346r
Return to citation in text: [1] -
Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708. doi:10.1021/cr800524f
Return to citation in text: [1] -
Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001. doi:10.1039/b807278g
Return to citation in text: [1] -
Diver, S. T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317. doi:10.1021/cr020009e
Return to citation in text: [1] -
Poulsen, C. S.; Madsen, R. Synthesis 2003, 1. doi:10.1055/s-2003-36243
Return to citation in text: [1] -
Li, J.; Lee, D. Eur. J. Org. Chem. 2011, 4269. doi:10.1002/ejoc.201100438
Return to citation in text: [1] -
O'Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563. doi:10.1002/chem.201000707
Return to citation in text: [1] -
Subrahmanyam, A. V.; Palanichamy, K.; Kaliappan, K. P. Chem.–Eur. J. 2010, 16, 8545. doi:10.1002/chem.201000482
Return to citation in text: [1] -
Kotha, S.; Meshram, M.; Tiwari, A. Chem. Soc. Rev. 2009, 38, 2065. doi:10.1039/b810094m
Return to citation in text: [1] -
Rosillo, M.; Domínguez, G.; Casarubios, L.; Amador, U.; Pérez-Castells, J. J. Org. Chem. 2004, 69, 2084. doi:10.1021/jo0356311
Return to citation in text: [1] -
Villar, H.; Frings, M.; Bolm, C. Chem. Soc. Rev. 2007, 36, 55. doi:10.1039/b508899m
Return to citation in text: [1] -
Maifeld, S. V.; Lee, D. Chem.–Eur. J. 2005, 11, 6118. doi:10.1002/chem.200500407
Return to citation in text: [1] -
Stragies, R.; Schuster, M.; Blechert, S. Angew. Chem., Int. Ed. Engl. 1997, 36, 2518. doi:10.1002/anie.199725181
The first example of CEYM was reported in early 1997.
Return to citation in text: [1] -
Kinoshita, A.; Sakakibara, N.; Mori, M. J. Am. Chem. Soc. 1997, 119, 12388. doi:10.1021/ja973134u
Return to citation in text: [1] -
Kinoshita, A.; Sakakibara, N.; Mori, M. Tetrahedron 1999, 55, 8155. doi:10.1016/S0040-4020(99)00297-5
Return to citation in text: [1] -
Grotevendt, A. G. D.; Lummiss, J. A. M.; Mastronardi, M. L.; Fogg, D. E. J. Am. Chem. Soc. 2011, 133, 15918. doi:10.1021/ja207388v
Return to citation in text: [1] -
Lloyd-Jones, G. C.; Margue, R. G.; de Vries, J. G. Angew. Chem., Int. Ed. 2005, 44, 7442. doi:10.1002/anie.200502243
Return to citation in text: [1] -
Pacheco, M. C.; Purser, S.; Gouverneur, V. Chem. Rev. 2008, 108, 1943. doi:10.1021/cr068410e
Return to citation in text: [1] -
Prakesch, M.; Grée, D.; Grée, R. Acc. Chem. Res. 2002, 35, 175. doi:10.1021/ar010055l
Return to citation in text: [1] -
Xu, B.; Mae, M.; Hong, J. A.; Li, Y.; Hammond, G. B. Synthesis 2006, 803. doi:10.1055/s-2006-926334
Return to citation in text: [1] -
Hammond, G. B. J. Fluorine Chem. 2006, 127, 476. doi:10.1016/j.jfluchem.2005.12.024
Return to citation in text: [1] -
Fustero, S.; Fernández, B.; Bello, P.; del Pozo, C.; Arimitsu, S.; Hammond, G. B. Org. Lett. 2007, 9, 4251. doi:10.1021/ol701811z
Return to citation in text: [1] [2] -
Arimitsu, S.; Fernández, B.; del Pozo, C.; Fustero, S.; Hammond, G. B. J. Org. Chem. 2008, 73, 2656. doi:10.1021/jo7025965
Return to citation in text: [1] [2] -
Fustero, S.; Bello, P.; Fernández, B.; del Pozo, C.; Hammond, G. B. J. Org. Chem. 2009, 74, 7690. doi:10.1021/jo9013436
Return to citation in text: [1] [2] -
Pujari, S. A.; Kaliappan, K. P.; Valleix, A.; Grée, D.; Grée, R. Synlett 2008, 2503. doi:10.1055/s-2008-1078179
As far as we know, only one example of CEYM involving fluorinated alkynes have been reported.
Return to citation in text: [1] -
Qing, F.-L.; Zheng, F. Synlett 2011, 1052. doi:10.1055/s-0030-1259947
See for a recent review that highlights the importance of gem-difluoro moiety-containing derivatives.
Return to citation in text: [1] -
Fustero, S.; Sanz-Cervera, J.-F.; Aceña, J. L.; Sánchez-Roselló, M. Synlett 2009, 525. doi:10.1055/s-0028-1087806
Return to citation in text: [1] -
Fustero, S.; Bello, P.; Miró, J.; Simón, A.; del Pozo, C. Chem.–Eur. J. 2012, 18, 10991. doi:10.1002/chem.201200835
Return to citation in text: [1] [2] [3] [4]
| 29. | Fustero, S.; Bello, P.; Miró, J.; Simón, A.; del Pozo, C. Chem.–Eur. J. 2012, 18, 10991. doi:10.1002/chem.201200835 |
| 23. | Fustero, S.; Fernández, B.; Bello, P.; del Pozo, C.; Arimitsu, S.; Hammond, G. B. Org. Lett. 2007, 9, 4251. doi:10.1021/ol701811z |
| 24. | Arimitsu, S.; Fernández, B.; del Pozo, C.; Fustero, S.; Hammond, G. B. J. Org. Chem. 2008, 73, 2656. doi:10.1021/jo7025965 |
| 25. | Fustero, S.; Bello, P.; Fernández, B.; del Pozo, C.; Hammond, G. B. J. Org. Chem. 2009, 74, 7690. doi:10.1021/jo9013436 |
| 29. | Fustero, S.; Bello, P.; Miró, J.; Simón, A.; del Pozo, C. Chem.–Eur. J. 2012, 18, 10991. doi:10.1002/chem.201200835 |
| 1. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746. doi:10.1021/cr9002424 |
| 2. | Lozano-Vila, A. M.; Monsaert, S.; Bajek, A.; Verpoort, F. Chem. Rev. 2010, 110, 4865. doi:10.1021/cr900346r |
| 3. | Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708. doi:10.1021/cr800524f |
| 14. |
Stragies, R.; Schuster, M.; Blechert, S. Angew. Chem., Int. Ed. Engl. 1997, 36, 2518. doi:10.1002/anie.199725181
The first example of CEYM was reported in early 1997. |
| 29. | Fustero, S.; Bello, P.; Miró, J.; Simón, A.; del Pozo, C. Chem.–Eur. J. 2012, 18, 10991. doi:10.1002/chem.201200835 |
| 12. | Villar, H.; Frings, M.; Bolm, C. Chem. Soc. Rev. 2007, 36, 55. doi:10.1039/b508899m |
| 13. | Maifeld, S. V.; Lee, D. Chem.–Eur. J. 2005, 11, 6118. doi:10.1002/chem.200500407 |
| 29. | Fustero, S.; Bello, P.; Miró, J.; Simón, A.; del Pozo, C. Chem.–Eur. J. 2012, 18, 10991. doi:10.1002/chem.201200835 |
| 7. | Li, J.; Lee, D. Eur. J. Org. Chem. 2011, 4269. doi:10.1002/ejoc.201100438 |
| 8. | O'Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563. doi:10.1002/chem.201000707 |
| 9. | Subrahmanyam, A. V.; Palanichamy, K.; Kaliappan, K. P. Chem.–Eur. J. 2010, 16, 8545. doi:10.1002/chem.201000482 |
| 10. | Kotha, S.; Meshram, M.; Tiwari, A. Chem. Soc. Rev. 2009, 38, 2065. doi:10.1039/b810094m |
| 11. | Rosillo, M.; Domínguez, G.; Casarubios, L.; Amador, U.; Pérez-Castells, J. J. Org. Chem. 2004, 69, 2084. doi:10.1021/jo0356311 |
| 26. |
Pujari, S. A.; Kaliappan, K. P.; Valleix, A.; Grée, D.; Grée, R. Synlett 2008, 2503. doi:10.1055/s-2008-1078179
As far as we know, only one example of CEYM involving fluorinated alkynes have been reported. |
| 4. | Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001. doi:10.1039/b807278g |
| 5. | Diver, S. T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317. doi:10.1021/cr020009e |
| 6. | Poulsen, C. S.; Madsen, R. Synthesis 2003, 1. doi:10.1055/s-2003-36243 |
| 27. |
Qing, F.-L.; Zheng, F. Synlett 2011, 1052. doi:10.1055/s-0030-1259947
See for a recent review that highlights the importance of gem-difluoro moiety-containing derivatives. |
| 28. | Fustero, S.; Sanz-Cervera, J.-F.; Aceña, J. L.; Sánchez-Roselló, M. Synlett 2009, 525. doi:10.1055/s-0028-1087806 |
| 21. | Xu, B.; Mae, M.; Hong, J. A.; Li, Y.; Hammond, G. B. Synthesis 2006, 803. doi:10.1055/s-2006-926334 |
| 22. | Hammond, G. B. J. Fluorine Chem. 2006, 127, 476. doi:10.1016/j.jfluchem.2005.12.024 |
| 24. | Arimitsu, S.; Fernández, B.; del Pozo, C.; Fustero, S.; Hammond, G. B. J. Org. Chem. 2008, 73, 2656. doi:10.1021/jo7025965 |
| 19. | Pacheco, M. C.; Purser, S.; Gouverneur, V. Chem. Rev. 2008, 108, 1943. doi:10.1021/cr068410e |
| 20. | Prakesch, M.; Grée, D.; Grée, R. Acc. Chem. Res. 2002, 35, 175. doi:10.1021/ar010055l |
| 25. | Fustero, S.; Bello, P.; Fernández, B.; del Pozo, C.; Hammond, G. B. J. Org. Chem. 2009, 74, 7690. doi:10.1021/jo9013436 |
| 17. | Grotevendt, A. G. D.; Lummiss, J. A. M.; Mastronardi, M. L.; Fogg, D. E. J. Am. Chem. Soc. 2011, 133, 15918. doi:10.1021/ja207388v |
| 18. | Lloyd-Jones, G. C.; Margue, R. G.; de Vries, J. G. Angew. Chem., Int. Ed. 2005, 44, 7442. doi:10.1002/anie.200502243 |
| 15. | Kinoshita, A.; Sakakibara, N.; Mori, M. J. Am. Chem. Soc. 1997, 119, 12388. doi:10.1021/ja973134u |
| 16. | Kinoshita, A.; Sakakibara, N.; Mori, M. Tetrahedron 1999, 55, 8155. doi:10.1016/S0040-4020(99)00297-5 |
| 23. | Fustero, S.; Fernández, B.; Bello, P.; del Pozo, C.; Arimitsu, S.; Hammond, G. B. Org. Lett. 2007, 9, 4251. doi:10.1021/ol701811z |
© 2013 Fustero et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)