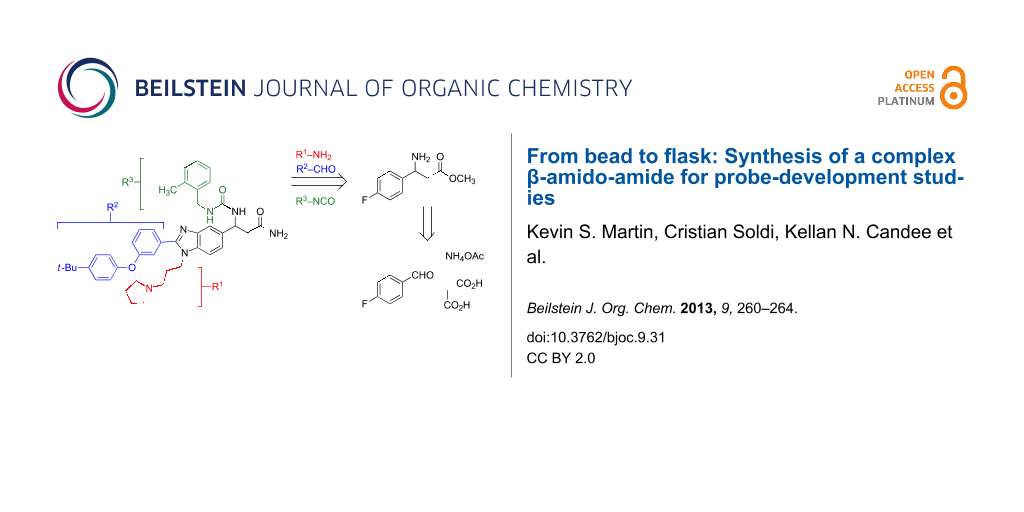
Abstract
A concise synthesis of benzimidazole-substituted β-amido-amide LLW62 is presented. The original synthesis of compounds related to LLW62 was developed on Rink resin as part of a “one-bead, one-compound” combinatorial approach for on-bead screening purposes. The current synthesis is carried out in solution and is amenable to scale-up for follow-up studies on LLW62 and investigations of related structures. The key step involves the use of a β-amino acid-forming three-component reaction (3CR), the scope of which defines its role in the synthetic strategy.

Graphical Abstract
Introduction
Library syntheses and high-throughput screening can often be combined to enable the discovery of new small-molecule probes that modulate biological phenomena [1]. Although the use of solid-phase, split-pool combinatorial synthesis for the preparation of solutions of small-molecule libraries has declined, the use of these compounds for on-bead screening has resulted in recent screening innovations [1,2]. The Lam and Kurth groups have published several “one-bead, one-compound” (OBOC) library syntheses of heterocyclic structures for a variety of screening endeavors [3-12]. Some of these compounds were identified as inhibitors of p21, which is a protein that modulates the activity of cyclin kinases [13-15]. One function of p21 is that it acts downstream of p53 to repair DNA-damaged cells and may function to convey anti-apoptotic activity to cancer cells (Figure 1) [13]. As such, an inhibitor of p21 could sensitize malignant cells to DNA-damaging chemical and radiation therapy by subverting this p21-mediated DNA repair process [14-17]. In this study, we developed a synthesis of LLW62 (1, Figure 2), which is a complex benzimidazole-substituted β-amido-amide similar in structure to inhibitors of p21 that were reported previously to support studies of this compound as a biological probe [14,15].
![[1860-5397-9-31-1]](/bjoc/content/figures/1860-5397-9-31-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: p21 determines the fate of DNA-damaged [13] cells.
Figure 1: p21 determines the fate of DNA-damaged [13] cells.
![[1860-5397-9-31-2]](/bjoc/content/figures/1860-5397-9-31-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of LLW62 (1) from acid 7 (A) or aldehyde 11 (B).
Figure 2: Retrosynthetic analysis of LLW62 (1) from acid 7 (A) or aldehyde 11 (B).
The synthesis of 1 emanates from a one-pot, three-component reaction (3CR) of an arylaldehyde, malonic acid (5), and ammonium acetate, which assembles the β-amino acid core (Figure 2) [14,15,18]. In the reported synthesis of 1, a protected β-amino acid core was attached to Rink-amide resin and carried through to 1 by a series of elaboration and tagging steps [14,15]. Synthetic intermediates in this route were not characterized, and 1 was ultimately purified by high-performance liquid chromatography and partially characterized by matrix-assisted laser desorption/ionization mass spectrometry [14]. In the current synthesis, we set out to develop a concise and scalable solution-phase route to 1 and provide characterization data for 1 and all intermediate compounds.
In our retrosynthetic analysis, we envisioned 1 coming from acylation of benzimidazole 3 with isocyanate 2 (Figure 2). We initially sought to avoid nitration, protection and deprotection steps and access this intermediate by performing a late-stage 3CR with benzimidazole 4, which would be available from nitrile 6 or acid 7 (Figure 2, A). Although synthesis of 4 proceeded without difficulty from acid 7, this route was unsuccessful at a late stage for a reason that we describe below. We next envisioned benzimidazole 3 emanating from β-amino ester 10, which could be accessed in a few steps starting with an early stage 3CR of aldehyde 11, malonic acid (5), and ammonium acetate (Figure 2, B). Gratifyingly, 10 was converted to the requisite benzimidizole 3 in three steps and carried through to 1.
Results and Discussion
Our initial target was benzimidazole 4, which we envisioned originating from nitrile 6 or acid 7, each of which is commercially available (Figure 2). We first attempted to synthesize 4 from 6, which would lead to the shortest possible synthesis of 1. Nitrile 6 was treated with N-(3-aminopropyl)pyrrolidine (8) to produce aniline 12 in 81% yield (Scheme 1) [19]. This compound was reduced to aniline 13 in 79% yield and converted to the benzimidizole 14 in 63% yield with aldehyde 9 under oxidative conditions. The resultant nitrile proved to be extremely insoluble and difficult to handle. Several reduction conditions were attempted to produce benzimidazole 4 with no success. In addition, attempts to use the nitrile in a Blaise-type reaction or similar nucleophilic addition were also unsuccessful (not shown). Although nitrile 6 would have provided the shortest, most direct entry into the requisite β-amino core structure, we turned our attention to another route to 4.
![[1860-5397-9-31-i1]](/bjoc/content/inline/1860-5397-9-31-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Attempted synthesis of aldehyde 4 from nitrile 6.
Scheme 1: Attempted synthesis of aldehyde 4 from nitrile 6.
Acid 7 was immediately more promising as a starting material for benzimidazole 4. Commercially available 7 was converted to methyl ester 15 in 90% yield, due to its ease of handling (Scheme 2) [20]. Next, SNAr displacement of the fluoride of 15 by N-(3-aminopropyl)pyrrolidine (8) proceeded in high yield, 99%, to give aniline 16 [20]. Reduction of the nitro group was nearly quantitative and subsequent benzimidazole formation with Oxone furnished benzimidazole 17 in 47% yield over two steps [20,21]. The ester of 17 was smoothly reduced to the alcohol 18, in 74% yield, and immediately oxidized to the aldehyde 4, in 87% yield. Unfortunately, 4 produced none of the desired β-amino acid 3 under several different variants of the 3CR with malonic acid (5) and ammonium acetate. Tan and Weaver demonstrated previously that the β-amino acid forming 3CR works best for electron-rich aldehydes and poorly for electron-deficient aldehydes [18]. Thus, we suspected that aldehyde 4 may be too electron poor for the 3CR to work efficiently. Our suspicions were supported by attempting 3CRs on aldehydes 19 and 20, each of which has a single nitro group, and neither was successful in this transformation.
![[1860-5397-9-31-i2]](/bjoc/content/inline/1860-5397-9-31-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (A) Synthesis of 4 from acid 7 and (B) attempted β-amino acid-forming 3CRs.
Scheme 2: (A) Synthesis of 4 from acid 7 and (B) attempted β-amino acid-forming 3CRs.
An early-stage 3CR enabled the use of the least electron-poor aldehyde in this key step (Scheme 3). Heating of 4-fluorobenzaldehyde (11), malonic acid and ammonium acetate under reflux proceeded smoothly, as previously described, to furnish β-amino acid 21 in 73% yield [18]. Methylation of 21 (80%) followed by nitration of 22 (67%), boc protection of 23 and SNAr displacement of the fluoride in 24 with amine 8 (71% over two steps), and finally reduction of the nitro group of 25 (63%) provided aniline 26 as our key intermediate for forming the benzimidazole core of 1. We next attempted to produce 27 under the higher yielding oxidative conditions described for the formation of nitrile containing benzimidazole 14. The yield for this reaction was significantly lower, less than 50%, compared to the reaction to produce 14, and we observed some transesterification of the methyl ester with butanol to produce a mixture of 27 and the butyl ester of 27 as the major products (not shown). We thus turned to using Oxone, and benzimidazole formation proceeded in acceptable yield (44%) from aniline 26 to furnish 27. Benzimidazole 27 was then saponified under basic conditions to give acid 28 (86%) [21]. Installation of the primary amide of 1 was then achieved in a single pot by treatment of 28 with ethyl chloroformate to make the mixed anhydride followed by displacement with ammonia gas to produce 29 in 65% yield [22]. Final Boc deprotection of 29 with TFA (87%) and subsequent acylation of the free amine of 30 with isocyanate 2 (75%) provided the desired compound 1 in 11 total steps and 3% overall yield.
![[1860-5397-9-31-i3]](/bjoc/content/inline/1860-5397-9-31-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of LLW62 by using an early stage 3CR.
Scheme 3: Synthesis of LLW62 by using an early stage 3CR.
Conclusion
We have completed a solution-phase synthesis of 1 and thus provided a common route to related compounds that may emerge from future on-bead screening experiments. The key step was the 3CR to form the β-amino acid core structure. Although the electronic requirements of this reaction limit it to electron-rich, or at least not excessively electron poor, aromatic aldehydes, application of this transformation early in the synthesis ultimately proved successful. Although this route is not suitable for large-scale production of 1, multigram quantities of this compound and benzimidazoles of comparable complexity are easily accessible for early stage studies of these compounds in vitro and in vivo using model organisms.
Supporting Information
| Supporting Information File 1: Experimental procedures and compound characterization. | ||
| Format: PDF | Size: 927.0 KB | Download |
Acknowledgements
This work was supported by the National Science Foundation (CAREER award to JTS) and the National Institutes of Health (NIGMS/P41GM089153). CS thanks CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and the Brazilian Ministry of Education for a postdoctoral fellowship. RHW acknowledges research support from the NIH (NCI/1R01CA135401; NIDDK/1R01DK082690) and the Medical Service of the US Department of Veterans’ Affairs. The authors thank Dr. Ruiwu Liu and Professor Kit S. Lam (Division of Hematology and Oncology, Department of Internal Medicine, University of California Davis Comprehensive Cancer Center) for helpful discussions.
References
-
Dolle, R. E.; Le Bourdonnec, B.; Worm, K.; Morales, G. A.; Thomas, C. J.; Zhang, W. J. Comb. Chem. 2010, 12, 765–806. doi:10.1021/cc100128w
Return to citation in text: [1] [2] -
Kodadek, T. Chem. Commun. 2011, 47, 9757–9763. doi:10.1039/c1cc12102b
Return to citation in text: [1] -
Aina, O. H.; Marik, J.; Liu, R.; Lau, D. H.; Lam, K. S. Mol. Cancer Ther. 2005, 4, 806–813. doi:10.1158/1535-7163.MCT-05-0029
Return to citation in text: [1] -
Dixon, S.; Ziebart, K. T.; He, Z.; Jeddeloh, M.; Yoo, C. L.; Wang, X.; Lehman, A.; Lam, K. S.; Toney, M. D.; Kurth, M. J. J. Med. Chem. 2006, 49, 7413–7426. doi:10.1021/jm0609869
Return to citation in text: [1] -
Zhang, H.; Aina, O. H.; Lam, K. S.; de Vere White, R.; Evans, C.; Henderson, P.; Lara, P. N.; Wang, X.; Bassuk, J. A.; Pan, C.-x. Urol. Oncol.: Semin. Orig. Invest. 2012, 30, 635–645. doi:10.1016/j.urolonc.2010.06.011
Return to citation in text: [1] -
Lam, K. S.; Lehman, A. L.; Song, A.; Doan, N.; Enstrom, A. M.; Maxwell, J.; Liu, R. Methods Enzymol. 2003, 369, 298–322. doi:10.1016/S0076-6879(03)69017-8
Return to citation in text: [1] -
Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.; Knapp, R. J. Nature 1991, 354, 82–84. doi:10.1038/354082a0
Return to citation in text: [1] -
Liu, G.; Fan, Y.; Zhao, Z.; Lam, K. S. Zhongguo Yaowu Huaxue Zazhi 2002, 12, 311–318.
Return to citation in text: [1] -
Miyamoto, S.; Liu, R.; Hung, S.; Wang, X.; Lam, K. S. Anal. Biochem. 2008, 374, 112–120. doi:10.1016/j.ab.2007.10.028
Return to citation in text: [1] -
Park, S. I.; Renil, M.; Vikstrom, B.; Amro, N.; Song, L.-w.; Xu, B.-l.; Lam, K. S. Lett. Pept. Sci. 2001, 8, 171–178. doi:10.1023/A:1016297601361
Return to citation in text: [1] -
Xiao, W.; Wang, Y.; Lau, E. Y.; Luo, J.; Yao, N.; Shi, C.; Meza, L.; Tseng, H.; Maeda, Y.; Kumaresan, P.; Liu, R.; Lightstone, F. C.; Takada, Y.; Lam, K. S. Mol. Cancer Ther. 2010, 9, 2714–2723. doi:10.1158/1535-7163.MCT-10-0308
Return to citation in text: [1] -
Dixon, S. M.; Milinkevich, K. A.; Fujii, J.; Liu, R.; Yao, N.; Lam, K. S.; Kurth, M. J. J. Comb. Chem. 2007, 9, 143–157. doi:10.1021/cc060090p
Return to citation in text: [1] -
Weiss, R. H. Cancer Cell 2003, 4, 425–429. doi:10.1016/S1535-6108(03)00308-8
Return to citation in text: [1] [2] [3] -
Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069
Return to citation in text: [1] [2] [3] [4] [5] -
Park, S.-H.; Park, J.-Y.; Weiss, R. H. J. Urol. 2008, 180, 352–360. doi:10.1016/j.juro.2008.02.038
Return to citation in text: [1] -
Weiss, R. H.; Borowsky, A. D.; Seligson, D.; Lin, P.-Y.; Dillard-Telm, L.; Belldegrun, A. S.; Figlin, R. A.; Pantuck, A. D. J. Urol. 2007, 177, 63–69. doi:10.1016/j.juro.2006.08.073
Return to citation in text: [1] -
Tan, C. Y. K.; Weaver, D. F. Tetrahedron 2002, 58, 7449–7461. doi:10.1016/S0040-4020(02)00824-4
Return to citation in text: [1] [2] [3] -
Göker, H.; Kuş, C.; Boykin, D. W.; Yildiz, S.; Altanlar, N. Bioorg. Med. Chem. 2002, 10, 2589–2596. doi:10.1016/S0968-0896(02)00103-7
Return to citation in text: [1] -
Dietrich, S. A.; Lindauer, R.; Stierlin, C.; Gertsch, J.; Matesanz, R.; Notararigo, S.; Diaz, J. F.; Altmann, K.-H. Chem.–Eur. J. 2009, 15, 10144–10157. doi:10.1002/chem.200901376
Return to citation in text: [1] [2] [3] -
Cellier, M.; Fabrega, O. J.; Fazackerley, E.; James, A. L.; Orenga, S.; Perry, J. D.; Salwatura, V. L.; Stanforth, S. P. Bioorg. Med. Chem. 2011, 19, 2903–2910. doi:10.1016/j.bmc.2011.03.043
Return to citation in text: [1] [2] -
Yan, S.; Larson, G.; Wu, J. Z.; Appleby, T.; Ding, Y.; Hamatake, R.; Hong, Z.; Yao, N. Bioorg. Med. Chem. Lett. 2007, 17, 63–67. doi:10.1016/j.bmcl.2006.09.095
Return to citation in text: [1]
| 22. | Yan, S.; Larson, G.; Wu, J. Z.; Appleby, T.; Ding, Y.; Hamatake, R.; Hong, Z.; Yao, N. Bioorg. Med. Chem. Lett. 2007, 17, 63–67. doi:10.1016/j.bmcl.2006.09.095 |
| 18. | Tan, C. Y. K.; Weaver, D. F. Tetrahedron 2002, 58, 7449–7461. doi:10.1016/S0040-4020(02)00824-4 |
| 21. | Cellier, M.; Fabrega, O. J.; Fazackerley, E.; James, A. L.; Orenga, S.; Perry, J. D.; Salwatura, V. L.; Stanforth, S. P. Bioorg. Med. Chem. 2011, 19, 2903–2910. doi:10.1016/j.bmc.2011.03.043 |
| 1. | Dolle, R. E.; Le Bourdonnec, B.; Worm, K.; Morales, G. A.; Thomas, C. J.; Zhang, W. J. Comb. Chem. 2010, 12, 765–806. doi:10.1021/cc100128w |
| 20. | Dietrich, S. A.; Lindauer, R.; Stierlin, C.; Gertsch, J.; Matesanz, R.; Notararigo, S.; Diaz, J. F.; Altmann, K.-H. Chem.–Eur. J. 2009, 15, 10144–10157. doi:10.1002/chem.200901376 |
| 21. | Cellier, M.; Fabrega, O. J.; Fazackerley, E.; James, A. L.; Orenga, S.; Perry, J. D.; Salwatura, V. L.; Stanforth, S. P. Bioorg. Med. Chem. 2011, 19, 2903–2910. doi:10.1016/j.bmc.2011.03.043 |
| 13. | Weiss, R. H. Cancer Cell 2003, 4, 425–429. doi:10.1016/S1535-6108(03)00308-8 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 15. | Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069 |
| 18. | Tan, C. Y. K.; Weaver, D. F. Tetrahedron 2002, 58, 7449–7461. doi:10.1016/S0040-4020(02)00824-4 |
| 3. | Aina, O. H.; Marik, J.; Liu, R.; Lau, D. H.; Lam, K. S. Mol. Cancer Ther. 2005, 4, 806–813. doi:10.1158/1535-7163.MCT-05-0029 |
| 4. | Dixon, S.; Ziebart, K. T.; He, Z.; Jeddeloh, M.; Yoo, C. L.; Wang, X.; Lehman, A.; Lam, K. S.; Toney, M. D.; Kurth, M. J. J. Med. Chem. 2006, 49, 7413–7426. doi:10.1021/jm0609869 |
| 5. | Zhang, H.; Aina, O. H.; Lam, K. S.; de Vere White, R.; Evans, C.; Henderson, P.; Lara, P. N.; Wang, X.; Bassuk, J. A.; Pan, C.-x. Urol. Oncol.: Semin. Orig. Invest. 2012, 30, 635–645. doi:10.1016/j.urolonc.2010.06.011 |
| 6. | Lam, K. S.; Lehman, A. L.; Song, A.; Doan, N.; Enstrom, A. M.; Maxwell, J.; Liu, R. Methods Enzymol. 2003, 369, 298–322. doi:10.1016/S0076-6879(03)69017-8 |
| 7. | Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.; Knapp, R. J. Nature 1991, 354, 82–84. doi:10.1038/354082a0 |
| 8. | Liu, G.; Fan, Y.; Zhao, Z.; Lam, K. S. Zhongguo Yaowu Huaxue Zazhi 2002, 12, 311–318. |
| 9. | Miyamoto, S.; Liu, R.; Hung, S.; Wang, X.; Lam, K. S. Anal. Biochem. 2008, 374, 112–120. doi:10.1016/j.ab.2007.10.028 |
| 10. | Park, S. I.; Renil, M.; Vikstrom, B.; Amro, N.; Song, L.-w.; Xu, B.-l.; Lam, K. S. Lett. Pept. Sci. 2001, 8, 171–178. doi:10.1023/A:1016297601361 |
| 11. | Xiao, W.; Wang, Y.; Lau, E. Y.; Luo, J.; Yao, N.; Shi, C.; Meza, L.; Tseng, H.; Maeda, Y.; Kumaresan, P.; Liu, R.; Lightstone, F. C.; Takada, Y.; Lam, K. S. Mol. Cancer Ther. 2010, 9, 2714–2723. doi:10.1158/1535-7163.MCT-10-0308 |
| 12. | Dixon, S. M.; Milinkevich, K. A.; Fujii, J.; Liu, R.; Yao, N.; Lam, K. S.; Kurth, M. J. J. Comb. Chem. 2007, 9, 143–157. doi:10.1021/cc060090p |
| 20. | Dietrich, S. A.; Lindauer, R.; Stierlin, C.; Gertsch, J.; Matesanz, R.; Notararigo, S.; Diaz, J. F.; Altmann, K.-H. Chem.–Eur. J. 2009, 15, 10144–10157. doi:10.1002/chem.200901376 |
| 1. | Dolle, R. E.; Le Bourdonnec, B.; Worm, K.; Morales, G. A.; Thomas, C. J.; Zhang, W. J. Comb. Chem. 2010, 12, 765–806. doi:10.1021/cc100128w |
| 2. | Kodadek, T. Chem. Commun. 2011, 47, 9757–9763. doi:10.1039/c1cc12102b |
| 20. | Dietrich, S. A.; Lindauer, R.; Stierlin, C.; Gertsch, J.; Matesanz, R.; Notararigo, S.; Diaz, J. F.; Altmann, K.-H. Chem.–Eur. J. 2009, 15, 10144–10157. doi:10.1002/chem.200901376 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 15. | Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069 |
| 18. | Tan, C. Y. K.; Weaver, D. F. Tetrahedron 2002, 58, 7449–7461. doi:10.1016/S0040-4020(02)00824-4 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 19. | Göker, H.; Kuş, C.; Boykin, D. W.; Yildiz, S.; Altanlar, N. Bioorg. Med. Chem. 2002, 10, 2589–2596. doi:10.1016/S0968-0896(02)00103-7 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 15. | Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 15. | Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069 |
| 16. | Park, S.-H.; Park, J.-Y.; Weiss, R. H. J. Urol. 2008, 180, 352–360. doi:10.1016/j.juro.2008.02.038 |
| 17. | Weiss, R. H.; Borowsky, A. D.; Seligson, D.; Lin, P.-Y.; Dillard-Telm, L.; Belldegrun, A. S.; Figlin, R. A.; Pantuck, A. D. J. Urol. 2007, 177, 63–69. doi:10.1016/j.juro.2006.08.073 |
| 14. | Weiss, R.; Park, S.-H.; Lam, K. S.; Liu, R. Inhibitors of Cyclin Kinase Inhibitor p21. WO Patent WO2010039668A2, April 8, 2010. |
| 15. | Park, S. H.; Wang, X.; Liu, R.; Lam, K. S.; Weiss, R. H. Cancer Biol. Ther. 2008, 7, 2015–2022. doi:10.4161/cbt.7.12.7069 |
© 2013 Martin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)